

Abstract
Caffeic acid phenethyl ester (CAPE) treatment suppressed proliferation, colony formation, and cell cycle progression in PC-3 human prostate cancer cells. CAPE decreased protein expression of cyclin D1, cyclin E, SKP2, c-Myc, Akt1, Akt2, Akt3, total Akt, mTOR, Bcl-2, Rb, as well as phosphorylation of Rb, ERK1/2, Akt, mTOR, GSK3α, GSK3β, PDK1; but increased protein expression of KLF6 and p21Cip1. Microarray analysis indicated that pathways involved in cellular movement, cell death, proliferation, and cell cycle were affected by CAPE. Co-treatment of CAPE with chemotherapeutic drugs vinblastine, paclitaxol, and estramustine indicated synergistic suppression effect. CAPE administration may serve as a potential adjuvant therapy for prostate cancer.
Introduction
Prostate cancer is one of the most common non-cutaneous carcinoma of men in western countries. More than 80% of patients died from prostate cancer developed bone metastases [1]–[3]. In 1941, Charles Huggins discovered that deprivation of androgen caused regression of hormone-responsive metastatic prostate cancer [4]. Since then, androgen ablation therapy has become the primary treatment for metastatic prostate cancer. However, most prostate cancer patients receiving androgen ablation therapy ultimately develop recurrent, castration-resistant tumors within 12–33 months after treatment. The median overall survival time is 1–2 years after tumor relapse [5], [6]. Chemotherapy is usually applied for treatment of metastatic hormone-refractory prostate cancer [7].
Commonly used chemotherapy drugs for metastatic prostate cancer include eoposide, paclitaxol, vinblastine, mitoxantrone, and estramustine. Etoposide and mitoxantrone are type II topoisomerase inhibitor [7], [8]. Estramustine is a derivative of estrogen with a nitrogen mustard-carbamate ester moiety [7]. Vinblastine binds tubulin and inhibits assembly of microtubules [7]. Paclitaxel disrupts mitotic spindle assembly, chromosome segregation, and cell division. Paclitaxel also stabilizes the microtubule polymer and thus protects it from disassembly [7]. Treatment with these chemotherapy drugs decreased prostate specific antigen (PSA) and radiographic response as well as improved pain and urinary symptoms in a sub-group of patients. However, they showed little effect on prolonging survival. Undesired side effects of these chemotherapeutic agents include toxic deaths, strokes, thrombosis, neutropenia, edema, dyspnea, malaise, and fatigue [7]. Co-treatment chemotherapy drugs with natural compounds with anticancer activity may reduce the dosage of chemotherapy drugs needed.
Caffeic acid phenethyl ester (CAPE), a bioactive component extracted from honeybee hive propolis, is a strong antioxidant [9], [10]. CAPE treatment in breast, prostate, and leukemic cancer cells causes inhibition of NF-κB activity [11], [12], induction of Bax [11], [13], activation of c-Jun N-terminal kinase (JNK) [11] and p38 mitogen-activated protein kinase (p38 MAPK) [11]. CAPE induces apoptosis via activation of caspase activity [11], [13] and down-regulation of Bcl-2, cIAP-1, cIAP-2, and XIAP [12], [13] in breast, prostate, and leukemic cancer cells. In addition, CAPE induces cell cycle arrest through suppression of cyclin D1 [14], [15], cyclin E [14], and c-Myc expression [15], as well as increases expression of the cyclin dependent kinase inhibitors p21cip1 [14], p27Kip1 [14], and p16INK4A [14] in colon and glioma cancer cells. These observations suggest that CAPE is a potential therapeutic agent for cancers.
PC-3 is one of the most commonly used prostate cancer cell lines established from bone-derived metastases. PC-3 cells do not express androgen receptor (AR) [16]. Mitoxantrone, estramustine, vinblastine, etoposide, and paclitaxel have been shown to induce proliferation inhibition, apoptosis, and cell cycle arrest in PC-3 cells in vitro [17]–[21], as well as to retard PC-3 xenografts growth in athymic nude mice [8], [21], [22]. Treatment with 88–176 µM of CAPE induced apoptosis in PC-3 cells via inhibition of NF-κB, cIAP-1, cIAP-2, and XIAP [12]. However, the achievable concentration of CAPE in human serum is around 5.0 µg/ml (17 µM) [23]. We thus examined if low concentration (0–20 µM) of CAPE can suppress the proliferation of PC-3 cells. We also determined if co-treatment of chemotherapy drugs with CAPE show synergistic inhibition effect on proliferation of PC-3 cells.
Results
CAPE treatment suppresses the proliferation and colony formation of PC-3 cells
Trypan blue staining indicated that CAPE dose-dependently inhibited proliferation of PC-3 cells with an EC50 around 20.4 µM (Fig. 1A). Hoescht dye-based 96-well proliferation assay showed that the growth inhibitory effect of CAPE happened within 24 hours following CAPE treatment at concentration as low as 2.5 µM (Fig. 1B). EC50 for growth inhibition of PC-3 cells was 51.4 µM, 30.7 µM, and 23.1 µM for 24, 48, and 72 h CAPE treatment, respectively, indicating that the suppressive effect of CAPE can be accumulated. Colony formation assay revealed that treatment of 10 µM and 20 µM CAPE efficiently inhibited the formation of PC-3 colonies in soft agar (Fig. 1C).
