

Abstract
Prostate tumors are complex entities composed of malignant cells mixed and interacting with nonmalignant cells. However, molecular analyses by standard gene expression profiling are limited because spatial information and nontumor cell types are lost in sample preparation. We scored 88 prostate specimens for relative content of tumor, benign hyperplastic epithelium, stroma, and dilated cystic glands. The proportions of these cell types were then linked in silico to gene expression levels determined by microarray analysis, revealing unique cell-specific profiles. Gene expression differences for malignant and nonmalignant epithelial cells (tumor versus benign hyperplastic epithelium) could be identified without being confounded by contributions from stroma that dominate many samples or sacrificing possible paracrine influences. Cell-specific expression of selected genes was validated by immunohistochemistry and quantitative PCR. The results provide patterns of gene expression for these three lineages with relevance to pathogenetic, diagnostic, and therapeutic considerations.
Keywords: microarray, expression profiles, linear regression, biomarkers, paracine
Prostate cancer is the most common malignancy in men and is the cause of considerable morbidity and mortality (1). There is therefore a major incentive to try to identify genes that could be reliable early diagnostic and prognostic markers and therapeutic targets. Recent studies have identified a number of genes that discriminate prostate tumor from nontumor samples (2–5). However, prostate tissue contains several cell types such as glandular epithelial cells, stromal cells, and cells of other supporting structures interspersed with neoplastic and hypertrophic epithelial cells. Gene expression differences derived from categorically labeled, tumor and nontumor, prostate samples may primarily reflect varying proportions of the nonneoplastic prostate components. Knowledge of which prostate “tumor” markers reflect true malignant versus nonmalignant cell differences as opposed to epithelial versus mesenchymal differences has been lacking. Finally, samples dissected free of nonneoplastic components are devoid of information about potentially important interactions. Here we employ a regression-based informatics approach for identification of cell-type-specific patterns of gene expression in prostate cancer. These results allow for the identification of genes that are differentially expressed in malignant versus nonmalignant prostate epithelial cells and further identify tumor-dependent changes in stromal cell gene expression.
Materials and Methods
Tissue Samples. Prostate samples were obtained from patients that were preoperatively staged as having organ-confined prostate cancer. Institutional Review Board-approved informed consent for participation in this project was obtained from all patients. Tissue samples were collected in the operating room, and specimens were immediately transported to institutional pathologists who provided fresh portions of grossly identifiable or suspected tumor tissue and separate portions of uninvolved tissues. All tissue was snap frozen upon receipt and maintained in liquid nitrogen until used for frozen section preparation at –22°C. Thirty-eight of the contributed cases contained carcinomas. An additional 50 additional samples, consisting of paired adjacent nontumor tissue and separate nontumor bearing cases, also were used, making a total of 88 specimens for analysis. Tissue for expression analysis was provided as 20-μm-thick serial cryosections sections (below).
Data Collection. Preoperative and follow-up demographic and clinical variables, histologic scoring, and DNA array data were collected into an internet accessible, secure Oracle database. Each physical object in the study was issued a unique identifier, and relationships between samples, subsamples, patients, and data were maintained. Tissue samples for expression analysis were prepared as 10- to 400-mm3 pieces, an amount that was found to be sufficient to yield 10 μg or more of total RNA. Before RNA preparation, 5-μm frozen sections were prepared at –22°C. The first section and a section every 200 μm thereafter were stained with hematoxylin and eosin for histopathological assessment, and all other intervening sections were prepared at 20-μm thickness for RNA extraction. Typically four to eight thin sections were examined per specimen by four pathologists. Preparative (20-μm) sections were lysed in RNA extraction buffer (RNeasy, Qiagen, Valencia, CA) and stored at –80°C. Thin sections were examined by four pathologists in a single session using a multihead microscope. Each pathologist assessed each specimen and completed a standardized form indicating the fraction of total area of the section occupied by the aggregate of all prostate carcinoma cells, benign prostatic hypertrophy (BPH) epithelial cells, dilated gland (dilated cystic atrophy) epithelial cells, and stromal cells. Clear spaces of glandular lumina, edema, defects, etc., were not considered, and minor proportions of neural, vascular, or other components were marked as “other” (median value, 3.1%). Average percentages of estimates from the four pathologists were calculated for epithelial cells of tumor, BPH, and cystic glands and total stromal cells for each sample.
