

Abstract
Parkinson's disease (PD) is a neurodegenerative disorder characterized by loss of dopamine-containing neurons, but the molecular pathways underlying its pathogenesis remain uncertain. Here, we show that by eliminating c-Jun N-terminal kinases (JNKs) we can prevent neurodegeneration and improve motor function in an animal model of PD. First, we found that c-Jun is activated in dopaminergic neurons from PD patients and in the 1-methyl-4-phenyl-1,2,4,6-tetrahydropyridine (MPTP) mouse model of PD. Examination of various JNK-deficient mice shows that both JNK2 and JNK3, but not JNK1, are required for MPTP-induced c-Jun activation and dopaminergic cell demise. Furthermore, we have identified cyclooxygenase (COX) 2 as a molecular target of JNK activation and demonstrated that COX-2 is indispensable for MPTP-induced dopaminergic cell death. Our data revealed that JNK2- and JNK3-induced COX-2 may be a principle pathway responsible for neurodegeneration in PD.
Parkinson's disease (PD) is a common neurodegenerative disorder characterized by selective and progressive demise of dopamine-containing neurons in the midbrain (1). Although mutated genes have been identified in some cases of inherited PD (2), genetic origin could not be found in most cases, which occur sporadically. Despite extensive investigations, the cause of PD remains unknown (3). Undoubtedly, the design of effective treatments for PD depends largely on our understanding of the molecular mechanisms leading to neurodegeneration, which is still incomplete. Yet, significant hints into PD pathogenesis have been yielded by the use of 1-methyl-4-phenyl-1,2,4,6-tetrahydropyridine (MPTP), a neurotoxin that replicates most of the neuropathological hallmarks of PD in humans, nonhuman primates, and other mammalian species, including mice (4). Although the MPTP mouse model departs from human PD in a few important ways, it offers a unique means to investigate, in vivo, molecular events underlying the demise of midbrain dopaminergic neurons (4).
Several lines of evidence indicate that excitotoxic mechanisms could play a role in dopaminergic neurodegeneration in PD (5), although the molecular pathway that leads to excitotoxic-mediated cell death remains elusive. We found previously that mice lacking the neuron-specific isoform of c-Jun N-terminal kinase (JNK), JNK3, display remarkable resistance to kainic acid-induced excitotoxicity (6). The JNK group of protein kinases phosphorylates the N-terminal activation domain of the transcription factor c-Jun, thereby regulating AP-1 complex transcriptional activity (7). Three genes (Jnk1, Jnk2, and Jnk3) with distinct expression patterns encode the JNK protein kinases (7). In contrast to JNK1 and JNK2, which are ubiquitously expressed, JNK3 is largely restricted to the brain (8). In the kainic acid-induced excitotoxicity model, the protective effect of JNK3 ablation in mutant animals is associated with marked reduction of c-Jun phosphorylation and decreased activity of the AP-1 transcription factor complex (6). A similar defect is observed in mice with a germ-line mutation of the c-jun gene that replaced the JNK phosphorylation sites (9). These data suggest that efficient transcription of JNK-dependent target genes is a necessary step in progression of neuronal cell death and prompted us to investigate JNK-mediated induction of deleterious gene(s) as a part of the cascade of events leading to neurodegeneration in parkinsonian syndromes.
Materials and Methods
Chemicals and Antibodies. MPTP·HCl (Sigma) was dissolved in 0.9% NaCl. The following antibodies were used: anti-phospho-c-Jun (Ser-73) (Cell Signaling Technology, Beverly, MA), c-Jun/AP-1 (N) and inhibitor of transcription factor NF-κB (IκBα; Santa Cruz Biotechnology), cyclooxygenase (COX) 2 (BD Biosciences, San Jose, CA), COX-1 (Cayman Chemical, Ann Arbor, MI), β-tubulin (Sigma), tyrosine hydroxylase (TH; Pel-Freez Biologicals), glial fibrillary acidic protein (GFAP; DAKO), and macrophage antigen complex 1 (MAC-1; Serotec). The Neuro-Trace 500/525 green-fluorescent Nissl stain solution was purchased from Molecular Probes.
Animals and Treatment. Two- to 3-month-old male mice were used. JNK1-, JNK2-, and JNK3-deficient mice were generated as described (8). C57BL/6J, COX-2–/– (Ptgs2Tm1Jed), and p50–/– (Nfkb1tm1Bal) mice were purchased from The Jackson Laboratory. All strains were backcrossed at least six times to the C57BL/6 background. For studies using knockout mice, WT littermates were used as controls. Mice were injected i.p. four times (2-h intervals over 1 day) with either 20 mg/kg MPTP·HCl or a corresponding volume of saline alone. At the indicated time points, animals were killed and their brains were processed for further analysis. This protocol was in accordance with the National Institutes of Health guidelines for the care and use of laboratory animals and was approved by the Yale Animal Care and Use Committee.
Human Brain Postmortem Study. Autopsy brainstem tissue from control subjects and PD patients (n = 3 and 4, respectively), which were well characterized clinically and neuropathologically, were obtained from the brain bank at the Institut National de la Santé et de la Recherche Médicale U289. PD patients and control subjects did not differ significantly in their mean age at death (controls, 78 ± 7 years; PD patients, 75 ± 5 years) or their mean interval from death to freezing of tissue (controls, 21.4 ± 5.6 h; PD patients, 18.8 ± 6.2 h). Brainstem tissue was fixed and processed for immunohistochemistry as described (10).