Figure 1. CAPE suppresses proliferation and colony formation of PC-3 cells.

Proliferation of PC-3 cell treated with increasing concentration of CAPE was determined by Trypan blue staining after 72 h treatment (A) or measuring total DNA content per well using Hoechst 33258 fluorescence by 96-well proliferation assay after 24, 48, and 72 h treatment (B). Relative cell numbers were normalized to the average cell number of the control (no CAPE treatment) of each cell line in each individual experiment. Columns represent mean for 18 replicates; bars represent standard deviation. Asterisk (*) represents cell number is statistically significantly different (p<0.05) compared to the control. Columns represent mean for 5 biological replicates; bars represent standard deviation. (C) Anticancer effect of CAPE was determined by colony formation assay of PC-3 cells treated with 0, 10, 20 µM for 14 days. Image is a representative result of three biological replicates.
Since CAPE was previously reported as an NF-κB inhibitor [10], we determined whether low dasage of CAPE can inhibit NF-κB activity using a plasmid-based luciferase reporter assay. Although CAPE treatment at 40 µM inhibited NF-κB activity, treatment with CAPE at concentration lower than 40 µM had no effect on NF-κB activity (Fig. 2A). This observation suggested that other mechanisms are responsible for CAPE's inhibitory effect at low dosage.
Figure 2. CAPE inhibits cell cycle progression in PC-3 cells.

(A) PC-3 cells transfected with a 4X NF-κB luciferase reporter plasmid for 24 hr were treated with increasing concentrations of CAPE for additional 24 h. Relative luciferase activity was determined to compare the effect of CAPE on NF-κB transcriptional activity. (B) PC-3 cells were treated with CAPE for 24, 48, or 72 h, harvested, and stained with propidium iodide dye for flow cytometric analysis for cell cycle distribution. (*) represents statistically significant difference (p<0.05) between the two group of cells being compared.
CAPE treatment disturbs cell cycle progression
Propidium iodide (PI) staining flow cytometry analysis revealed that treatment with 10–20 µM CAPE decreased the cell population in G1 phase and increased cell population in sub-G1 phase within 24 h in PC-3 cells. This effect was more dramatic at 72 h following CAPE treatment (Fig. 2B–2D). However, annexin V staining flow cytometry analysis indicated that 10–20 µM CAPE did not induce apoptosis in PC-3 cells (data not shown). Treatment with 20 µM CAPE for 72 h resulted in increase of cell cycle inhibitory proteins p21Cip1 and decrease of S-phase kinase-associated protein 2 (SKP2), phosphorylation of serine 807/811 on retinoblastoma (Rb), cycin D1, cyclin E, c-Myc, and phosphorylation of threonine 202/tyrosine 204 of extracellular signal-regulated kinase 1/2 (ERK1/2) (Fig. 3). No change in p27Kip1, total ERK1/2, or β-tubulin was observed. Compared to 24 h and 48 h treatment, 72 h treatment in general caused more change of protein expression level except for cyclin D1. This may explain the greater growth inhibition caused by CAPE at 72 h. Cyclin D1 increased after 24 h and 48 h treatment but decreased after 72 h treatment.
Figure 3. CAPE affects cell cycle regulating proteins in PC-3 cells.

Protein expression of c-Myc, cyclin D1, cyclin E, SKP2, phosho-Rb (S807/811), p27Kip1, p21Cip1, ERK1/2, pERK1/2 Thr202/Tyr204, β-tubulin, and β-actin in PC-3 cells treated with 20 µM CAPE for 24, 48, and 5, 10, 20 µM CAPE for 72 h were assayed by Western blotting.
CAPE treatment inhibits the abundance and activity of proteins in AKT-signaling pathway
Akt plays important role in survival and proliferation of prostate cancer cells [24]. We thus determined if CAPE treatment suppresses Akt signaling pathway. 72 h after 20 µM CAPE treatment decreased the abundance of total Akt, Akt1, Akt2, and Akt3 (Figure 4). CAPE treatment for 24–72 h significantly decreased the phosphorylation of Akt on serine 473 and threonine 308,. CAPE did not change the total abundance of phosphoinositide dependent kinase 1 (PDK1) (Fig. 4), however, phosphorylation of serine 241 on PDK1 was reduced by CAPE treatment. CAPE treatment also caused decrease of total mammalian target of rapamycin (mTOR) and slight reduction of phosphorylation on serine 2448 and 2481 of mTOR. CAPE treatment did not change the total abundance of GSK3α and GSK3β (Fig. 4). However, phosphosphorylation of GSK3α S21 and GSK3β S9 was increased after 24 h and 48 h of 20 µM CAPE treatment but decreased at 72 h of 20 µM CAPE treatment (Fig. 4). Bcl-2 is an anti-apoptosis factor downstream of Akt signaling. Overexpression of Bcl-2 has previously been reported to confer drug resistance of prostate cancers [5]. CAPE slightly decreased expression of Bcl-2.
Figure 4. CAPE inhibits Akt signaling-related proteins in PC-3 cells.