Amplification and GeneChip Hybridization. Total cellular RNA was isolated by using RNeasy kits (Qiagen) and quantified by RiboGreen fluorescent assay (Molecular Probes), and the quality of preparation was examined by using a BioAnalyzer 2100 (Agilent Technologies). Generation of cRNA was performed according to the standard Affymetrix protocol. Fifteen micrograms of the resulting biotinylated cRNA was fragmented and hybridized U95Av2 GeneChip arrays according to the Affymetrix protocol (outlined at www.affymetrix.com).
Data Analysis. Array images (.dat files) were digitized by using mas version 5 (Affymetrix). Gene expression values were generated from the resulting raw numerical data (.cel files) by the dchip program of Li and Wong (6). Most subsequent analyses were carried out by using the r environment and language including the gee-library for generalized estimated equations (7, 8). Differential expression between dichotomous variables (tumor/no tumor) was detected by a modification of the permutation method in Efron et al. (9). Class predictive genes were identified via the nearest shrunken centroids method by using the PAM package of r software (10).
Immunohistochemistry. Selected gene expression results were validated by the direct examination of the distribution of the protein in paraffin sections of five or more of the cases. Indirect immunohistochemistry was performed, as described (for details, see supporting information, which is published on the PNAS web site). The antibodies were obtained and used as follows: directed against desmin and prostate-specific membrane antigen (PSMA) (DAKO), keratin 15 and tubulin β4 (NeoMarkers), prostaglandin-D2 synthase (Cayman Chemical), and prostate-specific antigen (PSA) (Biodesign International).
Laser Capture Microdissection (LCM). Microdissection of freshly prepared frozen sections was performed by using an Arcturus (Mountain View, CA) Mark PixCell II LCM apparatus to isolate prostate cancer epithelium, stroma, and hypertrophic benign epithelial prostate cells. Total RNA was prepared from these samples and used in quantitative RT-PCR (qPCR) to validate cell-specific expression analysis as described in detail together with the gene list, primers, graphical relationships of Affymetrix (t statistic) to LCM (LCM/qPCR endpoint) in the supporting information.
Results
Eighty-eight tissue samples from 41 subjects undergoing prostatectomy for clinically early stage localized prostate carcinoma were independently scored by a panel of four pathologists for fractional composition of the four cell types. Agreement analysis on the continuous measures of fractional cell type as estimated by four pathologists were assessed as interobserver Pearson correlation coefficients. The average coefficients for tumor, stroma, BPH, and dilated gland cells were 0.92, 0.77, 0.73, and 0.49 (for graphs and details see supporting information), respectively, indicating reproducibility of scoring for the predominant cell types. The lesser reproducibility for the dilated gland category was due to the relative paucity of this cell type in the samples (median proportion = 5%). The samples were found to contain a wide range of relative tumor cell numbers ranging, in the case of tumor cells, from a low of 0.3% to a high of 100% tumor cells (Fig. 1).
Fig. 1.

Ternary graph of sample characteristics. Eighty-eight prostatectomy samples from 41 individuals comprising 50 nontumor and 38 tumor-containing specimens were scored for proportional content of tumor, BPH, stroma, and dilated cystic glands. Vertices represent pure tissue types. Epithelia of dilated cystic glands, nerves, and vessels are small components. Note the wide range of proportions of tumor and stromal cells. Estimated tumor percentages ranged from 0.3% through 100%. The proportions were used in the linear models (xkj in Eq. 1) for cell-associated gene expression.