Western Blot Analysis. Ventral midbrain and striatum were quickly dissected and lysed in whole-cell lysis buffer (0.4 M NaCl/25 mM Tris·HCl, pH 7.5/1% SDS/50 mM NaF/1 mM Na3VO4), plus protease inhibitors (Roche Diagnostics). Protein lysates (30 μg) were resolved on SDS/10% PAGE gels, electrotransferred to Immobilon-P (Millipore) membranes, and probed with antibodies to phospho-c-Jun (1:1,000 dilution), c-Jun (1:1,000 dilution), β-tubulin (1:50,000 dilution), COX-2 (1:250 dilution), COX-1 (1 μg/ml), or IκBα (1:250 dilution), followed by the appropriate horseradish peroxidase-conjugated secondary antibodies. Blots were visualized by chemiluminescence (SuperSignal; Pierce).
Immunohistochemistry. At the indicated time points, mice were fixed by transcardial perfusion of 4% paraformaldehyde/50 mM NaF/2 mM Na3VO4. Free-floating sections encompassing the entire midbrain and striatum were prepared by using a cryostat. For TH, GFAP, and MAC-1 immunostaining, tissue sections were incubated with primary antibodies overnight at 4°C. Biotin-conjugated secondary antibody incubation, avidin–biotin-conjugated peroxidase, and 3,3′-diaminobenzidine reactions were performed as described (11). Nissl counterstaining was performed by using standard procedures. Total numbers of TH- and Nissl-positive cells were counted in one hemi-brainstem by using stereological methods. For double immunofluorescent staining, sections were first incubated overnight with primary antibodies against phospho-c-Jun, followed by Cy3-conjugated secondary antibodies, and then with either NeuroTrace fluorescent Nissl stain for 20 min or primary antibodies against TH, GFAP, and MAC-1.
Measurement of Striatal Monoamine Levels. Mice were killed 7 days after MPTP or saline injections, and their striata were quickly dissected. Striatal tissue was then processed for HPLC measurement of dopamine and homovanillic acid content by means of electrochemical detection as described (11).
Measurement of Striatal 1-Methyl-4-phenylpyridinium Ion (MPP+) Levels. Mice were killed at 90 and 180 min after the last MPTP injection, and their striata were recovered and processed for HPLC using UV detection (295-nm wavelengh) to measure MPP+, as described elsewhere (11).
Rotarod Trial. The Rotarod apparatus (Ugo Basile, Trieste, Italy) consists of five rotating drums (3 cm in diameter) separated by flanges that enable five mice to be recorded simultaneously. Mice (at least 10 animals per group) were first pretrained three times (1 h apart) at 6–7 days after MPTP or saline injections by using an accelerating mode. After this conditioning period, the time on the rod (with a maximum recording time of 240 sec) was recorded for successive rotational speeds, and the overall rod performance of each animal was calculated by the trapezoidal method (12).
Gene Expression Analysis. WT and Jnk3–/– mice (three animals per group) received a single injection of kainic acid (30 mg/kg), and their hippocampi were dissected by 6 h after intoxication. Hippocampal tissue was then placed into TRIzol reagent (GIBCO/BRL) and homogenized, and total RNA was prepared according to the manufacturer's instructions. Probe synthesis and labeling, hybridization, and scanning were performed as reported (13). In this study, we used two different microarrays (Mu11K subA and subB) representing >10,000 mouse genes and expressed sequence tags. Results were analyzed by using genechip software (Version 3.1, Affymetrix, Santa Clara, CA).
COX-2 RT-PCR. Total RNA (2 μg), isolated from hippocampus by using TRIzol reagent, was reverse-transcribed by using SuperScript reverse transcriptase (GIBCO/BRL) in a total volume of 25 μl. From this reaction, 2 μl was used as a template for PCR amplification with Tsg DNA polymerase (Lamda Biotech, St. Louis). The COX-2 cDNA was amplified by using the following primers: 5′-AAAACCGTGGGGAATGTATGAGC-3′ and 5′-GATGGGTGAAGTGCTGGGCAAAG-3′. The PCR program consisted of 25 cycles at 94°C for 20 sec, 60°C for 40 sec, and 72°C for 40 sec.