Protein expression of Akt, Akt1, Akt2, Akt3, total Akt, phospho-Akt S473, phospho-Akt T308, mTOR, phospho-mTOR Ser2448 and Ser2481, GSK3α, GSK3β, phopho-GSK3α S21, phospho-GSK3β S9, PDK1, phospho-PDK1 Ser241, Bcl-2, KLF6, β-tubulin, and β-actin in PC-3 cells treated with 20 µM CAPE for 24, 48, and 5, 10, 20 µM CAPE for 72 h were assayed by Western blotting.
CAPE treatment affects genes regulating proliferation, survival, and death of PC-3 cells
We further studied the comprehensive change of gene expression in PC-3 cells treated with 20 µM CAPE for 24 h or 72 h by microarray. Genes with expression fold change >1.5 and P<0.05 were considered as genes significantly affected by CAPE treatment. CAPE affected expression of 69 unique genes after 24 h treatment (Table S1). 53 genes were up-regulated and 16 genes were down-regulated. Treatment with CAPE for 72 h altered expression of 147 unique genes (Table S2). 122 genes were up-regulated while 25 genes were down-regulated. 25 genes were commonly changed in both 24 h and 72 h treatment (Figure 5, Table S3). Among the 25 genes, 3 genes were down-regulated (CYP1B1, SCG5, PADI4) and 22 genes were up-regulated (LY96, LOC728285, TM4SF19, RGS2, PI3, AKR1C2, GDF15, HIST1H2BD, CCL20, CXCL5, RND3, KRT34, HIST2H2AA3, AKR1C4, KLF4, DUSP5, NOV, GK, CDKN1A, CXCL2, DUSP1, and HIST1H4H) (Fig. 5). Analysis of all the 191 gene probes affected by CAPE treatment either at 24 h or 72 h using Ingenuity Pathway Analysis (IPA) revealed that CAPE treatment affected genes involved in regulation of cell death, proliferation, and survival. Among the genes being affected by CAPE treatment, 52 genes involved in cell proliferation regulation (p value = 9.82×10−11), 41 genes involved in cell growth regulation (p value = 1.40×10−10), 68 genes involved in cell death regulation (p value = 1.40×10−12), and 27 genes involved in cell survival regulation (p = 3.43×10−6). Complete list of genes probes involved in these signaling pathways were shown in Table S4.
Figure 5. A scatter plot of log2 ratio (logR) for genes whose expression were significantly affected at either 24 h or 72 h post CAPE treatment.

Genes commonly affected at both time points are in red color, while those specifically affected at either time point are in black. IPA analysis of the unique genes (n = 191) genes changed either in 24 h or 72 h CAPE treatment indicated that group of genes regulating several cell functions, including cell proliferation (p-value 9.82×10−11, 52 genes), cell growth (p-value 1.40×10−10, 41 genes), cell death (p-value 1.40×10−12, 68 genes), and cell survival (p-value 1.40×10−6, 27 genes).
We validated some of the genes affected by CAPE treatment with quantitative real-time PCR (qRT-PCR). 17 out of 18 genes (GDF15, HIST1H2BD, CCL20, CXCL5, RND3, KLF4, DUSP5, NOV, CDKN1A, CXCL2, DUSP1, KLF6, TOP2A, PPP1R15A, CAV2, S100P, and GADD45A) tested by qRT-PCR showed similar alteration pattern following 24 h or 72 h CAPE treatment as compared to gene microarray. The only exception is TUBA1A. We did not observe any change of TUBA1A gene under CAPE treatment by qRT-PCR (Fig. 6). Western blotting assay indicated that protein level of KLF6 was increased by CAPE treatment (Fig. 4).
Figure 6. Validation of gene microarray result with qRT-PCR.

Gene expression level of GDF15, HIST1H2BD, CCL20, CXCL5, RND3, KLF4, DUSP5, NOV, CDKN1A, CXCL2, DUSP1, KLF6, TOP2A, PPP1R15A, CAV2, S100P, GADD45A, and TUBA1A in PC treated with 0 or 20 µM CAPE for 24 h or 72 h was determined by qRT-PCR.
Co-treatment of CAPE with chemotherapeutic drugs suppressed proliferation of PC-3 cells
Finally, we investigated if co-treatment of CAPE at serum-available dosage (0–20 µM) with commonly used chemotherapy drugs (etoposide, paclitaxol, vinblastine, mitoxantrone, and estramustin) can suppress growth of PC-3 cells more effectively than treatment with chemotherapy drugs alone. EC50 of CAPE, etoposide, paclitaxol, vinblastine, mitoxantrone, and estramustin for inhibiting proliferation of PC-3 cells was 18.3 µM, 1.7 µM, 3.0 nM, 2.1 nM, 5.9 nM, and 13.0 µM. Treatment of 20 µM CAPE suppressed growth of PC-3 cells more effectively than treatment with 1.0 µM etoposide, 2.5 nM paclitaxol, 5 nM mitoxantrone, 2 nM vinblastine, or 10 µM estramustine (Figure 7). When co-treatment with 20 µM CAPE, 0.5 µM etoposide, 1 nM paclitaxol, 1 nM vinblastine, 2.5 nM mitoxantrone, and 8 µM extramustine caused growth inhibition similar to the highest dosage we tested (Figure 7). Synergistic effect means the suppressive effect of two drugs being treated together is greater than the sum of their separate suppressive effect at the same doses, while antagonistic effects means the suppressive effect of two drugs is less than the sum of the effect of the two chemicals taken separately. According to the definition, co-treatment of CAPE showed synergistic effect with vinblastine, estramustine, or paclitaxol, and antagonistic effect with etoposide or mitoxantrone (Figure 7).