Despite inclusion of samples with very low tumor content, some 1,197 genes were identified as differentially expressed between tumor and nontumor samples (posterior probability >0.95, see supporting information) according to empirical Bayes estimates (9). Because tumor samples contained, on average, 53.4% cells of epithelial origin (tumor, BPH, dilated glands), and nontumor samples had an average epithelial composition of 24.7% (P = 3.5 × 10–11), we suspected that apparent differences in gene expression reflected stromal content. An illustrative subset of transcripts differentially expressed according to class was identified through nearest shrunken centroids discriminant analysis (10). Of 37 highly discriminant genes, 23 were predictive of nontumor and were mostly archetypal smooth muscle transcripts such as myosin, tropomyosin, actin, and others (supporting information). Thus, a corollary notion is that tumor markers identified through standard microarray studies may have little significance with respect to tumor-cell biology, being more reflective of fundamental differences between cells of epithelial versus mesenchymal lineage.
To assign gene expression to particular cell types within tumor specimens, a linear model was constructed in which it was assumed that the contribution to gene expression of any one cell type depends only on the proportion of that cell type and its corresponding characteristic cell-type expression level, βij, but not on the proportions of other cell types present. In Eq. 1, the average expression level Gjk of gene j in a sample k is the average of cell type expectations, βij, weighted by cell type fractions xki
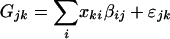 |
[1] |
Comparing “tumor versus no tumor” expression levels amounts to using two cell types whose proportions are taken as either 1 and 0 (all tumor) or 0 and 1 (no tumor) in model (Eq. 1) and taking the difference of the coefficients. A better procedure uses the proportions assessed by pathologists in a two-cell-type model. Coefficients, standard errors, and intercepts were calculated according to a two-cell type model (e.g., tumor vs. nontumor via simple linear regression of expression level on proportion of tumor cells) for each gene expression vector in 88 microarrays as a function of fractional content of tumor, then of stroma, and then of BPH. Thus, the expected cell type expression level is given as the regression coefficient, β, in the linear model (Eq. 1). Modified t statistics incorporating goodness of fit and effect size were calculated according to Tusher (11), where σβ is the standard error of the coefficient, and k is a small constant.
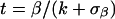 |
[2] |
For n = 88, a t statistic of 2.4 sets thresholds corresponding >4-fold expected differences in expression between the respective cell types (P < 0.02). By these criteria, many transcripts were found to have strong association with a particular cell type (Fig. 2). A global view of predicted cell-specific gene expression was obtained by hierarchical clustering of the t statistics from the linear model. A total of 3,384 transcripts displayed cell-type-associated gene expression patterns according to the criteria (Fig. 3). The procedure revealed that tumor- and nontumor-associated transcripts could be interpreted in terms of cell type specificity. Thus, 1,096 genes have strong “tumor” association, yet the majority (683) of these represent primarily differences in tumor–stroma gene expression (Fig. 3, tumor > stroma). Conversely, a large number of transcripts are predicted to be stroma associated (Fig. 3, stroma and stroma > tumor). Interestingly, a number of genes are strongly associated with BPH cell content (492). A subset of these (196) also showed a strong negative association with tumor cell content, indicating potential clinically useful markers of BPH. In addition, this analysis predicts 413 genes to be “tumor specific,” being strongly associated with tumor and displaying negative associations with both BPH and stroma (Fig. 3, tumor).
Fig. 2.