Results and Discussion
c-Jun Is Activated in Dopaminergic Neurons in both PD Brains and MPTP-Intoxicated Mice. JNK mediates activation of c-Jun by means of the phosphorylation of two serine residues (Ser-63 and Ser-73) located in the N-terminal domain. To test whether MPTP-induced dopaminergic cell death is associated with JNK-induced c-Jun activation, we first evaluated expression of phosphorylated c-Jun by Western blot analysis using an antibody raised against c-Jun phosphorylated at Ser-73. MPTP intoxication causes a significant increase of phosphorylated c-Jun in the ventral midbrain, which can be observed as early as 5 h after the first intoxication (Fig. 1A). It remains elevated for 1 day and returns to its basal level at 7 days. In the striatum, an increased level of phosphorylated c-Jun was observed also, albeit it appeared earlier (3 h postinjection) and was less sustained (data not shown). These data indicate that JNK-mediated c-Jun activation in dopaminergic nerve terminals precedes that in substantia nigra pars compacta (SNpc) cell bodies. Relevant to this interpretation is the finding that MPP+ (the toxic metabolite of MPTP) accumulates first in striatal dopaminergic terminals rather than in dopaminergic soma (14). To get further insights into the cellular localization of phosphorylated c-Jun, we examined its distribution by double immunofluorescent staining performed on mesencephalic brain sections. In saline-injected animals, no phospho-c-Jun-specific labeling could be detected in nigral neurons identified by Nissl staining (Fig. 1 B, G, and L). In contrast, by 12 h after MPTP treatment, there was a strong phospho-c-Jun immunoreactivity in some nigral neurons (Fig. 1 C, H, and M). These neurons were dopaminergic, as demonstrated by double labeling with anti-TH antibodies (Fig. 1 D, I, and N). Double staining using antibodies raised against glial cell markers (GFAP and MAC-1 for astrocytes and microglial cells, respectively) showed that c-Jun was phosphorylated exclusively in dopaminergic neurons (Fig. 1 E, J, and O for GFAP and F, K, and P for MAC-1). Interestingly, there was also a dynamic change in the subcellular distribution of phosphorylated c-Jun. By 8 h, phospho-c-Jun labeling was observed exclusively in the cytosol (Fig. 1H Inset), whereas it was nuclear by 12 h (Fig. 1 M Inset and N). These data indicate that phosphorylation of cytosolic c-Jun in dopaminergic neurons triggers its translocation from the cytoplasm to the nucleus possibly by means of the stabilization of the protein (15). Hence, phospho-c-Jun accumulation in the nucleus is consistent with its role in AP-1-dependent transcriptional activity.
Fig. 1.

c-Jun is activated in dopaminergic neurons after MPTP intoxication. (A) Western blot analysis for phospho-c-Jun in the mouse mesencephalon after MPTP injections (arrow). Compared with saline-injected mice (S), the phospho-c-Jun expression level increases in a time-dependent manner after MPTP intoxication. Data represent mean ± SEM. (n = 3–5). *, P < 0.01, compared with saline-injected mice (two-tailed t test). (B–P) Double immunofluorescent staining for phospho-c-Jun (G–K), Nissl (B and C), TH (D), GFAP (E), or MAC-1 (F) on ventral midbrain tissue sections. The expression of phospho-c-Jun in the SN (arrowheads in B) was virtually absent in saline-injected mice (G) but increased greatly by 8 h after MPTP treatment (H Inset). At 12 h, phospho-c-Jun expression (H) was still elevated in neuronal cells (M) and translocated to the nucleus (M Inset is a higher magnification of the area in the dotted rectangle). Neurons displaying phospho-c-Jun immunoreactivity were positive for the dopaminergic cell-specific marker TH (N). Phospho-c-Jun staining never colocalized with the astrocytic marker GFAP (O) or the microglial cell marker MAC-1 (P). [Scale bar represents 60 μm (B, C, G, H, L, and M), 10 μm (D, I, and N), and 20 μm (H and M Insets).]
To investigate whether dopaminergic cell death in PD might be associated with similar subcellular redistribution of c-Jun, we next surveyed the expression pattern of c-Jun/AP-1 transcription factor in the mesencephalon of postmortem human brain from patients with idiopathic PD and matched control subjects. Midbrain sections were subjected to immunohistochemistry using a polyclonal antibody raised against the N-terminal domain of c-Jun. At the cellular level, c-Jun/Ap-1 staining (Fig. 2A, arrows) was observed throughout the mesencephalon in dopaminergic neurons identified by their neuromelanin content (Fig. 2 A, arrowheads). In control subjects, c-Jun/AP-1 immunoreactivity was detected exclusively in the perikarya and processes of dopaminergic neurons (Fig. 2 A), whereas nuclear staining could be observed occasionally in pigmented dopaminergic neurons from PD brains (Fig. 2 B and C, arrows). Specificity of labeling was confirmed by absorption of the antiserum with the control peptide. In these conditions, no staining was observed (data not shown). Because the phosphorylation of c-Jun induces its translocation into the nucleus, our postmortem data suggest that c-Jun/AP-1 is activated in some dopaminergic neurons in the pathology. Quantitative analysis of c-Jun-positive pigmented neurons in the SNpc demonstrated that the proportion of melanized neurons with nuclear c-Jun staining was increased significantly (Mann–Whitney U test, P < 0.005) in the PD patient group compared with control subjects (mean proportion in percentage ± SEM: 1.5 ± 0.08 and 0.02 ± 0.01 for PD patients and control subjects, respectively), suggesting that c-Jun activation may be related to PD physiopathology.
Fig. 2.

Immunohistochemical detection of c-Jun/AP-1 in transverse sections of control (A) and parkinsonian (B and C) SNpc. c-Jun immunoreactivity (arrows) was observed in perikarya and processes of dopaminergic neurons, identified by their neuromelamin content (arrowheads). (B and C) Note examples of melanized dopaminergic neuron displaying strong immunoreactivity in the nucleus (arrow), which can be distinguished clearly from neuromelanin pigments (arrowheads). [Scale bar represents 40 μm(A and B), 15 μm (B Inset), and 10 μm (C).]
