

Abstract
Nuclear factor erythroid 2 related factor 2 (Nrf2) is a transcription factor that mediates the upregulation of a battery of cytoprotective genes in response to cell stress. Recent studies have shown that Nrf2 also modulates immune responses and exhibits anti-inflammatory activity. In this report, we demonstrate that a common food preservative, tBHQ, can activate Nrf2 in T cells, as evidenced by Nrf2 binding to the antioxidant response element (ARE) and the subsequent upregulation of Nrf2 target genes. The activation of Nrf2 suppresses IFNγ production, while inducing the production of the Th2 cytokines, IL-4, IL-5, and IL-13. Nrf2 activation also suppresses T-bet DNA binding and promotes GATA-3 DNA binding. Collectively, the present studies suggest that Nrf2 activation skews CD4+ T cells toward Th2 differentiation and thus represents a novel regulatory mechanism in CD4+ T cells. Further studies will be needed to determine whether the commercial use of Nrf2 activators as food preservatives promotes food allergies in humans.
Introduction
Th1/Th2 differentiation of CD4+ T cells is a critical process in tailoring an adaptive immune response to a specific pathogen, which allows for flexibility in T cell function and downstream immune activity. However, there is also an inherent potential danger in pathological responses due to Th1/Th2 imbalance. Th1 responses are vital in cell-mediated immunity, which is important in host defense against numerous bacterial and viral pathogens (1). Conversely, Th2 responses are thought to be important in host defense against larger parasites, such as helminths. Th2 responses also play a major pathological role in the development of allergy and asthma (1, 2). Although the incidence of allergy and asthma has sharply increased in the U.S. and worldwide over the last several decades, the underlying cause is currently unknown (3–5).
Nuclear factor erythroid 2-related factor 2 (Nrf2) is a basic leucine zipper transcription factor that is activated by cell stress from various sources, including oxidative as well as electrophilic stimuli (6, 7). In the absence of oxidants and electrophiles, Nrf2 is sequestered in the cytosol by its repressor protein, Kelch ECH associating protein 1 (Keap1), and is subsequently ubiquitinated and degraded (8, 9). During cell stress, ubiquitination of Nrf2 by Keap1 is disrupted, allowing Nrf2 to translocate to the nucleus, and up-regulate genes containing an antioxidant response element (ARE) in their promoter regions (10–12). Nrf2-regulated genes facilitate a variety of functions, including, but not limited to, antioxidative activity, detoxification and transport of xenobiotics, as well as glutathione homeostasis (13–15). In immune cells, Nrf2 has been reported to up-regulate numerous genes such as heme oxygenase-1 (Hmox1), NAD(P)H quinone oxidoreductase (Nqo1), and γ-glutamylcysteine ligase catalytic subunit (Gclc). Therefore, these genes are useful markers of Nrf2 activation in leukocytes (16, 17).
The synthetic antioxidants, tert-butylhydroquinone (tBHQ) and butylated hydroxyanisole (BHA), are widely used experimentally as Nrf2 activators. Specifically, it has been shown that tBHQ modifies the thiol groups of cysteine (Cys)-273 and Cys-288 on the Keap1 protein, which ultimately inhibits the repression of Nrf2 by Keap1 and facilitates Nrf2 signaling (18). With respect to commercial use, both tBHQ and BHA are food preservatives used to stabilize oils and fats against oxidative deterioration (19). While banned for use in food in Japan and other countries, tBHQ and BHA are currently used as preservatives in numerous types of food in the U.S., including cereals, crackers, potato chips, noodles, grains, vegetable oils, margarine, and others.
Recent studies have demonstrated anti-inflammatory effects of Nrf2 activation and conversely, the pro-inflammatory effects of Nrf2 deletion in a number of different models. For instance, Nrf2-null mice have been shown to have increased mortality in models of sepsis (20), more severe pathology in EAE (21), increased production of inflammatory mediators in LPS-induced neuroinflammation (22) and worsened injury in a model of T cell-mediated hepatitis (23). With respect to lung inflammation, Nrf2-null mice have increased susceptibility to ovalbumin-induced asthma (24), allergic airway inflammation (25), and hyperoxia-induced acute lung injury (26). Accordingly, there has been considerable interest in developing anti-inflammatory therapeutics targeting Nrf2 (16).
Further evidence for an important role of Nrf2 in immune regulation is derived from the development of autoimmune disease in Nrf2-null mice. Several papers have characterized the development of multi-organ autoimmune inflammation in Nrf2-null mice, which resembles systemic lupus erythematosus (SLE) in humans (14, 27, 28). Like SLE in humans, the autoimmune disease in Nrf2-null mice is found predominantly in adult females, progresses with age, is characterized by the production of antibodies against dsDNA and ultimately results in glomerulonephritis. Moreover, it has been reported that in addition to the SLE-like disease, 14-month-old Nrf2-null female mice develop hemolytic anemia, which often occurs in human SLE patients (29). Notably, a recently-identified NRF2 polymorphism has been shown to be associated with autoimmune nephritis in female SLE patients (30).
In addition to its general anti-inflammatory effects, a number of studies suggest that Nrf2 activation may also alter Th1/Th2 balance specifically. Expression of IL-4 and IL-13 is elevated in lungs of Nrf2-null mice in models of asthma and pulmonary fibrosis (24, 31). In addition, Nrf2-deficient lung dendritic cells (DC) have been shown to have heightened oxidative stress that confers a Th2-like immunoresponsiveness upon stimulation with ambient particulate matter (32). With respect to Th1 responses, activation of Nrf2 by sulforaphane has been shown to restore age-related decrease of DC response in a model of contact hypersensitivity (33). Collectively, these studies initiated the investigation of Nrf2 in Th1/Th2 responses and served as the basis for the current studies.
The present studies investigated the role of Nrf2 in T cell function and differentiation. These studies indicate that Nrf2 is activated by tBHQ in T cells, as evidenced by the binding of Nrf2 to its response element, ARE, and subsequent up-regulation of Nrf2 target genes. The current studies also demonstrate that Nrf2 activation inhibits Th1 cytokine production and T-bet activity, while concurrently promoting Th2 cytokine production and GATA-3 binding activity. In addition, the Nrf2 activator, tBHQ, induced Il4-regulated firefly luciferase (Fluc) activity and Il13-regulated renilla luciferase (Rluc) activity in CD4+ T cells isolated from Il4-Fluc/Il13-Rluc transgenic mice, suggesting that Nrf2 promotes transcriptional activity in both the Il4 and Il13 genes.