Statistical modeling. (A) Regression on cell type. The expected cell type expression levels are the coefficients β in models of gene expression as a linear function of fractional cell type (Eq. 1) and were calculated by using the lsfit function in r. Modified t statistics were calculated as t = β/(0.0029 + βse), where βse is the standard error of the coefficient. “Volcano plot” representations of the data reveal genes associated with the tumor cell type with high confidence in the upper right portion of the graph. Similar plots for the BPH and stroma cell types are omitted (see supporting information). (B) Multiple regression on percentage stroma, BPH, and tumor allows direct identification of tumor–BPH differences beyond the effect of stroma. Posterior probabilities akin to those in Efron et al. (9) used an estimating equations approach (gee library for r) (10). BPH-specific gene expression is in the upper left (note CK15), and tumor-specific gene expression is in the upper right (tubulin-β) of the graph. (C) Tumor–stroma interaction model. Inclusion of cross-product terms in the linear model identifies genes in which the contribution of a cell may be more or less than in another tissue environment; i.e., the contributions of individual cell types to the overall profile depend on the proportions of other types present. Data show tumor-stroma cross-product t statistics versus probabilities (y axis), which were calculated as in B by comparing actual with permuted t statistics. The upper left portion of graph represents a large number of stroma-associated genes with a high likelihood deviation from a strictly linear model. The right portion of the graph reveals a number of tumor-associated genes that appear to deviate from linearity, albeit with considerably less confidence. Among these is TCRγ, which is among the most discriminant tumor/no tumor genes even at low proportions of tumor; i.e., the expression of TCRγ is greater than that predicted by proportion of tumor cells alone. The stromal gene with the greatest deviation was TGF-β2, a candidate paracrine signaling molecule in prostate cancer.
Fig. 3.

Global view of cell-type-associated gene expression. The t statistics calculated from two-cell-type linear models embody the direction and magnitude as well as goodness of fit of the coefficients. t statistics were filtered to include genes with >4-fold predicted changes in between pure and 0% specific cell type sample composition and an absolute correlation coefficient of >0.25. By these criteria, 3,387 transcripts displayed cell-type-associated gene expression (see supporting information), and the t statistics are visualized here by hierarchical clustering. Red corresponds to a positive correlation, and green corresponds to a negative correlation between cell type (B, BPH; S, stroma; T, tumor) and gene expression. Representative genes from each group are at right. Previously available tumor/no tumor distinction is represented by middle labels. The analysis provides for further classification of “no tumor” markers into stromal (the vast majority) and BPH-associated genes. Likewise, “tumor” markers can be subdivided. Markers of the tumor–stroma difference may reflect epithelial mesenchymal differences in gene expression. Genes that differ according to the tumor–BPH distinction may reflect changes between malignant and nonmalignant states of prostate epithelium.
The transcript groups were characterized by distinct personalities in terms of gene function. The BPH cell-associated groups (B > S, B > T) included a number of previously identified nonmalignant prostate epithelial markers including 15-lipoxygenase-II, CD38, and p63 (12–14). This group contained a number of neuroendocrine markers such as cystatin-A, chromogranin-A, cholecystokinin, and cholecystokinin receptor (15). Notably, the BPH group of genes included IL-1β convertase. IL-1 is a putative neuroendocrine morphogen in prostate (15). The stroma cell compartment was dominated by archetypal smooth muscle and connective tissue-associated genes: vimentin, myosins, actin, and dystrophin. Other strong stroma associations included participants in transforming growth factor (TGF)-β and fibroblast growth factor signaling pathways (Fig. 3).
Transcripts with strong tumor associations that were also anticorrelated with other cell types included hepsin, macmarcks, LIM protein, and α-methyl CoA racemase, as noted (3, 5, 16, 17). Interestingly, a number of enzymes involved in O- and N-linked glycosylation were strongly tumor-specific, including UDP N-acetylglucosamine pyrophosphorylase-1, which in this study carried the third highest cell-type-associated t statistic of 7.2 (see supporting information). Also noted were several genes participating in small GTP protein signaling pathways. The set of transcripts that were associated with both tumor and BPH cell content included, not surprisingly, PSA. In fact, six separate GeneChip probe sets for this gene present on the Affymetrix arrays segregated into this group.
Specific differences between BPH and tumor cell expression are of interest diagnostically and may shed light on pathogenesis. A four-cell-type model (via multiple regression of expression level on the tissue proportions using no intercept) allows direct and unbiased estimates of differences in expression between two cell types. Simultaneous regression holding the effect of stroma constant accounts for the fact that in the prostate, cell-type-associated differences in gene expression were dominated by the inverse relationship between fractional content of tumor cells and stromal cells. Because multiple samples are used from some subjects, the estimating equations approach implemented in the gee library for r was used (8). The procedure identified a number of transcripts predicted to be specific for either BPH or tumor cells (Fig. 3B). Cytokeratin-15 (CK15) expression was predicted with high confidence to be associated with the BPH cell type. Other putative BPH epithelial cell markers included the intermediate filament protein NF-H, histone H2A1B, CD38, and 15-lipoxygenase (see Fig. 3). Transcripts predicted to be specifically expressed in tumor as opposed to BPH cells included β-tubulin, UDP N-acetyl glucosamine pyrophosphorylase 1, and SGP-28, among others.