JNK-Deficient Mice Are More Resistant to MPTP. Data from postmortem material and the MPTP mouse model suggest that JNK-mediated c-Jun activation might be implicated in PD-associated neurodegeneration. To test this hypothesis, we compared MPTP toxicity in mutant mice deficient in JNK1, JNK2, or JNK3 (Jnk–/–) with that of their WT littermates. In saline-injected animals, there were no differences in dopaminergic cell content between WT and individual JNK knockout mice (Fig. 3 A and B, histological data not shown for Jnk1–/–). This finding suggests that ablation of a single JNK isoform has no impact on the normal development of the nigrostriatal pathway, a result consistent with some of our investigations on other brain regions (6, 8). However, whereas MPTP induced an 80% loss of TH-positive nigral cells in WT animals, the number of dopaminergic neurons was reduced by only 50% in both Jnk2–/– and Jnk3–/– intoxicated animals (Fig. 3B). Nissl staining and counting of neurons indicate that TH-positive cell loss after MPTP treatment is due not to MPTP-induced down-regulation of the TH marker but rather to dopaminergic cell destruction (see Table 1, which is published as supporting information on the PNAS web site). In contrast, despite the fact that JNK1 is expressed in the mesencephalon (8), MPTP-treated Jnk1–/– mice displayed as much dopaminergic cell death as their WT littermates (Fig. 3B). These observations indicate that JNK2 and JNK3 are the main JNK isoforms implicated in stress-induced dopaminergic cell death in the MPTP mouse model.
Fig. 3.

Comparison of MPTP-induced nigrostriatal pathway injury in WT and Jnk–/– mice. (A) Peroxidase immunohistochemistry for TH on midbrain sections from saline- and MPTP-injected WT and JNK-deficient mice. (Scale bar represents 200 μm.) (B) Stereological counts of TH-positive cells in the SNpc at 7 days after MPTP intoxication. JNK2 or JNK3 ablation elicits sparing of TH-positive neurons after MPTP treatment, as compared with WT mice. Dual deletion of JNK2 and JNK3 increases the protective effect further. *, P < 0.01, compared with MPTP-injected WT mice (Mann–Whitney U test). (C and D) Motor performance of saline- or MPTP-treated WT and JNK-deficient mice measured on a Rotarod. The mean times on the rod recorded for increasing rod-rotation speeds (8–15 mice per group; C) and the mean overall rod performance (ORP, see Materials and Methods) for each group of mice (D) show that MPTP-intoxicated JNK-deficient animals display significant improvement of motor functions compared with MPTP-treated WT mice. *, P < 0.05, by Mann–Whitney U test.
The partial effect of Jnk2 or Jnk3 ablation opens the question whether these two JNK isoforms could compensate for each other. To investigate this issue, we generated compound mutants of JNK2/JNK3 knockouts. Unlike compound mutants JNK1/JNK2 (8), the JNK2/JNK3-null mice develop normally with no apparent nigrostriatal pathway abnormalities. Yet, the protective effect afforded by the dual deletion of the Jnk2 and Jnk3 genes was more pronounced than that found in mice carrying a deletion of either one of these JNK isoforms (only 15% of TH-positive cells were lost in Jnk2/Jnk3–/– mice; Fig. 3 A and B). It appears, therefore, that both JNK2 and JNK3 are required for MPTP-induced dopaminergic cell death in vivo. In addition to the sparing of nigral dopaminergic cell bodies, we observed that striatal dopaminergic nerve terminals in JNK-deficient mice were also more preserved than in their WT counterparts. Indeed, whereas MPTP induced a 94% reduction in striatal dopamine content in WT animals, it reached only 55% in double JNK2/JNK3 null mice (see Table 2, which is published as supporting information on the PNAS web site). In JNK2- or JNK3-deficient mice, the levels of dopamine were also markedly attenuated after MPTP treatment, albeit less than in WT animals but more than in JNK2/JNK3 compound mutants (90% and 80% reduction in Jnk2–/– and Jnk3–/–, respectively). Consistent with the dopaminergic cell-count results, MPTP-treated JNK1-deficient mice showed striatal dopamine depletion similar to that of WT animals, and therefore, there is no protective effect of JNK1 ablation in this model of PD (data not shown).
The symptomatic manifestations in PD (motor dysfunction) are due to a profound reduction in striatal dopamine content caused by the loss of dopaminergic nerve fibers in the striatum (1). Because JNK deficiency is associated with a protective effect on the nigrostriatal pathway after MPTP intoxication, we sought to determine whether JNK mutant animals have attenuated motor deficits. The locomotory ability of mice was evaluated 6–7 days after saline or MPTP treatment by using a Rotarod (12). We found that MPTP induces a profound reduction of motor performance in WT animals even at low speeds (15–25 rpm; Fig. 3C). This defect could be reversed when the animals were given l-dopa (levodopa) and benserazide (50 and 2.5 mg/kg, respectively, twice a day for 3 days), suggesting that the observed motor dysfunction is a direct consequence of nigrostriatal pathway injury (data not shown). Yet, MPTP-treated Jnk2–/– and Jnk3–/– mice show a much better ability to perform the test but only at low speeds, as evidenced by the fact that their performance dramatically dropped at rod rotations >30 rpm (Fig. 3C). Consequently, the overall rod performance score was not statistically different between MPTP-treated WT and JNK2 or JNK3 knockout animals (Fig. 3D). In sharp contrast, MPTP-intoxicated double JNK mutants perform as well as saline-injected mice regardless of the speed of the rod (Fig. 3 C and D). Thus, these results show that ablation of JNK2 and JNK3 not only protects dopaminergic neurons against MPTP-induced neurodegeneration but also improves the motor function in this animal model of PD.