Figure 7. Combined treatment of CAPE with chemotherapy drugs shows synergistic and antagonistic inhibition on proliferation of PC-3 cells.

Proliferation of PC-3 cells treated with increasing dosage (0, 5, 10, 20 µM) of CAPE in combination with increasing concentration of etoposide (A), paclitaxol (B), vinblastine (C), mitoxantrone (D), and estramustine (E) for 72 h was determined by 96-well proliferation assay. The right part of the figure show the ratio of expected cell number/observed cell number. For example, treatment of 5 µM of CAPE or 1 nM vinblastine decreases cell number of PC-3 to 80.9% and 88.7%, respectively, compared to the control (no treatment). The expected cell number of treatment combining 5 µM of CAPE and 1 nM vinblastine is 0.809*0.887 = 71.8%. The observed cell number is 48.8% compared to the control. So the ratio is 0.718/0.488 = 1.5. Ratio larger than one represents synergy of growth inhibition, while ratio smaller than one represents antagonistic effect.
According to our observation, p21Cip1 plays important role in regulation of growth inhibition induced by CAPE treatment. To confirm this, we knocked down p21Cip1 in PC-3 and determined if these PC-3 cells become more resistant to CAPE treatment. As expected, following 24 h of CAPE treatment, PC-3 cells with less p21Cip1 protein expression were more resistant to growth inhibition caused by CAPE treatment (Fig. 8).
Figure 8. Growth response to CAPE treatment of PC-3 and PC-3 p21Cip1 siRNA cells.

Protein levels of wild type PC-3, PC-3 cells transfect with scramble control (20 nM), and PC-3 cells transfected with p21Cip1 siRNA (20 nM) were determined by Western blotting assay. Proliferation of these PC-3 cells treated with 20 µM CAPE for 24 h was determined by 96-well plate proliferation assay as described in Material and Methods.
Discussion
Our observation suggested that caffeic acid phenethyl ester (CAPE) can inhibit proliferation and colony formation of PC-3 human prostate cancer cells at concentration 10–20 µM. These observations suggested that the achievable concentration of CAPE in human serum, (17 µM) [23], is possibly to cause growth inhibition in prostate tumors in patients.
Cyclin-dependent kinase inhibitor p21Cip1 binds and inhibits the kinase activities of Cdk2/cyclin A, Cdk2/cyclin E, Cdk4/cyclin D, and Cdk6/cyclin D complexes [25]. p21Cip1 can interact with proliferating cell nuclear antigen (PCNA), a DNA polymerase accessory factor, and plays a regulatory role in S phase DNA replication and DNA damage repair [26]. SKP2 is a member of the F-box protein family. SKP2 constitutes one of the four subunits of ubiquitin protein ligase complex called SCFs (SKP, cullin, F-box containing complex), which functions in phosphorylation-dependent ubiquitination. SKP2 is an essential element of the cyclin A-Cdk2 S-phase kinase [27]. Reduction in phosphorylation of Rb restricts cell proliferation by inhibiting E2F activity [28]. ERK1 and ERK2 are involved in the control of many fundamental cellular processes including cell proliferation, survival, differentiation, apoptosis, motility and metabolism. ERK1/2 play important roles in canonical MAPK (Mitogen-Activated Protein Kinase) signaling pathway and are critical regulators of the growth and survival [29]. CAPE induced p21Cip1 and reduced cyclin D, cyclin E, SKP2, and phosphorylation of Rb and ERK1/2 (Fig. 3). CAPE may thus suppress the growth of PC-3 cells via these proteins [30].
Akt is a serine/threonine protein kinase regulating inhibition of apoptosis and stimulation of cell proliferation. Up-regulation of PI3K/Akt activity is associated with poor clinical outcome of prostate cancer [31]. There are three mammalian isoforms of this enzyme, Akt1, Akt2, and Akt3 [32], [33]. Protein abundance and activity of Akt3 have previously been suggested to contribute to the more aggressive clinical phenotype of androgen non-responsive prostate and breast cancers [34]. Akt3 enzymatic activity was approximately 20-60-fold higher in AR-negative PC-3 and DU-145 cells compared to the AR-positive LNCaP prostate cancer cells [34], [35]. We observed that CAPE suppressed Akt signaling-related proteins, including Akt1, Akt2, Akt3, total Akt, mTOR, Bcl-2, pAkt Ser 473, pAKt Thr 308, pmTOR Ser 2448/2481, pGSK3α Ser21, pGSK3β Ser9, and pPDK1 Ser241. CAPE was recently reported to suppress phosphorylation of Akt in other human cells, such as CD4+ T cells [36], coronary smooth muscle cell [37], and A549 lung cancer cells [38]. Phosphatase and tensin homolog (PTEN) protein acts as a phosphatase to dephosphorylate phosphatidylinositol (3,4,5)-trisphosphate. PTEN negatively controls the phosphoinositide 3-kinase/Akt signaling pathway [39]. PC-3 cells acquire a homozygous deletion