Materials and Methods
Materials
BHA, tBHQ, and all other reagents were purchased from Sigma Aldrich (St. Louis, MO) unless otherwise noted.
Nrf2-null mice
Nrf2-null mice on a mixed C57BL/6 and AKR background were generated as described previously and received from Dr. Jefferson Chan (6). The mice were subsequently back-crossed 8 generations onto the C57BL/6 background and are 99% congenic (analysis performed by Jackson Laboratories, Bar Harbor, ME). Female mice, 8–14 weeks of age, were used for the current studies. Wild-type female C57BL/6 mice, 8 weeks of age, were purchased from Charles River Laboratories (Wilmington, MA). Mice were given food and water ad libitum. All animal studies were conducted in accordance with the Guide for the Care and Use of Animals as adopted by the National Institutes of Health, and were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Kansas Medical Center.
T cell isolation
Single-cell suspensions from spleens and lymph nodes were washed and filtered, after which CD3+ or CD4+ T cells were isolated with magnetic beads by negative selection following the manufacturer’s instructions (Miltenyi Biotec, Auburn, CA). The total number of cells isolated varied, depending on the specific experiment. Cellular viability was >90% as determined by trypan blue exclusion. The purity of the isolated cell populations was >90% as determined by flow cytometric analyses.
Cell culture
Cells were cultured in DMEM (with L-glutamine, sodium bicarbonate and D-glucose) supplemented with 100 units penicillin/ml, 100 units streptomycin/ml, 50 μM 2-mercaptoethanol, and 10% fetal calf serum. For most studies, cells were treated with either tBHQ or BHA at various concentrations for 30 min prior to T cell activation. The duration of T cell activation ranged from 6 h (mRNA quantification of Nrf2 target genes) to 96 h or more (cytokine analysis), depending on the specific endpoint measured (noted in the figure legends). T cells were activated with purified hamster anti-mouse CD3e (500A2, 1.5 μg/ml), purified hamster anti-mouse CD28 (37.51, 1.5 μg/ml), and an F(ab′)2 fragment specific for anti-Syrian hamster IgG that was used to cross-link CD3 and CD28 to enhance activation. Anti-CD3 and anti-CD28 were purchased from BD Biosciences (San Jose, CA), and the F(ab′)2 cross-linker was purchased from Jackson ImmunoResearch Laboratories (West Grove, PA).
mRNA quantification by branched-DNA (bDNA) Assay
For studies not restricted by limited amounts of total RNA, bDNA analysis was performed for detection of mRNA. Cells were treated as described, after which they were lysed with diluted lysis buffer. The cell lysis and bDNA assays were performed using a commercially-available kit (Quantigene® 1.0, Panomics, Fremont, CA) following the manufacturer’s protocol as described previously (34). The specific oligonucleotide probe sets used for the quantification of Ifng are shown in Supplemental data (Supp. Table 1). The probe sets were designed using ProbeDesigner software (Bayer Corp., Diagnostics Div., Tarrytown, NY) as described previously (35). Probe sets were synthesized by Integrated DNA Technologies (Coralville, IA).
Cytokine protein quantification: IFNγ ELISA
For single-plex analysis, IFNγ protein was quantified by a sandwich ELISA method using a commercially-available kit following the manufacturer’s protocol (E-Bioscience, San Diego, CA).
mRNA quantification. Real-time PCR
For studies for which limited total RNA was available, real-time PCR was used for mRNA quantification. Total RNA was isolated using TRI reagent® (Sigma Aldrich, St. Louis, MO), following the manufacturer’s protocol. The isolated RNA was then reverse-transcribed after which relative expression levels of the target genes were determined by Sybr Green real-time PCR. Relative mRNA expression for the target genes were normalized to ribosomal protein L13a and calculated using the DDCt method as previously described (36). Primers for rpL13a were designed using Primer3 software (http://primer3.sourceforge.net), were synthesized by Integrated DNA Technologies (Coralville, IA), and are as follows: rpL13a forward primer, 5′-ACAAGAAAAAGCGGATGGTG-3′ and rpL13a reverse primer, 5′-TTCTCCTCCAGAGTGGCTGT-3′. The primers for Nfe2l2 (Nrf2), Hmox1, Nqo1, and Gclc have been published (28, 37, 38).
Nuclear protein isolation
Nuclear protein was isolated using two different commercially available kits, depending on the downstream application. For transcription factor binding ELISA, nuclear protein was isolated using the Nuclear Extract kit from Active Motif (Carlsbad, CA), following the manufacturer’s protocol. For transcription factor array, nuclear protein was isolated using the Procarta TF Nuclear Extraction kit (Panomics, Fremont, CA). Protein concentrations were determined using the BioRad protein assay (BioRad, Hercules, CA), which is based on the Bradford method of protein quantification.
ELISA-based DNA/protein binding assay
Nrf2 (ARE), T-bet, and GATA-3 binding activities were quantified using TransAM™ assay kits from Active Motif (Carlsbad, CA), following the manufacturer’s protocol. The wild-type oligonucleotide (provided by the manufacturer) is an unlabelled free consensus oligonucleotide that competes with the plate-bound oligonucleotide to confirm specificity. In addition to the wild-type oligonucleotide, a mutated oligonucleotide was also used as an additional control. The Cos-7 (Nrf2-transfected), Cos-7 (T-bet-transfected), and Jurkat nuclear extracts served as positive controls for the Nrf2, T-bet, and GATA-3 binding assays, respectively.
Cytokine protein quantification: Multiplex suspension array
For multiplex cytokine analysis, cytokine concentrations were determined using a MilliPlex™ mouse cytokine kit for the detection of IFNγ, TNFα, IL-4, IL-5, IL-13 and IL-10 following the manufacturer’s protocol (Millipore, Billerica, MA). The cytokine concentrations were quantified using a Bio-Plex™ 200 System (Bio-Rad Laboratories, Hercules, CA). PE and xMAP bead fluorescence was detected by a dual-laser detector (532 and 635nm) and quantified by Bio-Plex Manager 5.0 software (Bio-Rad Laboratories, Hercules, CA).