Including a term dependent on both the tumor cell proportion and the stroma cell proportion (i.e., the cross-product xkTxkS) in Eq. 1 for the four-cell-type multiple regression model, we calculated the gene expression in stroma (or tumor) cells which is not independent but, rather, dependent on the proportion of tumor (or stroma) (Fig. 2C). Many genes displayed expression profiles with high tumor–stroma cross product terms including TGF-β2, which in the linear model is predicted to be in stroma. Also among stroma-associated genes with high cross products was desmin. Immunohistochemical staining (see below) supports this finding. High cross-product tumor-associated genes were also identified and included the T cell receptor γ (TCRγ) transcript (Affymetrix probe set 41468_at). In this instance, the high cross-product appears to be the result of TCRγ transcript being a very highly discriminant tumor marker. That is, even relatively low percentage tumor samples display high expression, an exception to the linear model consistent with stromal modulation of tumor TCRγ expression.
Immunohistochemical Validation. Selected predicted cell-type-specific gene expression patterns were tested by examining the distribution of gene expression on the protein level by using immunohistochemistry. At least five cases of tumor-bearing tissue with adjacent BPH, stroma, and dilated glands were examined with each antibody. β-Tubulin is predicted to be a strongly tumor-associated gene. Immunohistochemical staining revealed uniform expression in tumor cells (Fig. 4a, arrows) of crowded gland-like structures of the tumor but negative in stroma or epithelial cells of adjacent BPH and dilated glands. Prostaglandin-D2 synthase (PD2S) is predicted to be a moderately tumor-associated gene. Apical surfaces of the epithelial cells of tumor-gland structures were highly immunoreactive (double arrows), whereas BPH glands displayed little or no immunoreactivity (arrow) (Fig. 4b). Prostate-specific membrane antigen (PSMA) is predicted to be strongly tumor associated. Staining revealed strong immunoreactivity that was strictly confined to the apical membranes of tumor gland cells (double arrows), but only weak reactivity was observed in adjacent BPH cells (Fig. 4c, arrow). Desmin is predicted to be a stromal gene with high likelihood of tumor–stroma cell interaction. Numerous desmin-positive spindle shaped cells forming files and parallel clusters fill the stroma tissue component, whereas all epithelial cells are negative (Fig. 4d). Interestingly the stroma within zones of tumor (T) is distinct from adjacent normal (N) stroma in that the desmin-positive spindle cell population is sparse, suggesting a distinct remodeling of cells in the tumor-associated stroma. CK15 is predicted to be strongly associated with BPH. Uniform labeling of most cells of myoepithelial of hyperplastic epithelium (Fig. 4e, arrow) is apparent, whereas no expression could be detected in adjacent tumor cells of the same cases (Fig. 4e, double arrows). PSA is predicted to be present in BPH and tumor cells. Strong immunoreactivity was noted in both tumor and BPH glands (Fig. 4f, left and right, respectively). These observations provide direct confirmation of the cell-type-specific expression of proteins as predicted on the basis of the dissection of transcript expression described here.
Fig. 4.

Validation by immunohistochemistry. The cell-type-specific expression of representative proteins was examined by immunohistochemistry. (a) β-tubulin. (b) Prostaglandin-D2 synthase (PD2S). (c) Prostate-specific membrane antigen (PSMA). (d) Desmin. (e) CK15. (f) PSA. Single and double arrows indicate sites of differential expression (see text). T and N indicate tumor-infiltrated and normal stroma, respectively.