of PTEN, thus Akt is constantly active. There are two phosphorylation sites on Akt, threonine 308 and serine 473. Phosphorylation of Thr308 on Akt is activated by PDK1 [40]. Phosphorylation of serine 473 is activated by mTOR kinase, its associated protein rector, and SIN1/MIP1 [41], [42]. CAPE phosphorylation of serine 241 on PDK1 and attenuated the phosphorylation of serine 2448 and 2481 on mTOR (Fig. 4). Reduction of PDK1 and mTOR activity may therefore contribute to the decrease of phsphorylation on Akt. The activities of glycogen synthase kinase 3 alpha (GSK3α and GSK3β are known to be inhibited by Akt-mediated phosphorylation at Ser21 and Ser9 respectively, limiting their ability to phosphorylate cell cycle regulating proteins, such as cyclin D1 and p21Cip1 [43], [44]. Phosphosphorylation of GSK3α S21 and GSK3β S9 was increased after 24 h and 48 h of 20 µM CAPE treatment but decreased at 72 h of 20 µM CAPE treatment (Fig. 4). Increased phosphorylation of GSK3α S21 and GSK3β S9 may contribute to the increase of p21Cip1 at 24 h and 48 h after CAPE treatment. GSK3β-dependent phosphorylation of cyclin D1 mediated nuclear export and rapid degradation within the cytoplasm of cyclin D1 [45]. Reduction of GSK3βactivity due to increase of phosphorylation (Fig. 4) resulted in less phosphorylation of cyclin D1 and therefore accumulation of cyclin D1 at 24 h and 48 h (Fig. 3). Increase of GSK3βactivity due to decrease of phosphorylation (Fig. 4) would therefore decrease the abundance of cyclin D1 at 72 h (Fig. 3). Decreased phosphorylation of GSK3α and GSK3β at 72 h was consistent with the decreased phosphorylation of Akt. Suppression of Akt signaling by CAPE may contribute to the inhibition of survival and growth in PC-3 cells.
We noticed that genes affected by CAPE at 24 h and 72 h post treatment was moderately correlated (r = 0.56, Fig. 5). There were only 25 significantly affected genes in common between these two time points. Since the growth inhibition and cell cycle perturbation caused by CAPE treatment started within 24 h and the suppressive effect accumulated over time, we hypothesized that the most important target genes for anticancer activity of CAPE were these 25 common genes. Krüppel-like factor 4 (KLF4) transactivates the p21Cip1 promoter and inhibits proliferation through activation of p21Cip1 as well as direct suppression of cyclin D1 and cyclin B1 gene expression [46]–[48]. Nov gene encodes protein CCN3 (Nov) which inhibits cell proliferation via Notch/p21Cip1 pathway [49]. Elevation of KLF4 and Nov genes may suppress PC-3 growth via p21Cip1. Growth/differentiation factor-15(GDF-15) is a divergent TGFβ family member that has been implicated in inhibition of tumor growth and increased tumor invasiveness [50]. A few genes are cytokines involved in inflammation response, such as CCL20 [51], CXCL2 [52], CXCL5 [53]. They were found up-regulated, suggesting that CAPE induces inflammation response in PC-3 cells. In addition, CAPE treatment increases RhoE/Rnd3. Up-regulation of the small G-protein RhoE/Rnd3/Rho8 inhibits the proliferation of prostate cancer cells by promoting apoptosis and inhibiting cell cycle progression [54].
Besides the 25 commonly changed genes, some differentially expressed genes specifically after 24 h or 72 h treatment also regulate cell survival, proliferation, or cell death. CAPE treatment increased KLF6, S100P, GADD45A, PPP1R15A, S100P, but decreased TOP2A and CAV2. Kruppel-like factor 6 (KLF6) is a zinc finger transcription factor and functions as tumor suppressor gene in human prostate cancer [55]. KLF6 up-regulates p21Cip1 in a p53-independent manner and significantly reduces cell proliferation [55]. S100P protein regulates calcium signal transduction, cytoskeletal interaction, protein phosphorylation, transcriptional control, cell cycle progression, and differentiation. Elevation of S100P in PC3 cells promoted cell growth, increased the percentage of S-phase cells, decreased basal apoptosis rate, promoted anchorage independent growth in soft agar, and confer resistance to chemotherapy [56]. GADD45A protein responds to environmental stresses by mediating activation of the p38/JNK pathway. The Gadd45 protein has been described to form a complex with p21Cip1. The p21Cip1-binding domain of GADD45A also encodes the Cdc2-binding activity. GADD45A interacts with Cdc2, dissociates the Cdc2-cyclin B1 complex, alters cyclin B1 nuclear localization, and thus inhibits the activity of Cdc2/cyclin B1 kinase [57]–[59]. PPP1R15A (Protein phosphatase 1 regulatory subunit 15A, also known as GADD34) has been shown to induce growth arrest and apoptosis. PPP1R15A up-regulation enhances p21Cip1 protein expression and induces p21Cip1 promoter activity [60].