Generation of the Il4-Fluc/Il13-Rluc transgenic mouse
A 184 Kb BAC clone (RP24-259D13) that includes both the Il4 and Il13 genes from chromosome 5 was selected from a BAC library and used to generate the Il4-Fluc/Il13-Rluc transgenic mouse (Manuscript in preparation). In addition to the Il4 and Il13 genes, the BAC clone also includes the Il5 gene as well as the proximal regulatory regions of Il5 and thus includes the majority of the Th2 locus (it is missing distal regulatory regions for the Il5 gene, however). The first exon of the Il4 gene was replaced with Fluc and the first exon of the Il13 gene was replaced with Rluc by homologous recombination. The purified BAC DNA was then introduced into fertilized murine oocytes by pronuclear microinjection. The fertilized oocytes were then transferred into pseudopregnant foster mothers. The copy number of the transgene in the pups was determined by quantitative real-time PCR as previously described (39). A detailed description of the generation of the Il4-Fluc/Il13-Rluc Tg mice is currently being prepared for publication.
Statistical Analysis
The mean ± standard error was determined for each treatment group in the individual experiments. Homogeneous data were evaluated by one-way or two-way parametric analysis of variance. When significant differences were observed, either Dunnett’s (for one-way ANOVA) or the Holm-Sidak (for two-way ANOVA) post-hoc test was used to compare treatment groups to the vehicle (VH) control using SigmaStat 3.01a software from Systat Software, Inc. (Chicago, IL).
Results
Inhibition of IFNγ production by the Nrf2 activator, tBHQ
Although Nrf2 has been described as anti-inflammatory in various in vivo models of inflammation, the role of Nrf2 in T cells remains largely uncharacterized. Accordingly, the effects of Nrf2 activation on T cell activity were investigated. In our initial studies, a mixed splenocyte population was used in combination with a T cell-specific activator, anti-CD3/anti-CD28. A marked concentration-dependent inhibition of IFNγ production by the Nrf2 activator, tBHQ, was observed in CD3/CD28-activated splenocytes (Fig. 1a). It is notable that even low concentrations of tBHQ (0.01 μM) substantially decreased IFNγ secretion (58%), whereas higher concentrations completely abolished IFNγ production. Treatment with another Nrf2 activator, BHA, also significantly reduced IFNγ production by T cells, causing a marked decrease in IFNγ secretion at 1 μM (data not shown). Neither tBHQ nor BHA significantly affected viability (data not shown). Consistent with its effect on protein secretion, tBHQ also inhibited IFNγ mRNA expression in wild-type splenocytes, whereas splenocytes from Nrf2-null mice were relatively refractory to this effect (Fig. 1b). In addition, markedly higher mRNA expression of IFNγ was observed in anti-CD3/anti-CD28-activated splenocytes derived from Nrf2-null mice relative to wild-type (Fig. 1c), suggesting Nrf2 plays an endogenous role in the regulation of IFNγ. There was no difference in the ratios of CD4+, CD8+, CD19+, CD4+CD25+CD62LhiCD44lo (naïve), CD4+CD25+CD62L loCD44hi (effector memory) or CD4+CD25− populations (Supp. Figs. 1, 2). These studies demonstrate that activation of Nrf2 can inhibit IFNγ production by activated T cells and that Nrf2 plays a critical role in the endogenous regulation of IFNγ.
Fig. 1.

Nrf2 inhibits IFNγ mRNA expression and protein secretion in splenocytes activated with anti-CD3/anti-CD28. Freshly-isolated splenocytes were pretreated with vehicle (VH) or tBHQ for 30 min prior to activation with anti-CD3/anti-CD28. A.) The cell supernatants were then collected after 24 h and cytokines analyzed. B.) Total RNA was isolated 6 h after activation and IFNγ mRNA expression quantified by real-time PCR. C.) Fold difference in IFNγ mRNA levels between wild-type and Nrf2-null splenocytes, 6 h after activation. * denotes p<0.05 as compared to wild-type control. † denotes p<0.05 as compared to Nrf2-null control. ‡ represents a significant difference (p<0.05) between the wild-type and Nrf2-null genotypes.
Up-regulation of Nrf2 and its target genes in CD3+ cells by the Nrf2 activator, tBHQ
Because activation of Nrf2 inhibited IFNγ production by anti-CD3/anti-CD28-activated splenocytes (Fig. 1), the expression of Nrf2 and its ability to be activated was assessed in the pan T cell population. Similar to other cell types, Nrf2 expression in T cells is up-regulated after treatment with the Nrf2 activator, tBHQ (40), suggesting that Nrf2 upregulates its own expression (Fig. 2a). In addition, treatment of wild-type T cells with tBHQ resulted in increased binding of Nrf2 to the antioxidant response element (ARE), whereas decreased binding of Nrf2 to the ARE was observed in T cells derived from Nrf2-null mice (Fig. 2b). The increased binding of Nrf2 to the ARE correlated with increased mRNA expression of the Nrf2-target genes, Hmox1, Nqo1, and Gclc in tBHQ-treated wild-type T cells. In contrast, treatment of Nrf2-null T cells with tBHQ did not up-regulate Nqo1 or Gclc and only modestly induced Hmox1 expression (Fig. 2c–e). Collectively, these data indicate that Nrf2 is expressed, inducible, and capable of gene transactivation in wild-type T cells as evidenced by the detection and induction of Nrf2 mRNA levels, binding of Nrf2 to the ARE, as well as the Nrf2-dependent up-regulation of the target genes Hmox1, Nqo1, and Gclc.
Fig. 2.