MPTP Metabolism, Microglial Cell Activation, and c-Jun Phosphorylation in JNK-Deficient Mice. One of the first rate-limiting factors in MPTP toxicity is the conversion of MPTP into MPP+ by means of the monoamine oxidase B enzymatic activity (3). To confirm that resistance of JNK-deficient mice was due to ablation of Jnk genes and not to alteration of MPTP metabolism, we measured striatal levels of MPP+ after systemic injection of the toxin. JNK-deficient and WT mice exhibit similar striatal MPP+ content (see Table 3, which is published as supporting information on the PNAS web site). Thus, poor drug delivery or metabolism was not likely to account for the lower susceptibility of Jnk–/– mice to MPTP toxicity. Microglial cell activation associated with neurodegenerative processes represents another important component of MPTP toxicity (11, 16). These cells can exert deleterious effects by means of multiple pathways, including production of nitric oxide (17). It has been reported that the microglial response to MPTP arises as early as 12 h postinjection, a time point that is significantly earlier than the active phase of dopaminergic neuron degeneration (18). Moreover, recent evidence has suggested that the JNK pathway is crucial for microglial cell activation (19). It is, therefore, conceivable that targeted deletion of Jnk may protect dopaminergic neurons from MPTP toxicity indirectly by means of the inhibition of microglial cell activation. To test this hypothesis, we surveyed midbrain expression of MAC-1. Jnk–/– and WT mice exhibited a comparable level of activated microglial cells distributed throughout the SN (Fig. 4A). This finding suggests not only that Jnk–/– and WT mice were subjected to an equal noxious stress but also that JNK ablation does not interfere with microglial cell activation. Similar results were obtained regarding astrocytosis (Fig. 4A). These findings are consistent also with the observation that MPTP-induced JNK/c-Jun activation is confined strictly to neurons (Fig. 1). Thus, it is unlikely that JNK deficiency confers protection by means of the inhibition of glial cell-associated deleterious mechanisms. Yet, early (8 h) and late (24 h) MPTP-induced c-Jun phosphorylation in the mesencephalon was decreased markedly in JNK2- and JNK3-deficient mice and inhibited completely in compound JNK2/JNK3 mutants (Fig. 4B). Thus, these data demonstrate that disruption of the Jnk2 and Jnk3 genes suppressed MPTP-induced phosphorylation of c-Jun and presumably AP-1 transcriptional activity in nigral dopaminergic neurons in vivo.
Fig. 4.

Comparison of glial reaction and c-Jun activation in MPTP-intoxicated WT and JNK-knockout (KO) mice. (A) Immunohistochemical analysis for MAC-1 and GFAP indicates that MPTP-induced glial reaction is not altered in JNK-null mice as compared with WT animals. (Scale bar represents 300 μm.) (B) Western blot analysis for phospho-c-Jun expression in the mesencephalon after MPTP treatment shows that JNK2 and JNK3 are required for MPTP-induced c-Jun phosphorylation. Data represent mean ± SEM for three or four mice per group. *, P < 0.05; **, P < 0.01, compared with MPTP-treated WT mice (two-tailed t test).
COX-2 Is a Target Gene of JNK Activation. It has been suggested that, at least in certain conditions, JNK-induced neuronal cell death is mediated by sustained levels of AP-1 transcriptional activity rather than by JNK effects on nontranscriptional targets. This contention is supported by the finding that kainic acid-induced neuronal cell death in vivo required functional c-Jun phosphorylation sites (9). Efficient transcription of target genes seems, therefore, to be required for JNK-induced neuronal cell death. To identify such genes and evaluate whether they might be implicated in JNK-mediated neurodegeneration in vivo, we took advantage of the kainic acid-induced excitotoxicity model that we used previously to study and characterize the phenotype of the Jnk3–/– mice (6). We compared gene expression profiles between kainic acid-treated WT and Jnk3–/– mice by using GeneChip microarrays. We found ≈50 genes to be differentially expressed with higher expression levels in WT mice than in JNK3-deficient animals (fold increase ranging from 2.2 to 22; data not shown). Importantly, expression of the Jnk3 gene was found to be missing in Jnk3–/– mice but was present in WT animals, thus attesting that the experiment had been conducted successfully. Focusing on genes that have been implicated previously in neuronal cell death processes, we found that expression of Cox-2 was up-regulated in WT but not in Jnk3–/– mice. To confirm this finding, we monitored hippocampal Cox-2 mRNA expression by RT-PCR. As expected, kainic acid-treated WT mice exhibited a strong induction of Cox-2 mRNA, whereas Jnk3–/– showed virtually no induction (Fig. 5A), suggesting that JNK3 is essential for Cox-2 induction during excitotoxicity.
Fig. 5.

JNK signaling pathway is required for COX-2 induction after neuronal noxious stress. (A) Expression of Cox-2 mRNA in the hippocampus of kainic acid-treated mice, as assayed by RT-PCR, is abolished in Jnk3-null mice. (B) Western blot analysis for COX-2 expression in the mesencephalon after MPTP treatment in WT mice for the indicated times. *, P < 0.01, compared with saline-injected mice. (C) MPTP-induced COX-2 expression in the mesencephalon is attenuated in JNK-deficient mice. *, P < 0.05; **, P < 0.01, compared with MPTP-treated WT mice (two-tailed t test). In B and C, data represent mean ± SEM for three or four mice per group. (D) Unlike COX-2 expression, COX-1 expression is not altered after MPTP treatment.