Vinblastine, paclitaxol, and CAPE affect gene expression of α-tubulin and β-tubulin (Figure S1, S2), while etoposide, mitoxantrone, and CAPE affect gene expression of type II topoisomerase (Figure S3). However, etoposide induces p21Cip1 via p53 and down-regulation of c-Myc in cancer cells [61], [62]. Mitoxantrone induces p21Cip1 [63]. Vinblastine induces apoptosis via reduction of p21Cip1 [64]. Paclitaxol induces an Akt-dependent phosphorylation on p21Cip1 leading to an association of p21Cip1 with 14-3-3 and thus accumulation of the phosphorylated form of p21Cip1 in cytoplasm which prevents the inhibitory effect of p21Cip1 [65]. No study reports the relationship between p21Cip1 and estramustine. Since CAPE treatment increases both mRNA and protein level of p21Cip1(Fig. 3) and knockdown of p21Cip1 in PC-3 cells made cells more resistant to CAPE treatment (Fig. 8), CAPE may suppress growth and survival of PC-3 cells more similar to etoposide and mitoxantrone, but less similar to vinblastine, paclitaxol, and estramustine. Besides CDKN1A (p21Cip1 gene), CAPE treatment also increased gene expression of KLF4, KLF6, Nov, GADD45A, PPP1R15A. These genes all suppress proliferation via p21Cip1. Therefore, although co-treatment with CAPE suppressed more PC-3 cells than treatment with chemotherapy drug alone, CAPE only showed synergistic suppressive effect with vinblastine, paclitaxol, and estramustine (Fig. 7). CAPE treatment also suppressed abundance and phosphorylation of Akt, as well as upstream and downstream signaling proteins in Akt signaling. We therefore believe that p21Cip1 induction and suppression of Akt signaling both play important role in growth inhibition caused by CAPE treatment in PC-3 cells. We summarize the Akt/p21Cip1 signaling pathway network being affected by CAPE treatment in PC-3 in Figure 9.
Figure 9. Putative model of anticancer effect of CAPE in PC-3 human prostate cancer cells.

Protein abundance or activity being stimulated by CAPE treatment are labeled with red upward arrows, while those being suppressed by CAPE treatment are labeled with blue downward arrows.
In conclusion, our observations provided insight into the molecular mechanism of CAPE's anti-proliferative effect in PC-3 prostate cancer cells. Our data suggested that CAPE administration may be useful as a potential adjuvant therapy in combination with chemotherapies for metastatic prostate cancer.
Materials and Methods
Chemicals
Caffeic aicd phenethyl ester, etoposide, paclitaxol, vinblastine, estramustine, and mitoxantrone were purchased from Sigma (St. Louis, MO, U.S.A.).
Cell Culture
PC-3 cells were generous gift from Dr. Shutsung Liao's lab (The University of Chicago) and were maintained in DMEM (Gibco/Invitrogen, Carlsbad, CA, U.S.A.) supplemented with 10% fetal bovine serum (FBS; Atlas Biologicals, Fort Collins, CO, U.S.A.), penicillin (100 U/ml), and streptomycin (100 ug/ml).
Cell Proliferation Assay
Relative cell number was analyzed by measuring DNA content of cell lysates with the fluorescent dye Hoechst 33258 (Sigma) as described previously [66]–[69]. EC50 (concentration of drug to cause 50% growth inhibition) of drugs on PC-3 cells was determined by an Excel add-in program ED50V10.
Soft Agar Colony Formation Assay
8000 cells were suspended in 0.3% low melting agarose (Lonza, Allendale, NJ, U.S.A.) with 10% fetal bovine serum in DMEM medium and then layered on top of 3 ml of 0.5% low melting agarose plus 10% fetal bovine serum in DMEM medium in 6 cm dishes. Cells were allowed to grow at 37°C with 5% CO2 for 14 days. The plates were stained with 0.005% crystal violet in 30% ethanol for 6 h.
Luciferase-reporter Assay
PC-3 cells were seeded at 1.9×105 cells/well in a 12-well plate in DMEM containing 10% FBS. 24 h after plating, PC-3 cells were transfected with pRL-TK-Renilla luciferase plasmid (normalization vector; 8 ng/well), 4X NF-κB (reporter gene vector; 800 ng/well) using the PolyJet™ in vitro DNA transfection reagent (SignaGen Laboratories, Rockville, MD). 24 h after transfection, cells were treated with increasing concentrations of CAPE. After an additional 24 hr, cells were lysed in 100 µL passive lysis buffer (Promega, Madison, WI, U.S.A.) and luciferase activity was measured using a Dual-Luciferase kit (Promega) in a 20/20n luminometer Turner Biosystems.
Flow Cytometric Analysis
Cells were seeded in 6 cm dishes in 4.5 mL of media and CAPE was added 24 h after plating. After indicated time (24, 48, 72 hours) of culture in the presence of various concentrations of CAPE, cells were removed with trypsin and fixed in 70% ethanol in PBS overnight at −20°C. Fixed cells were washed with PBS, treated with 0.1 mg/mL RNase A in PBS for 30 min, and then suspended in 50 µg/mL propidium iodide in PBS. Cell cycle profiles and distributions were determined by flow cytometric analysis of cells using a BD Facscan flow cytometer (BD Biosciences, San Jose, CA, U.S.A.) as previously described [69].