LCM–qPCR Validation. Five independent specimens and one specimen used for expression analysis were used for isolation of tumor, BPH, and stromal cells by LCM. Primer sets for 28 genes, including several genes validated by immunohistochemistry, were examined by qPCR, such as PSA, β-tubulin, desmin, and Cytokeratin-15, 504 PCR runs in all (for gene list and details, see supporting information). The overall pattern of qPCR results exhibited a clear correlation with the expression level based on cell type. To quantitatively examine the relationship, the Pearson correlation coefficient and associated probability (see supporting information) for each cell type was calculated between qPCR end points from the LCM samples, and the corresponding t statistics derived from the in silico dissection for the same cell type across the 20 genes with complete data. This analysis yielded correlation coefficients of 0.689 (P = 0.004), 0.609 (P = 0.0042), and 0.524 (P = 0.0144) for the tumor, BPH, and stroma cell types, respectively. Thus, all correlation coefficients are statistically significant (see also supporting information). It is apparent, therefore, that for all three cell types there is a significant correlation between these two independent and multistep methods of cell-type-specific analysis for the genes examined.
Discussion
Our analysis was conducted to discriminate true markers of tumor cells, BPH cells, and stromal cells of prostate cancer. Conventional least squares regression using individual cell-type proportions produces clear predictions of cell-specific expression for a large number of genes. Many of these predictions are readily accepted on the basis of prior knowledge of prostate gene expression and biology, which provide confidence in the method. These are strikingly illustrated by numerous genes predicted to be preferentially expressed by stromal cells that are characteristic of connective tissue and only poorly expressed or absent in epithelial cells.
This analysis allows segregation of molecular “tumor” and “nontumor” markers into more discrete and informative groups. Thus, genes identified as tumor-associated may be further categorized into tumor versus stroma (epithelial versus mesenchymal) and tumor versus BPH (perhaps reflecting true differences between the malignant cell and its hyperplastic counterpart). A recent meta-analysis produced a list of 500 genes up-regulated in prostate cancer (5). Of these 338 (unique Unigene identifiers) were identified in our analysis as tightly correlated with the presence of tumor (supporting information). The method presented here indicates that 157 of these “tumor-associated” transcripts represent a tumor–stroma dichotomy. Another 26 appear to be associated with BPH cells and tumor cells, and 89 are relatively unique to tumor cells. Notably, only two transcripts associated by our method with stroma were classified as tumor-associated in the meta-analysis. Conversely, 296 of 500 genes identified in the meta-analysis as indicative of “normal” prostate can be divided into 271 stromal genes and only 15 genes associated with BPH cells and not malignant cells. Thus, the vast majority of markers associated with “normal” prostate tissues in recent microarray-based studies appear to relate to cells of the stroma. This result is not surprising given that, at least here, normal samples are composed of a relatively greater proportion of stromal cells.
The strongest single discriminator between BPH cells and tumor cells in this study was CK15, a result confirmed by immunohistochemistry (Fig. 4e). CK15 has previously received little attention in this context, but BPH markers play an important role in the diagnosis of ambiguous clinical cases (18). The clinical utility of CK15 and other predicted BPH markers will require further study.
It was expected that not all genes would be expressed as a linear function of cell-type. Transcripts with high cross-products in the covariance matrix suggest that expression in one cell type was not independent of the proportion of another tissue, as would be expected in a paracrine mechanism. The stroma transcript with the highest dependence on tumor percentage was TGF-β2, a cytokine previously identified as important in prostate cell proliferation (19). Another such stroma cell gene for which immunohistochemistry was practical was desmin, which showed considerably altered staining in the tumor-associated stroma. In fact, a large number of typical stroma cell genes displayed dependence on the proportion of tumor, adding evidence to the speculation that tumor-associated stroma differs fundamentally from nonassociated stroma (20). Tumor–stroma paracrine signaling may be reflected in peritumor “halos” of altered gene expression that may be present a much bigger “target” for detection than the tumor cells alone.