MPTP-Induced COX-2 Expression Requires Functional JNK but Not NF-κB Pathway. COX, also known as prostaglandin H synthase, is a rate-limiting enzyme for prostanoid synthesis that is present in two main isoforms: COX-1 and COX-2. COX-1 is expressed constitutively in many cell types, whereas COX-2 expression is generally induced by cytokines and other stress-induced stimuli (20). In brain, COX-2 is present in selected neurons and its expression is up-regulated in numerous pathological conditions, including Alzheimer's disease (21). Relevant to the present study are the findings that Cox-2–/– mice are strongly resistant to excitotoxicity-induced neuronal cell death (22) and that COX-2 expression is up-regulated in PD brains (23). These data suggest that JNK-mediated COX-2 induction might represent an important step in the cascade of molecular events leading to neurodegeneration in parkinsonian syndromes. To test this hypothesis we first examined whether MPTP induces COX-2 expression in mouse brain. Western blot analysis revealed that COX-2 is barely expressed in the mesencephalon of saline-injected animals but strongly induced after MPTP challenge (Fig. 5B). In contrast, change in COX-2 expression was at no time detected in the striatum (data not shown). In the mesencephalon, induction of COX-2 expression became noticeable by 1 day, peaked at 2 to 4 days, and persisted for at least 6 days. These data indicate that JNK/c-Jun activation after MPTP injection precedes COX-2 induction by at least 20 h, a result consistent with the hypothesis that COX-2 expression depends on JNK-induced AP-1 transcriptional activity. If this hypothesis is true, COX-2 up-regulation should be attenuated in MPTP-injected JNK-deficient mice. We tested this possibility by comparing the expression level of COX-2 in WT and Jnk–/– mice killed at 2 or 4 days after MPTP treatment. COX-2 expression was moderately attenuated in single JNK knockout mice and almost abolished in compound JNK mutants (Fig. 5C). These data further support the assertion that MPTP-induced COX-2 expression requires a JNK2/JNK3-mediated signaling pathway. Furthermore, the recent finding that MPTP-induced COX-2 expression is confined strictly to dopaminergic neurons is in agreement with the present results (23). Interestingly, JNK-induced COX expression was specific to the COX-2 isoform because brain COX-1 expression was not changed after MPTP treatment and was not altered in JNK-deficient mice, as compared with WT mice (Fig. 5D).
The regulation of COX-2 expression by the JNK signaling pathway described here is consistent with several reports describing the presence of putative AP-1 sites in the Cox-2 gene promoter region (24, 25). However, other transcriptional regulatory elements also have been characterized and shown to be essential for the transcriptional induction of Cox-2 in different cell types and experimental conditions. Among these other transcriptional regulatory elements, NF-κB sites are thought to play a central role (20, 26). To investigate whether the NF-κB signaling pathway might be involved in MPTP-induced COX-2 expression, we first analyzed activation of this signaling pathway in WT mice after MPTP treatment. The degradation of IκBα, taken as an index of NF-κB activation, was analyzed by using the time points used previously to study the phosphorylation of c-Jun (Fig. 1 A). IκBα degradation was seen by 12 h after MPTP treatment, became maximal by 2 days, and was not observed further by 7 days (see Fig. 6A, which is published as supporting information on the PNAS web site). Thus, activation of the JNK/c-Jun signaling pathway slightly precedes that of NF-κB. Importantly, IκBα degradation in MPTP-treated JNK-deficient mice was similar to that in WT mice, suggesting that JNK ablation does not interfere with the NF-κB signaling pathway. We next tested whether MPTP-induced COX-2 expression was altered in mice with a defective NF-κB signaling pathway. The NF-κB transcription factor is constituted, for the most part, of the p50 (NF-κB1) and p65 (RelA) polypeptides. Because disruption of the mouse RelA locus leads to embryonic lethality, we used p50–/– mice that develop normally but display functional defects in immune responses (27). In contrast to JNK-deficient mice (Fig. 5C), COX-2 expression in p50–/– mice after MPTP was similar to that of WT mice (Fig. 6B). Importantly, analysis of histological and neurochemical parameters in MPTP-lesioned p50–/– mice showed that the magnitude of the nigrostriatal pathway injury is identical to that of WT mice (data not shown). Thus, these results indicate that COX-2 induction occurs independently of the NF-κB transcriptional pathway and that this transcription factor has no significant role in the MPTP-induced neurodegenerative process.
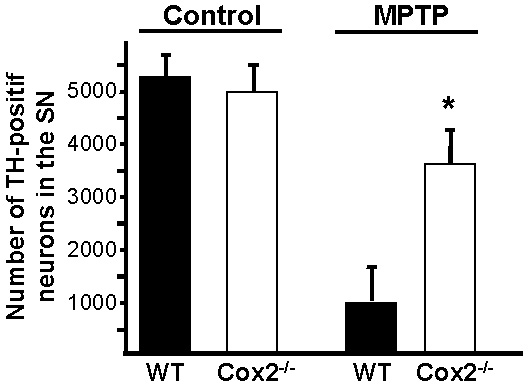
COX-2 Is Instrumental in MPTP-Induced Neurodegeneration. To evaluate the impact of COX-2 up-regulation on dopaminergic cell death, we then compared the neurotoxic effect of MPTP in Cox-2–/– mice and their WT littermates. TH-positive cell counts in the SN revealed that Cox-2–/– mice were more resistant (2.6-fold increase in TH-positive neuron survival) to MPTP-induced dopaminergic cell death than WT mice (see Fig. 7, which is published as supporting information on the PNAS web site), suggesting the importance of this JNK-dependent target molecule in neuronal cell death.