Activation of Nrf2 by tBHQ in isolated T cells. Magnetically isolated CD3+ T cells were left untreated (BKG) or pretreated with tBHQ or vehicle (VH) for 30 min prior to activation with anti-CD3 and anti-CD28. Cells were cultured for either 6 h (mRNA analysis, panels A and C–E) or 4 h (ARE binding, panel B). A) Nrf2 mRNA was detected by real-time PCR from total RNA. B) ARE binding was determined from nuclear protein extracts and quantified spectrophotometrically by measuring absorbance at 450 nm. C–E) Nqo1, Hmox1, and Gclc mRNA expression was detected by real-time PCR from total RNA. * denotes p<0.05 as compared to VH. ‡ denotes p<0.05 as compared to Nrf2-null T cells.
Nrf2 activation skews CD4+ T cells toward Th2 differentiation
The early effects of tBHQ on IFNγ production prompted an investigation into the effects of Nrf2 activation in CD4+ T cell polarization. Similar to the early effects shown in Fig. 1, treatment of CD4+ T cells with tBHQ over 96 h resulted in inhibition of IFNγ secretion, whereas substantially increased IFNγ production was observed in CD4+ T cells from Nrf2-null mice (Fig. 3a). In correlation, tBHQ treatment increased IL-4 secretion in wild-type CD4+ T cells, whereas decreased IL-4 production was observed in CD4+ T cells from Nrf2-null mice (Fig. 3b). Treatment with tBHQ also increased IL-5 and IL-13 secretion by CD4+ T cells, and accordingly, the absence of the Nrf2 gene resulted in decreased IL-5 and IL-13 production (Fig. 3c, 3d). The decreased IFNγ production and increased IL-4, IL-5, and IL-13 production by tBHQ were also observed in CD4+ T cells from AhRd mice (which have a defective AhR, Ah receptor), suggesting the effects are independent of AhR (Supp. Fig. 3). There was little difference in TNFα and IL-10 secretion between the Nrf2-null and wild-type genotypes, suggesting that the effects of Nrf2 activation are relatively selective for Th1/Th2 cytokines (Fig. 3e, 3f). Furthermore, the effects of Nrf2 activation at 96 h were also observed in restimulated CD4+ T cells in which wild-type CD4+ T cells treated with tBHQ exhibited decreased IFNγ production and increased IL-4, IL-5, and IL-13 secretion (Fig. 4). Conversely, IL-4, IL-5, and IL-13 levels were low in restimulated CD4+ T cells from Nrf2-null mice. Collectively, the data suggest that activation of Nrf2 promotes Th2 differentiation of CD4+ T cells, while inhibiting Th1 differentiation.
Fig. 3.

Effect of Nrf2 activation on cytokine production in activated CD4+ cells. Magnetically-isolated CD4+ cells from Nrf2-null and wild-type mice were pretreated with vehicle (VH) or tBHQ for 30 min prior to activation with anti-CD3/anti-CD28. The cells were then cultured for 96 h after which the cell supernatants were harvested. Cytokine concentrations in the supernatants were quantified by multiplex suspension assay. * denotes p<0.05 as compared to wild-type control. ‡ represents a significant difference (p<0.05) between the wild-type and Nrf2-null genotypes.
Fig. 4.

Effect of Nrf2 activation on cytokine production in restimulated CD4+ cells. Magnetically-isolated CD4+ cells were treated as in Fig. 3 and then cultured for 5 d after which the cells were harvested, resuspended and restimulated. The supernatants were harvested 24 h after restimulation and cytokines quantified. * denotes p<0.05 as compared to wild-type control. ‡ represents a significant difference (p<0.05) between the wild-type and Nrf2-null genotypes.
Nrf2 activation potentiates GATA-3 DNA binding, while concurrently suppressing T-bet DNA binding
Because cytokine analyses suggest that tBHQ promotes Th2 differentiation, the effect of Nrf2 activation on transcription factors involved in Th1/Th2 differentiation (T-bet and GATA-3) was assessed. Consistent with the cytokine analyses, activation of Nrf2 suppressed binding of T-bet to its consensus sequence and concurrently potentiated GATA-3 binding to its consensus sequence (Fig. 5). Furthermore, CD4+ T cells from Nrf2-null mice exhibited significantly increased T-bet DNA binding and suppressed GATA-3 DNA binding. Collectively, these studies provide further evidence that Nrf2 positively regulates Th2 differentiation, which is associated with enhanced GATA-3 binding activity.
Fig. 5.

Effect of Nrf2 activation on T-bet and GATA-3 binding activity in restimulated CD4+ cells. Magnetically-isolated CD4+ cells were treated and restimulated as in Fig. 4. The cells were harvested 24 h after restimulation. Nuclear protein was then isolated and T-bet/GATA-3 binding activity quantified. * denotes p<0.05 as compared to wild-type control. ‡ represents a significant difference (p<0.05) between the wild-type and Nrf2-null genotypes.
tBHQ drives IL-4-regulated and IL-13-regulated luciferase activity in a transgenic mouse model
Recently, we engineered a transgenic mouse model with which to study the molecular events that occur during Th2 differentiation (manuscript in preparation). A 184 kb BAC clone (RP24-259D13) from mouse chromosome 5 with Il4 and Il13 at center and flanked by kif3a and rad50 genes was used to generate BAC transgenic mice. The BAC clone was modified by excising the first exon of the Il4 gene and replacing it with the firefly luciferase (Fluc) gene. In addition, the first exon of the Il13 gene was replaced with the renilla luciferase (Rluc) gene. The modified Th2 locus was then inserted into the mouse genome to create the transgenic mouse. This unique model allows for focused investigation into the molecular modifications that occur in the Th2 locus during CD4+ T cell differentiation. Treatment of CD4+ T cells from this transgenic mouse with tBHQ caused a robust increase in both firefly and renilla luciferase activities, suggesting increased transcriptional activity in both the Il4 and Il13 genes (Fig. 6).
Fig. 6.