Gene Microarray Analysis
Total RNAs were isolated from PC-3 cells treated with 20 µM CAPE or control vehicle for 24 or 72 hours using RNeasy mini kit (Qiagen, Valencia, CA, U.S.A.). The quantity of total RNA was determined by NanoDrop 2000 (Thermo Fisher Scientific, Waltham, MA, U.S.A.). The quality of total RNA samples were examined by Bioanalyzer 2100 (Agilent, Santa Clara, CA, U.S.A.) to avoid seriously degraded RNA. RNA samples with RNA integrity numbers (RIN) of <7 were excluded from this study. Complementary RNA targets were synthesized, amplified, labeled, and purified using the TargetAmp Nano-G Bioti-aRNA Labeling kit (Epicentre, Madison, WI, U.S.A.) according to the manufacturer's instruction [70]. Hybridization of labeled probe to Illumina BeadChips Human HT-12v3 was conducted according to protocol recommended by Illumina (San Diego, CA, U.S.A.). Each HT-12 chip has totally 48,804 unique 50-mer oligonucleotides probes with 15-fold feature redundancy in average [70]. Beadchips were scanned on the Illumina BeadArray 500GX reader and image processed by Illumina BeadScan software. Illumina BeadStudio software was used for preliminary data analysis [70]. All data is MIAME compliant and that the raw data has been deposited to the MIAMEExpress database (http://www.ebi.ac.uk/miamexpress/) (MIAMEExpress array databse accession number: E-MTAB-773).
Western Blotting Analysis
Proteins were separated on 6–12% SDS-PAGE gels and expression levels were determined by Western blotting using following antibodies: Total Akt, Akt2, β-actin and PDK1 were from Novus (Littleton, CO, U.S.A.). Cyclin D1, Cyclin E, p-Akt (Ser 473), p-Akt (Thr 308), p-ERK1/2, GSK3α, GSK3β, p-GSK3α, p-GSK3β, mTOR, p-mTOR(Ser2481), p-PDK1(Ser241), Rb, and p-Rb(Ser807/811) were from Cell Signaling (Danvers, MA, U.S.A.). c-Myc was purchased from Epitomics (Burlingame, CA, U.S.A.). p21Cip1, p27Kip1 and SKP2 were purchased from Santa Cruz (Santa Cruz, CA, U.S.A.). KLF6 was from Abnova (Taipei, Taiwan). Akt1, Akt3, Bcl-2, ERK1/2, p-mTOR(Ser2448) and β-tubulin were from Millpore (Billerica, MA, U.S.A.). Anti-rabbit and anti-mouse IgG secondary antibodies were from Santa Cruz. β-actin was used as loading control.
Quantitative real-time PCR
PC3 cells seeded in 10 cm dish were treated with 0 or 20 µM CAPE for 24 h or 72 h. Total RNA was isolated with RNeasy Mini Kit (Qiagen, Venlo, Hilden, Germany). cDNA was synthesized from total RNA using RevertAid H Minus First Strand cDNA Synthesis Kit (Fermentas, Waltham, Massachusetts, U.S.A.). Real-time PCR was performed on an ABI PRISM 7000 system (Applied Biosystems, Foster City, California, U.S.A.) using Maxima SYBR Green/ROX qPCR Master Mix (Fermentas). The sequences of primers are as following: CAV2 primers, 5′-agctgtctgcacatctggatt-3′(forward) and 5′-tcgtacacaatggagcaatga-3′(reverse); CCL20 primers, 5′- gaatcagaagcagcaagcaac-3′(forward) and 5′-cgtgtgaagcccacaataaat -3′(reverse); CDKN1A primers, 5′-caaaaactaggcggttgaatg-3′(forward) and 5′-aaaaggagaacacgggatgag-3′(reverse); CXCL2 primers, 5′-cttattggtggctgttcctga-3′(forward) and 5′-tcaaacacattaggcgcaatc -3′(reverse); CXCL5 primers, 5′- atctgcaagtgttcgccatag-3′(forward) and 5′-caaatttccttcccgttcttc-3′(reverse); DUSP1 primers, 5′-accatctgccttgcttacctt-3′(forward) and 5′-tgaagctgaagttgggagaga-3′(reverse); DUSP5 primers, 5′-ttgggtccaatgaggtagttg-3′(forward) and 5′-ccaaagtccaaggtcagtgaa-3′(reverse); GADD45A primers, 5′-gcagatggaaagaggtgaaaa-3′(forward) and 5′-agttttccttcctgcatggtt-3′(reverse); GDF15 primers, 5′-ctacaatcccatggtgctcat-3′(forward) and 5′-agtggcagtctttggctaaca-3′(reverse); HIST1H2BD primers, 5′-ggaagtctcatctgcctgaaa-3′(forward) and 5′-ttagttccttcccctcggtaa-3′(reverse); KLF4 primers, 5′-aagaacagatggggtctgtga-3′(forward) and 5′-ccttggcattttgtaagtcca-3′(reverse); KLF6 primers, 5′-taacggctgcaggaaagttta-3′(forward) and 5′-ccttcccatgagcatctgtaa-3′(reverse); NOV primers, 5′- ctctattggctccctttttgg -3′(forward) and 5′-ttgaagagctgcatgtttcct-3′(reverse); PPP1R15A primers, 5′-tgatgatgatggcatgtatgg-3′(forward) and 5′-ttatcagaaggctgggagaca-3′(reverse); RND3 primers, 5′-aagcggaacaaatcacagaga-3′(forward) and 5′-tcttcgctttgtcctttcgta-3′(reverse); S100P primers, 5′-gaaggcaggactcaaatgatg-3′(forward) and 5′-cctaggggaataattgccaac-3′(reverse); TOP2A primers, 5′-tgtcccagctctcatatttgg-3′(forward) and 5′-catttcgaccacctgtcactt-3′(reverse); TUBA1A primers, 5′-cttccaccctgagcaacttatc-3′(forward) and 5′-atctccttgccaatggtgtagt-3′(reverse).
siRNA knockdown of p21Cip1
Human p21Cip1(CDKN1A) antisense and randomly scrambled sequence control were purchased from Thermo (Waltham, Massachusetts, U.S.A.). The transfection procedure was performed using lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA, U.S.A.) according to the manufacturer's recommended protocal. 20 nM RNA were used for both scramble and p21Cip1 knockdown.