Conclusion
Our study provides genetic evidence for the regulation of COX-2 by the JNK signaling pathway in mammals in vivo. Moreover, we have shown here that JNK-mediated COX-2 transcriptional induction is essential for MPTP-induced dopaminergic cell death. Although JNK-mediated COX-2 induction appears to be necessary for MPTP-induced neurodegeneration, it remains uncertain whether it is sufficient. Indeed, we cannot rule out the possibility that other JNK deleterious molecular targets may participate in this mechanism also. Yet, we believe that this possibility might not be the case because pharmacological inhibition of JNK activation in MPTP-treated Cox-2–/– mice does not mitigate the neurodegenerative processes further (23). Importantly, our results are consistent with previous studies. However, one major advantage to our gene targeting approach, as compared with pharmacological inhibition (28) or gene transfer of the JIP1-derived JNK binding domain inhibitor (29), is the ability to investigate the role of a single JNK isoform independently of inhibition of its counterparts. This aspect is of major importance; mounting evidence indicates that different JNK isoforms have distinct biological functions and might not all be implicated in stress-induced neuronal cell death (7). Thus, our study demonstrates also that not all JNK isoforms contribute equally to stress-induced dopaminergic cell death. Indeed, although JNK2 and JNK3 mediate MPTP-associated stress response in dopaminergic neurons, JNK1 does not. Yet, this finding is consistent with recent in vitro investigations using cultured cerebellar neurons in which both JNK2 and JNK3, but not JNK1, were found to be activated selectively by stress and responsible for c-Jun activation (30). Thus, our data further support the idea that various JNK isoforms are likely to exert different functions. For instance, acute activation of JNKs after noninvasive environmental stimuli (such as physical restraint) does not result in neurodegeneration, suggesting that the JNK signaling pathway may play important physiological roles in normal neuronal function (31). These data underline, therefore, the importance of developing specific JNK inhibitors for therapeutic applications because long-term pan-JNK inhibition in the nervous system might not be desirable. Finally, postmortem examination of parkinsonian brain tissue suggests that the deleterious signaling pathway described here may be activated and may induce dopaminergic cell death in the human disease; evidence of c-Jun activation and up-regulated expression of COX-2 in dopaminergic neurons was found in PD patients (23). Altogether, our data indicate that JNK2/JNK3 and/or COX-2 represent promising molecular targets for the development of future therapeutic intervention in PD.
Supplementary Material
Acknowledgments
We thank J. Stein and Drs. Vernice Jackson-Lewis and Ali Naini for technical assistance; T. S. Zheng, C. Dong, and C.-Y. Kuan for helpful discussions; and F. Manzo for manuscript preparation. This work was supported by grants from the National Institutes of Health National Institute on Aging (to P.R.), the National Institute of Neurological Disorders and Stroke (to S.P.), the U.S. Department of Defense (to S.P. and M.V.), the Lowenstein Foundation (to S.P. and M.V.), the Lillian Goldman Charitable Trust (to S.P.), the Parkinson's Disease Foundation (to R.A.F. and S.P.), the American Parkinson's Disease Association (to M.V.), and the National Parkinson Foundation (to E.C.H. and M.V.). S.H. was a recipient of the French Foreign Office Lavoisier Program Fellowship. M.V. was a recipient of a fellowship from the Human Frontier Science Program Organization. P.T. was a recipient of Scholarship DFGTE 343/1-1 from the German Research Foundation.
Abbreviations: COX, cyclooxygenase; GFAP, glial fibrillary acidic protein; JNK, c-Jun N-terminal kinase; MAC-1, macrophage antigen complex 1; MPP+, 1-methyl-4-phenylpyridinium ion; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; PD, Parkinson's disease; SN, substantia nigra; SNpc, SN pars compacta; TH, tyrosine hydroxylase.
References
- 1.Fahn, S. & Przedborski, S. (2000). in Merritt's Neurology, ed. Rowland, L. P. (Lippincott Williams & Wilkins, New York), pp. 679–693.