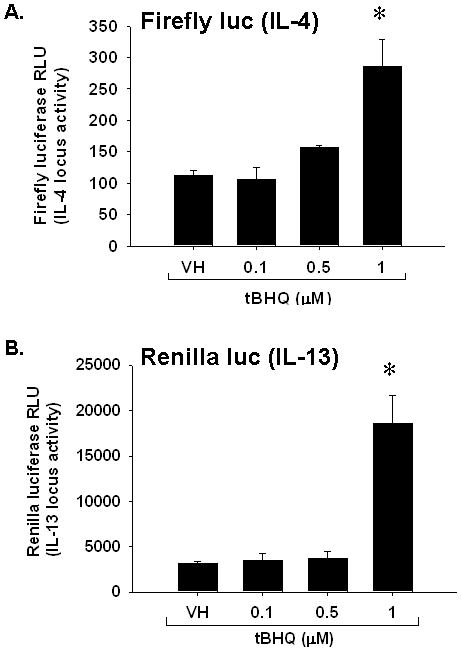
Effect of Nrf2 activation by tBHQ on il4/il13-regulated luciferase activity in restimulated CD4+ cells. CD4+ cells were isolated from Il4-Fluc/Il13-Rluc transgenic mice after which the cells were treated and restimulated as in Fig. 5. The cells were harvested 24 h after restimulation, lysed, substrate added and chemiluminescence quantified. Quantification of A.) il4-regulated firefly luciferase activity and B.) il13-regulated renilla luciferase activity. * denotes p<0.05 as compared to VH control.
Discussion
Previously published studies have demonstrated that Nrf2 plays a significant immunomodulatory role in a number of different models of inflammation (20, 22, 24). Although many of these studies have suggested that Nrf2 can affect various aspects of T cell biology, its specific role in T cell function is currently incompletely understood (17, 21, 23, 24, 33). Accordingly, the purpose of the present study was to determine the role of Nrf2 in peripheral T cell differentiation.
Our data demonstrate that Nrf2 is expressed and is able to be activated by tBHQ in T cells, as determined by binding of Nrf2 to the ARE and induction of Nrf2 target genes. Importantly, these studies indicate that activation of Nrf2 inhibits production of the Th1 cytokine, IFNγ, while concurrently promoting the secretion of Th2 cytokines by wild-type CD4+ T cells during both primary and secondary activation. Conversely, CD4+ T cells from Nrf2-null mice produce increased IFNγ and decreased IL-4, IL-5, and IL-13 than wild-type T cells. In addition, the present studies demonstrate that tBHQ suppresses T-bet DNA binding and enhances GATA-3 DNA binding in wild-type, but not Nrf2-null, CD4+ T cells. In Il4/Il13-regulated luciferase transgenic mice, tBHQ treatment induced firefly luciferase, regulated by the Il4 gene, as well as renilla luciferase, regulated by the Il13 gene. Collectively, these studies indicate that Nrf2 activation in CD4+ T cells results in Th2 skewing, whereas lack of functional Nrf2 results in Th1 skewing. Overall, these studies suggest that the Nrf2 signaling pathway represents a novel regulatory mechanism in Th1/Th2 differentiation that warrants further investigation.
It is interesting to note that Nrf2-null female mice develop an autoimmune disease that resembles SLE in humans (27, 28). The autoimmunity in the Nrf2-null mice is characterized by production of autoantibodies against dsDNA, deposition of IgM, IgG, and C3 in the glomeruli and the development of glomerulonephritis. Although autoantibody production in SLE may suggest a Th2 response, there is considerable evidence to indicate that IFNγ contributes to the pathology of lupus nephritis in both humans and mice (41–48). Neither IFNγ−/− MRL/lpr nor IFNγ+/− MRL/lpr mice develop the overt renal disease that is typically associated with the MRL/lpr model of SLE (41). In addition, SLE patients with diffuse proliferative lupus nephritis (DPLN) have increased numbers of IFNγ-producing cells (42, 43). Increased IFNγ sera concentrations have also been reported in SLE patients (44, 45). A higher incidence of snp mutations in both IFNγR1 and IFNγR2 have been found in SLE patients (46). Importantly, patients who were treated with IFNγ for rheumatoid arthritis or myeloproliferative disorder developed a severe SLE-like disease, suggesting that IFNγ plays a causative role in SLE in some patients (47, 48). Consistent with the aforementioned studies, our data demonstrate that CD4+ T cells derived from the Nrf2-null mice, which develop an SLE-like autoimmunity, produce markedly increased amounts of IFNγ compared to wild-type.
The food additive, tBHQ, belongs to a family of phenolic antioxidant preservatives, which also includes BHA and butylated hydroxytoluene (BHT), all of which are added to a variety of foods to avert and/or slow spoilage. Because there has been considerable concern that many of the phenolic antioxidants may cause cancer, the primary toxicological focus has been the carcinogenicity of these chemicals (19). In this regard, the phenolic antioxidants have been widely studied and appear to be non-carcinogenic to humans. In contrast, the immunotoxicity of the phenolic antioxidants is not well characterized. It has been reported that BHA inhibits IL-2 production by T cells (49), however the current studies are the first to report the mechanism by which this may occur. This is the first report of the effects of tBHQ on T cell function and differentiation.
The effects of the food preservatives, tBHQ and BHA, on CD4+ T cell function occur at concentrations that are of physiological relevance to humans. Concentrations of tBHQ as low as 10 nM produce significant inhibition of IFNγ secretion, whereas BHA inhibits IFNγ at 1 μM (Fig. 1 and data not shown). Notably, human volunteers administered tBHQ orally (100 – 150 mg) had serum concentrations of tBHQ ranging from 24 – 222.5 μM, concentrations at least 2 – 20 fold higher than those used in the current study (50). Thus, the modulation of Th1/Th2 differentiation by tBHQ occurs at concentrations to which humans are likely exposed. Given that humans are exposed to tBHQ by ingestion, and the present studies suggest that tBHQ can enhance production of Th2 cytokines by CD4+ T cells, it seems possible that ingestion of tBHQ and potentially other phenolic antioxidants may create an environment in the gut that is permissive to food allergy or other Th2-skewed imbalances. Further studies will be needed to investigate the role of tBHQ in oral tolerance and atopic responses.
Inhibition of IFNγ transcription by tBHQ appears to be largely Nrf2-dependent. However, at higher concentrations, tBHQ can also suppress IFNγ production in a Nrf2-independent fashion (Fig. 1b). This suppressive activity may be due to impaired calcium influx, as has been described to occur with di-tBHQ, a structurally-related congener of tBHQ (51).