In an elegant gene profiling study of prostate tumor, a group of genes was identified that correlated with Gleason score and clinical outcome (17). These studies were restricted to specimens with very high proportions of tumor cells. Therefore, in contrast to our study, Singh et al. (17) could not assess the role of cells neighboring the cancer, which may participate in the gene expression “signature” of tumor and, possibly, its biology. With increased sample numbers, it will be of interest to determine how cell-type-specific gene expression patterns, derived from routine, undissected prostate specimens, will correlate to various clinical parameters, such as Gleason score, disease relapse, and subsequent therapeutic response.
In summary, we have used a straightforward bioinformatics approach using simple and multiple linear regression to identify genes whose expression in prostate tissue is specifically correlated with either tumor cells, BPH epithelial cells or stromal cells. These results confirm a variety of previous observations and importantly identify a large number of gene candidates as specific products of various cells involved in prostate cancer pathogenesis. Context-dependent expression that is not readily attributable to single cell types is also recognized. The investigative approach described here is also applicable to a wide variety of tumor marker discovery investigations in other organs.
Supplementary Material
Acknowledgments
We thank the following participating physicians for recruiting patients, for providing tissue, and for consultation: Michael Albo, M.D., Octavio Armas, M.D., Martin Bastuba, M.D., Jonnathan Bernie, M.D., Mohammed Bidair, M.D., Michael M. Bonin, M.D., Ph.D., Don Boychuk, M.D., Stephen Bridge, M.D., Scott Brown, M.D., Naomi Buckwalter, M.D., Daniel Cosgrove, M.D., Douglas Darlin, M.D., Lamia Gabal-Shehab, M.D., Jeffrey Gaines, M.D., Franklin Gaylis, M.D., Lee Harbach, M.D., Ardath Huffaker, M.D., Warren O. Kessler, M.D., Wilfred Kearse, M.D., Huathin Khaw, M.D., Ronald MacIntyre, M.D., Lachlan Macleay, M.D., Kenneth Lasser, M.D., Sharon Mair, M.D., John Martin, M.D., Arturo Martinez, M.D., Ph.D., Edgar Masmilla, M.D., Robert Masters, M.D., James McMurtry, M.D., Wayne Muller, M.D., Peter Nanigian, M.D., Mahmood Nazari, M.D., Kenneth Nitahara, M.D., Kevin O'Brien, M.D., Julia Phillipson, M.D., Sapan Polepalle, M.D., Oleg Ratner, M.D., Ph.D., Eugene Rhee, M.D., Bruce Robbins, M.D., Howard Robin, M.D., Carol Salem, M.D., Joseph D. Schmidt, M.D., Michael Seiba, M.D., Ahmed Shabaik, M.D., Brian Shay, M.D., Ralph Shishido, M.D., Daniel Smiley, M.D., Robbin Smith, M.D., George Szollar, M.D., Evan Vapnek, M.D., Fred Walker, M.D., and Noel Weidner, M.D. We thank Connie White for Clinical Coordination activities. This work was supported by U.S. Public Health Service Grants UO1 CA84998 and CA84107 from the National Institutes of Health/National Cancer Institute. We thank the Veterans Affairs San Diego GeneChip Core Facility and the John and Rebecca Moores UCSD Cancer Center Microarray Shared Resource for assistance with the gene expression assays.
Abbreviations: TGF, transforming growth factor; TCR, T cell receptor; BPH, benign prostatic hypertrophy; qPCR, quantitative PCR; LCM, laser capture microdissection; CK15, cytokeratin-15; PSA, prostate-specific antigen.
Note Added in Proof. During the review of this manuscript, a report by Liu et al. (21) appeared, which utilizes a linear combination model akin to that described here.