- 2.Lansbury, P. T., Jr., & Brice, A. (2002) Curr. Opin. Cell Biol. 14, 653–660. [DOI] [PubMed] [Google Scholar]
- 3.Olanow, C. W. & Tatton, W. G. (1999) Annu. Rev. Neurosci. 22, 123–144. [DOI] [PubMed] [Google Scholar]
- 4.Dauer, W. & Przedborski, S. (2003) Neuron 39, 889–909. [DOI] [PubMed] [Google Scholar]
- 5.Beal, M. F. (1992) FASEB J. 6, 3338–3344. [PubMed] [Google Scholar]
- 6.Yang, D. D., Kuan, C. Y., Whitmarsh, A. J., Rincon, M., Zheng, T. S., Davis, R. J., Rakic, P. & Flavell, R. A. (1997) Nature 389, 865–870. [DOI] [PubMed] [Google Scholar]
- 7.Davis, R. J. (2000) Cell 103, 239–252. [DOI] [PubMed] [Google Scholar]
- 8.Kuan, C. Y., Yang, D. D., Samanta Roy, D. R., Davis, R. J., Rakic, P. & Flavell, R. A. (1999) Neuron 22, 667–676. [DOI] [PubMed] [Google Scholar]
- 9.Behrens, A., Sibilia, M. & Wagner, E. F. (1999) Nat. Genet. 21, 326–329. [DOI] [PubMed] [Google Scholar]
- 10.Hunot, S., Brugg, B., Ricard, D., Michel, P. P., Muriel, M. P., Ruberg, M., Faucheux, B. A., Agid, Y. & Hirsch, E. C. (1997) Proc. Natl. Acad. Sci. USA 94, 7531–7536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liberatore, G., Jackson-Lewis, V., Vukosavic, S., Mandir, A. S., Vila, M., McAuliffe, W. J., Dawson, V. L., Dawson, T. M. & Przedborski, S. (1999) Nat. Med. 5, 1403–1409. [DOI] [PubMed] [Google Scholar]
- 12.Rozas, G., López-Martín, E., Guerra, M. J. & Labandeira-García, J. L. (1998) J. Neurosci. Methods 83, 165–175. [DOI] [PubMed] [Google Scholar]
- 13.Sandberg, R., Yasuda, R., Pankratz, D. G., Carter, T. A., del Rio, J. A., Wodicka, L., Mayford, M., Lockhart, D. J. & Barlow, C. (2000) Proc. Natl. Acad. Sci. USA 97, 11038–11043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herkenham, M., Little, M. D., Bankiewicz, K., Yang, S. C., Markey, S. P. & Johannessen, J. N. (1991) Neuroscience 40, 133–158. [DOI] [PubMed] [Google Scholar]
- 15.Musti, A. M., Treier, M. & Bohmann, D. (1997) Science 275, 400–402. [DOI] [PubMed] [Google Scholar]
- 16.Kurkowska-Jastrzebska, I., Babiuch, M., Joniec, I., Przybylkowski, A., Czlonkowski, A. & Czlonkowska, A. (2002) Int. Immunopharmacol. 2, 1213–1218. [DOI] [PubMed] [Google Scholar]
- 17.Vila, M., Jackson Lewis, V., Guégan, C., Wu, D. C., Teismann, P., Choi, D.-K., Tieu, K. & Przedborski, S. (2001) Curr. Opin. Neurol. 14, 483–489. [DOI] [PubMed] [Google Scholar]
- 18.Jackson-Lewis, V., Jakowec, M., Burke, R. E. & Przedborski, S. (1995) Neurodegeneration 4, 257–269. [DOI] [PubMed] [Google Scholar]
- 19.Hidding, U., Mielke, K., Waetzig, V., Brecht, S., Hanisch, U., Behrens, A., Wagner, E. & Herdegen, T. (2002) Biochem. Pharmacol. 64, 781–788. [DOI] [PubMed] [Google Scholar]
- 20.Smith, W. L., DeWitt, D. L. & Garavito, R. M. (2000) Annu. Rev. Biochem. 69, 145–182. [DOI] [PubMed] [Google Scholar]
- 21.Pasinetti, G. M. (1998) J. Neurosci. Res. 54, 1–6. [DOI] [PubMed] [Google Scholar]
- 22.Iadecola, C., Niwa, K., Nogawa, S., Zhao, X., Nagayama, M., Araki, E., Morham, S. & Ross, M. E. (2001) Proc. Natl. Acad. Sci. USA 98, 1294–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Teismann, P., Tieu, K., Choi, D.-K., Wu, D.-C., Naini, A., Hunot, S., Vila, M., Jackson-Lewis, V. & Przedborski, S. (2003) Proc. Natl. Acad. Sci. USA 100, 5473–5478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Okada, Y., Voznesensky, O., Herschman, H., Harrison, J. & Pilbeam, C. (2000) J. Cell. Biochem. 78, 197–209. [PubMed] [Google Scholar]
- 25.Ogasawara, A., Arakawa, T., Kaneda, T., Takuma, T., Sato, T., Kaneko, H., Kumegawa, M. & Hakeda, Y. (2001) J. Biol. Chem. 276, 7048–7054. [DOI] [PubMed] [Google Scholar]
- 26.Mestre, J. R., Mackrell, P. J., Rivadeneira, D. E., Stapleton, P. P., Tanabe, T. & Daly, J. M. (2001) J. Biol. Chem. 276, 3977–3982. [DOI] [PubMed] [Google Scholar]
- 27.Sha, W. C., Liou, H. C., Tuomanen, E. I. & Baltimore, D. (1995) Cell 80, 321–330. [DOI] [PubMed] [Google Scholar]
- 28.Saporito, M. S., Brown, E. M., Miller, M. S. & Carswell, S. (1999) J. Pharmacol. Exp. Ther. 288, 421–427. [PubMed] [Google Scholar]
- 29.Xia, X. G., Harding, T., Weller, M., Bieneman, A., Uney, J. B. & Schulz, J. B. (2001) Proc. Natl. Acad. Sci. USA 98, 10433–10438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coffey, E. T., Smiciene, G., Hongisto, V., Cao, J., Brecht, S., Herdegen, T. & Courtney, M. J. (2002) J. Neurosci. 22, 4335–4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu, X., Raber, J., Yang, D., Su, B. & Mucke, L. (1997) Proc. Natl. Acad. Sci. USA 94, 12655–12660. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}