The current studies suggest that activation of Nrf2 in CD3/CD28-stimulated CD4+ T cells skews differentiation toward a Th2 response. This effect was unexpected as previous studies suggest that Nrf2 may serve to limit or regulate Th2 responses in models of asthma (24, 32, 52). The reason for the differential effects of Nrf2 on Th2 cytokine responses is currently unclear, but may due to a number of different factors. The current studies report the effects of Nrf2 activation on isolated CD3/CD28-stimulated CD4+ T cells, whereas asthma and experimental models of asthma are complex with numerous cell types involved. In addition, the environment of the lung itself appears to be particularly conducive to Th2 responses, whereas the present studies are likely to be more neutral with respect to Th1/Th2 differentiation.
An important implication of these studies is that activation of Nrf2 by food additives, such as tBHQ and BHA, may compromise cell-mediated immunity by impairing the production of the signature Th1 cytokine, IFNγ. Furthermore, induction of the Th2 cytokines, IL-4, IL-5, and IL-13, may create an environment that is conducive for the development of atopy or other Th2-mediated conditions. Because humans are exposed to tBHQ through ingestion of food, the development of food allergies may be of particular concern. Notably, there has been a rise in reports of food allergy that seems to correlate with the increased use of tBHQ and other phenolic antioxidants as food preservatives (53–55). Collectively, the current findings suggest that tBHQ and other food preservatives may modulate immune responses, which warrants further investigation.
Supplementary Material
Acknowledgments
The authors are grateful to Dr. Jefferson Chan for generously providing the Nrf2-null mice and Dr. Scott Reisman, Dr. Jie Liu, and Kai Wu for technical assistance with the animals used in the current studies. In addition, Dr. Joyce Slusser and Michael Stewart provided technical assistance with the flow cytometry studies. The authors would also like to thank Dr. Bryan Copple for kindly providing materials used in these studies and the post-docs and students of the Klaassen lab for careful reading of the manuscript.
Abbreviations used in this paper
- ARE
antioxidant response element
- BAC
bacterial artificial chromosome
- bDNA
branched DNA
- BHA
butylated hydroxyanisole
- BKG
background
- DC
dendritic cell
- EAE
experimental autoimmune encephalitis
- GATA-3
GATA-binding protein 3
- Gclc
γ-glutamylcysteine ligase catalytic subunit
- Hmox1
heme oxygenase-1
- Keap1
Kelch ECH associating protein 1
- Nqo1
NAD(P)H quinone oxidoreductase
- Nrf2
nuclear factor erythroid 2-related factor 2
- SLE
systemic lupus erythematosus
- T-bet
T-box expressed in T cells
- tBHQ
tert-butylhydroquinone
Footnotes
This work was supported by the following National Institutes of Health grants: K99 ES018885 (C.E.R.), R01 DK081461 (C.D.K.), T32 ES007079 and P20 RR016475.
References
- 1.Abbas AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature. 1996;383:787–793. doi: 10.1038/383787a0. [DOI] [PubMed] [Google Scholar]
- 2.Del Prete G. Human Th1 and Th2 lymphocytes: their role in the pathophysiology of atopy. Allergy. 1992;47:450–455. doi: 10.1111/j.1398-9995.1992.tb00662.x. [DOI] [PubMed] [Google Scholar]
- 3.Branum AM, Lukacs SL. Food allergy among U.S. children: trends in prevalence and hospitalizations. NCHS Data Brief. 2008:1–8. [PubMed] [Google Scholar]
- 4.Moorman JE, Rudd RA, Johnson CA, King M, Minor P, Bailey C, Scalia MR, Akinbami LJ. National surveillance for asthma--United States, 1980–2004. MMWR Surveill Summ. 2007;56:1–54. [PubMed] [Google Scholar]
- 5.Bousquet J, Van Cauwenberge P, Khaltaev N. Allergic rhinitis and its impact on asthma. J Allergy Clin Immunol. 2001;108:S147–334. doi: 10.1067/mai.2001.118891. [DOI] [PubMed] [Google Scholar]
- 6.Chan K, Lu R, Chang JC, Kan YW. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc Natl Acad Sci U S A. 1996;93:13943–13948. doi: 10.1073/pnas.93.24.13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, Yamamoto M, Talalay P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci U S A. 2002;99:11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McMahon M, Itoh K, Yamamoto M, Hayes JD. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J Biol Chem. 2003;278:21592–21600. doi: 10.1074/jbc.M300931200. [DOI] [PubMed] [Google Scholar]
- 10.Venugopal R, Jaiswal AK. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc Natl Acad Sci U S A. 1996;93:14960–14965. doi: 10.1073/pnas.93.25.14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y-i. An Nrf2/Small Maf Heterodimer Mediates the Induction of Phase II Detoxifying Enzyme Genes through Antioxidant Response Elements. Biochemical and Biophysical Research Communications. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 12.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 13.Lee JM, Li J, Johnson DA, Stein TD, Kraft AD, Calkins MJ, Jakel RJ, Johnson JA. Nrf2, a multi-organ protector? Faseb J. 2005;19:1061–1066. doi: 10.1096/fj.04-2591hyp. [DOI] [PubMed] [Google Scholar]
- 14.Li J, Stein TD, Johnson JA. Genetic dissection of systemic autoimmune disease in Nrf2-deficient mice. Physiol Genomics. 2004;18:261–272. doi: 10.1152/physiolgenomics.00209.2003. [DOI] [PubMed] [Google Scholar]