References
- 1.Howe, H. L., Wingo, P. A., Thun, M. J., Ries, L. A. G., Rosenberg, H. M., Feigal, E. G. & Edwards, B. K. (2001) J. Natl. Cancer Inst. 93, 824–842. [DOI] [PubMed] [Google Scholar]
- 2.DeVita, V. T., Jr., & Bleickardt, E. W. (2001) Cancer J. 7, Supp. 1, S2–S13. [PubMed] [Google Scholar]
- 3.Dhanasekaran, S. M., Barrette, T. R., Ghosh, D., Shah, R., Varambally, S., Kurachi, K., Pienta, K. J., Rubin, M. A. & Chinnaiyan, A. M. (2001) Nature 412, 822–826. [DOI] [PubMed] [Google Scholar]
- 4.Ho, S. M. & Lau, K. M. (2002) Curr. Urol. Rep. 3, 53–60. [DOI] [PubMed] [Google Scholar]
- 5.Rhodes, D. R., Barrette, T. R., Rubin, M. A., Ghosh, D. & Chinnaiyan, A. M. (2002) Cancer Res. 62, 4427–4433. [PubMed] [Google Scholar]
- 6.Li, C. & Wong, W. H. (2001) Proc. Natl. Acad. Sci. USA 98, 31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iheka, R. & Gentleman, R. (1996) J. Comput. Graph. Stat. 5, 299–314. [Google Scholar]
- 8.Zeger, S. L. & Liang, K. Y. (1986) Biometrics 42, 121–130. [PubMed] [Google Scholar]
- 9.Efron, B., Tibshirani, R., Storey, J. D. & Tusher, V. (2001) J. Am. Stat. Assoc. 96, 1151–1160. [Google Scholar]
- 10.Tibshirani, R., Hastie, T., Narasimhan, B. & Chu, G. (2002) Proc. Natl. Acad. Sci. USA 99, 6567–6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tusher, V. G., Tibshirani, R. & Chu, G. (2001) Proc. Natl. Acad. Sci. USA 98, 5116–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Di Como, C. J., Urist, M. J., Babayan, I., Drobnjak, M., Hedvat, C. V., Teruya-Feldstein, J., Pohar, K., Hoos, A. & Cordon-Cardo, C. (2002) Clin. Cancer Res. 8, 494–501. [PubMed] [Google Scholar]
- 13.Kramer, G., Steiner, G., Fodinger, D., Fiebiger, E., Rappersberger, C., Binder, S., Hofbauer, J. & Marberger, M. (1995) J. Urol. 154, 1636–1641. [PubMed] [Google Scholar]
- 14.Nie, D., Che, M., Grignon, D., Tang, K. & Honn, K. V. (2001) Cancer Metastasis Rev. 20, 195–206. [DOI] [PubMed] [Google Scholar]
- 15.Chiao, J. W., Hsieh, T. C., Xu, W., Sklarew, R. J. & Kancherla, R. (1999) Int. J. Oncol. 15, 1033–1037. [DOI] [PubMed] [Google Scholar]
- 16.Welsh, J. B., Sapinoso, L. M., Su, A. I., Kern, S. G., Wang-Rodriguez, J., Moskaluk, C. A., Frierson, H. F., Jr., & Hampton, G. M. (2001) Cancer Res. 61, 5974–5978. [PubMed] [Google Scholar]
- 17.Singh, D., Febbo, P. G., Ross, K., Jackson, D. G., Manola, J., Ladd, C., Tamayo, P., Renshaw, A. A., D'Amico, A. V., Richie, J. P., et al. (2002) Cancer Cell 1, 203–209. [DOI] [PubMed] [Google Scholar]
- 18.Sherwood, E. R., Berg, L. A., Mitchell, N. J., McNeal, J. E., Kozlowski, J. M. & Lee, C. (1990) J. Urol. 143, 167–171. [DOI] [PubMed] [Google Scholar]
- 19.Blanchere, M., Mestayer, C., Saunier, E., Broshuis, M. & Mowszowicz, I. (2001) Prostate 46, 311–318. [DOI] [PubMed] [Google Scholar]
- 20.Tuxhorn, J. A., Ayala, G. E., Smith, M. J., Smith, V. C., Dang, T. D. & Rowley, D. R. (2002) Clin. Cancer Res. 8, 2912–2923. [PubMed] [Google Scholar]
- 21.Liu, P., Nakorchevskiy, A. & Marcotte, E. M. (2003) Proc. Natl. Acad. Sci. USA 100, 10370–10375. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.