- 15.Maher JM, Dieter MZ, Aleksunes LM, Slitt AL, Guo G, Tanaka Y, Scheffer GL, Chan JY, Manautou JE, Chen Y, Dalton TP, Yamamoto M, Klaassen CD. Oxidative and electrophilic stress induces multidrug resistance-associated protein transporters via the nuclear factor-E2-related factor-2 transcriptional pathway. Hepatology. 2007;46:1597–1610. doi: 10.1002/hep.21831. [DOI] [PubMed] [Google Scholar]
- 16.Thimmulappa RK, Scollick C, Traore K, Yates M, Trush MA, Liby KT, Sporn MB, Yamamoto M, Kensler TW, Biswal S. Nrf2-dependent protection from LPS induced inflammatory response and mortality by CDDO-Imidazolide. Biochem Biophys Res Commun. 2006;351:883–889. doi: 10.1016/j.bbrc.2006.10.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim HJ, Nel AE. The role of phase II antioxidant enzymes in protecting memory T cells from spontaneous apoptosis in young and old mice. J Immunol. 2005;175:2948–2959. doi: 10.4049/jimmunol.175.5.2948. [DOI] [PubMed] [Google Scholar]
- 18.Li W, Kong AN. Molecular mechanisms of Nrf2-mediated antioxidant response. Mol Carcinog. 2009;48:91–104. doi: 10.1002/mc.20465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shahidi F. Antioxidants in food and food antioxidants. Nahrung. 2000;44:158–163. doi: 10.1002/1521-3803(20000501)44:3<158::AID-FOOD158>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 20.Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, Biswal S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest. 2006;116:984–995. doi: 10.1172/JCI25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson DA, Amirahmadi S, Ward C, Fabry Z, Johnson JA. The absence of the pro-antioxidant transcription factor Nrf2 exacerbates experimental autoimmune encephalomyelitis. Toxicol Sci. 2010;114:237–246. doi: 10.1093/toxsci/kfp274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Innamorato NG, Rojo AI, Garcia-Yague AJ, Yamamoto M, de Ceballos ML, Cuadrado A. The transcription factor Nrf2 is a therapeutic target against brain inflammation. J Immunol. 2008;181:680–689. doi: 10.4049/jimmunol.181.1.680. [DOI] [PubMed] [Google Scholar]
- 23.Osburn WO, Yates MS, Dolan PD, Chen S, Liby KT, Sporn MB, Taguchi K, Yamamoto M, Kensler TW. Genetic or pharmacologic amplification of nrf2 signaling inhibits acute inflammatory liver injury in mice. Toxicol Sci. 2008;104:218–227. doi: 10.1093/toxsci/kfn079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rangasamy T, Guo J, Mitzner WA, Roman J, Singh A, Fryer AD, Yamamoto M, Kensler TW, Tuder RM, Georas SN, Biswal S. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J Exp Med. 2005;202:47–59. doi: 10.1084/jem.20050538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ishii Y, Itoh K, Morishima Y, Kimura T, Kiwamoto T, Iizuka T, Hegab AE, Hosoya T, Nomura A, Sakamoto T, Yamamoto M, Sekizawa K. Transcription factor Nrf2 plays a pivotal role in protection against elastase-induced pulmonary inflammation and emphysema. J Immunol. 2005;175:6968–6975. doi: 10.4049/jimmunol.175.10.6968. [DOI] [PubMed] [Google Scholar]
- 26.Reddy NM, Kleeberger SR, Kensler TW, Yamamoto M, Hassoun PM, Reddy SP. Disruption of Nrf2 impairs the resolution of hyperoxia-induced acute lung injury and inflammation in mice. J Immunol. 2009;182:7264–7271. doi: 10.4049/jimmunol.0804248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoh K, Itoh K, Enomoto A, Hirayama A, Yamaguchi N, Kobayashi M, Morito N, Koyama A, Yamamoto M, Takahashi S. Nrf2-deficient female mice develop lupus-like autoimmune nephritis. Kidney Int. 2001;60:1343–1353. doi: 10.1046/j.1523-1755.2001.00939.x. [DOI] [PubMed] [Google Scholar]
- 28.Ma Q, Battelli L, Hubbs AF. Multiorgan autoimmune inflammation, enhanced lymphoproliferation, and impaired homeostasis of reactive oxygen species in mice lacking the antioxidant-activated transcription factor Nrf2. Am J Pathol. 2006;168:1960–1974. doi: 10.2353/ajpath.2006.051113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee JM, Chan K, Kan YW, Johnson JA. Targeted disruption of Nrf2 causes regenerative immune-mediated hemolytic anemia. Proc Natl Acad Sci U S A. 2004;101:9751–9756. doi: 10.1073/pnas.0403620101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cordova EJ, Velazquez-Cruz R, Centeno F, Baca V, Orozco L. The NRF2 gene variant, -653G/A, is associated with nephritis in childhood-onset systemic lupus erythematosus. Lupus. 2010;19:1237–1242. doi: 10.1177/0961203310367917. [DOI] [PubMed] [Google Scholar]
- 31.Kikuchi N, Ishii Y, Morishima Y, Yageta Y, Haraguchi N, Itoh K, Yamamoto M, Hizawa N. Nrf2 protects against pulmonary fibrosis by regulating the lung oxidant level and Th1/Th2 balance. Respir Res. 2010;11:31. doi: 10.1186/1465-9921-11-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Williams MA, Rangasamy T, Bauer SM, Killedar S, Karp M, Kensler TW, Yamamoto M, Breysse P, Biswal S, Georas SN. Disruption of the transcription factor Nrf2 promotes pro-oxidative dendritic cells that stimulate Th2-like immunoresponsiveness upon activation by ambient particulate matter. J Immunol. 2008;181:4545–4559. doi: 10.4049/jimmunol.181.7.4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim HJ, Barajas B, Wang M, Nel AE. Nrf2 activation by sulforaphane restores the age-related decrease of T(H)1 immunity: role of dendritic cells. J Allergy Clin Immunol. 2008;121:1255–1261. doi: 10.1016/j.jaci.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang YK, Yeager RL, Klaassen CD. Circadian expression profiles of drug-processing genes and transcription factors in mouse liver. Drug Metab Dispos. 2009;37:106–115. doi: 10.1124/dmd.108.024174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hartley DP, Klaassen CD. Detection of chemical-induced differential expression of rat hepatic cytochrome P450 mRNA transcripts using branched DNA signal amplification technology. Drug Metab Dispos. 2000;28:608–616. [PubMed] [Google Scholar]
- 36.Farraj AK, Harkema JR, Kaminski NE. Allergic rhinitis induced by intranasal sensitization and challenge with trimellitic anhydride but not with dinitrochlorobenzene or oxazolone in A/J mice. Toxicol Sci. 2004;79:315–325. doi: 10.1093/toxsci/kfh112. [DOI] [PubMed] [Google Scholar]
- 37.Hagemann TL, Gaeta SA, Smith MA, Johnson DA, Johnson JA, Messing A. Gene expression analysis in mice with elevated glial fibrillary acidic protein and Rosenthal fibers reveals a stress response followed by glial activation and neuronal dysfunction. Hum Mol Genet. 2005;14:2443–2458. doi: 10.1093/hmg/ddi248. [DOI] [PubMed] [Google Scholar]
- 38.Leung L, Kwong M, Hou S, Lee C, Chan JY. Deficiency of the Nrf1 and Nrf2 transcription factors results in early embryonic lethality and severe oxidative stress. J Biol Chem. 2003;278:48021–48029. doi: 10.1074/jbc.M308439200. [DOI] [PubMed] [Google Scholar]
- 39.Chandler KJ, Chandler RL, Broeckelmann EM, Hou Y, Southard-Smith EM, Mortlock DP. Relevance of BAC transgene copy number in mice: transgene copy number variation across multiple transgenic lines and correlations with transgene integrity and expression. Mamm Genome. 2007;18:693–708. doi: 10.1007/s00335-007-9056-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kwak MK, Itoh K, Yamamoto M, Sutter TR, Kensler TW. Role of transcription factor Nrf2 in the induction of hepatic phase 2 and antioxidative enzymes in vivo by the cancer chemoprotective agent, 3H-1, 2-dimethiole-3-thione. Mol Med. 2001;7:135–145. [PMC free article] [PubMed] [Google Scholar]
- 41.Balomenos D, Rumold R, Theofilopoulos AN. Interferon-gamma is required for lupus-like disease and lymphoaccumulation in MRL-lpr mice. J Clin Invest. 1998;101:364–371. doi: 10.1172/JCI750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Masutani K, Akahoshi M, Tsuruya K, Tokumoto M, Ninomiya T, Kohsaka T, Fukuda K, Kanai H, Nakashima H, Otsuka T, Hirakata H. Predominance of Th1 immune response in diffuse proliferative lupus nephritis. Arthritis Rheum. 2001;44:2097–2106. doi: 10.1002/1529-0131(200109)44:9<2097::AID-ART360>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 43.Uhm WS, Na K, Song GW, Jung SS, Lee T, Park MH, Yoo DH. Cytokine balance in kidney tissue from lupus nephritis patients. Rheumatology (Oxford) 2003;42:935–938. doi: 10.1093/rheumatology/keg255. [DOI] [PubMed] [Google Scholar]
- 44.Tucci M, Lombardi L, Richards HB, Dammacco F, Silvestris F. Overexpression of interleukin-12 and T helper 1 predominance in lupus nephritis. Clin Exp Immunol. 2008;154:247–254. doi: 10.1111/j.1365-2249.2008.03758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.al-Janadi M, al-Balla S, al-Dalaan A, Raziuddin S. Cytokine profile in systemic lupus erythematosus, rheumatoid arthritis, and other rheumatic diseases. J Clin Immunol. 1993;13:58–67. doi: 10.1007/BF00920636. [DOI] [PubMed] [Google Scholar]
- 46.Nakashima H, Inoue H, Akahoshi M, Tanaka Y, Yamaoka K, Ogami E, Nagano S, Arinobu Y, Niiro H, Otsuka T, Niho Y. The combination of polymorphisms within interferon-gamma receptor 1 and receptor 2 associated with the risk of systemic lupus erythematosus. FEBS Lett. 1999;453:187–190. doi: 10.1016/s0014-5793(99)00701-2. [DOI] [PubMed] [Google Scholar]
- 47.Machold KP, Smolen JS. Interferon-gamma induced exacerbation of systemic lupus erythematosus. J Rheumatol. 1990;17:831–832. [PubMed] [Google Scholar]
- 48.Wandl UB, Nagel-Hiemke M, May D, Kreuzfelder E, Kloke O, Kranzhoff M, Seeber S, Niederle N. Lupus-like autoimmune disease induced by interferon therapy for myeloproliferative disorders. Clin Immunol Immunopathol. 1992;65:70–74. doi: 10.1016/0090-1229(92)90250-r. [DOI] [PubMed] [Google Scholar]
- 49.Dornand J, Gerber M. Inhibition of murine T-cell responses by anti-oxidants: the targets of lipo-oxygenase pathway inhibitors. Immunology. 1989;68:384–391. [PMC free article] [PubMed] [Google Scholar]
- 50.WHO. WHO Food Additives Series. World Health Organization; Geneva: 1975. Toxicological evaluation of some food colours, thickening agents, and certain other substances. [PubMed] [Google Scholar]
- 51.Kass GE, Duddy SK, Moore GA, Orrenius S. 2,5-Di-(tert-butyl)-1,4-benzohydroquinone rapidly elevates cytosolic Ca2+ concentration by mobilizing the inositol 1,4,5-trisphosphate-sensitive Ca2+ pool. J Biol Chem. 1989;264:15192–15198. [PubMed] [Google Scholar]
- 52.Li YJ, Takizawa H, Azuma A, Kohyama T, Yamauchi Y, Takahashi S, Yamamoto M, Kawada T, Kudoh S, Sugawara I. Disruption of Nrf2 enhances susceptibility to airway inflammatory responses induced by low-dose diesel exhaust particles in mice. Clin Immunol. 2008;128:366–373. doi: 10.1016/j.clim.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 53.Sicherer SH, Munoz-Furlong A, Godbold JH, Sampson HA. US prevalence of self-reported peanut, tree nut, and sesame allergy: 11-year follow-up. J Allergy Clin Immunol. 2010;125:1322–1326. doi: 10.1016/j.jaci.2010.03.029. [DOI] [PubMed] [Google Scholar]
- 54.Sicherer SH, Sampson HA. Food allergy. J Allergy Clin Immunol. 2010;125:S116–125. doi: 10.1016/j.jaci.2009.08.028. [DOI] [PubMed] [Google Scholar]
- 55.Grundy J, Matthews S, Bateman B, Dean T, Arshad SH. Rising prevalence of allergy to peanut in children: Data from 2 sequential cohorts. J Allergy Clin Immunol. 2002;110:784–789. doi: 10.1067/mai.2002.128802. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.