Data Analysis
Data are presented as the mean +/− SD of at least three independent experiments. Student's t test (two-tailed, unpaired) was used to evaluate the statistical significance of results from proliferation assay experiments.
Supporting Information
A network enriched by IPA analysis with drug targets (TUBA) of docetaxel and vinblastine (colored in orange) indicated. The union of differentially expressed genes (DEGs) at 24 h and 72 h post CAPE treatment was input to IPA. Upregulated genes are colored in red, and downregulated genes in green. Values of log ratio of expression change were also shown in the bottom of DEGs.
(JPG)
{kind=link}
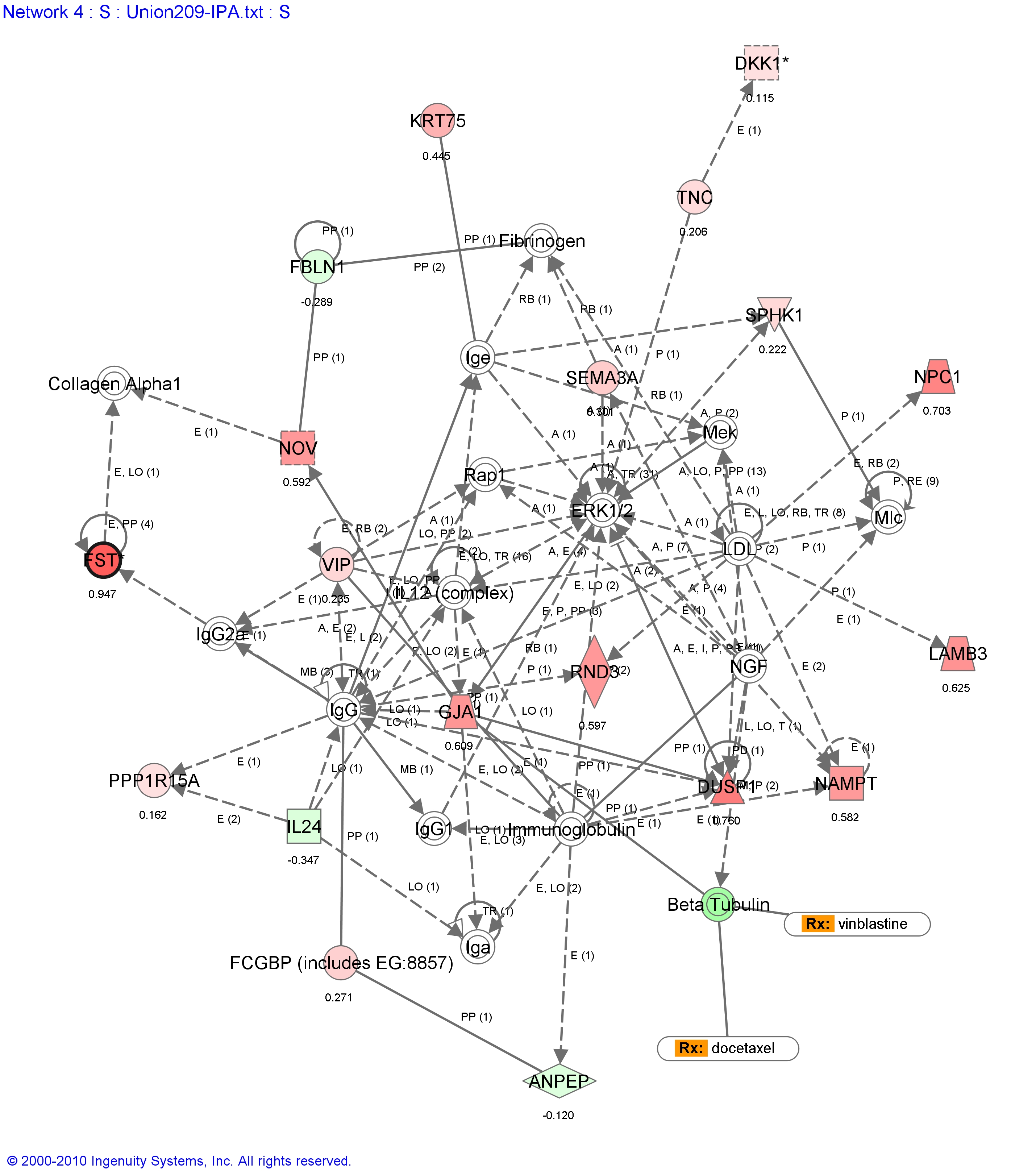
A network enriched by IPA analysis with drug targets (beta tublin) of docetaxel and vinblastine (colored in orange) indicated. The input of IPA analysis and its display is the same as in Figure S1.
(JPG)
{kind=link}
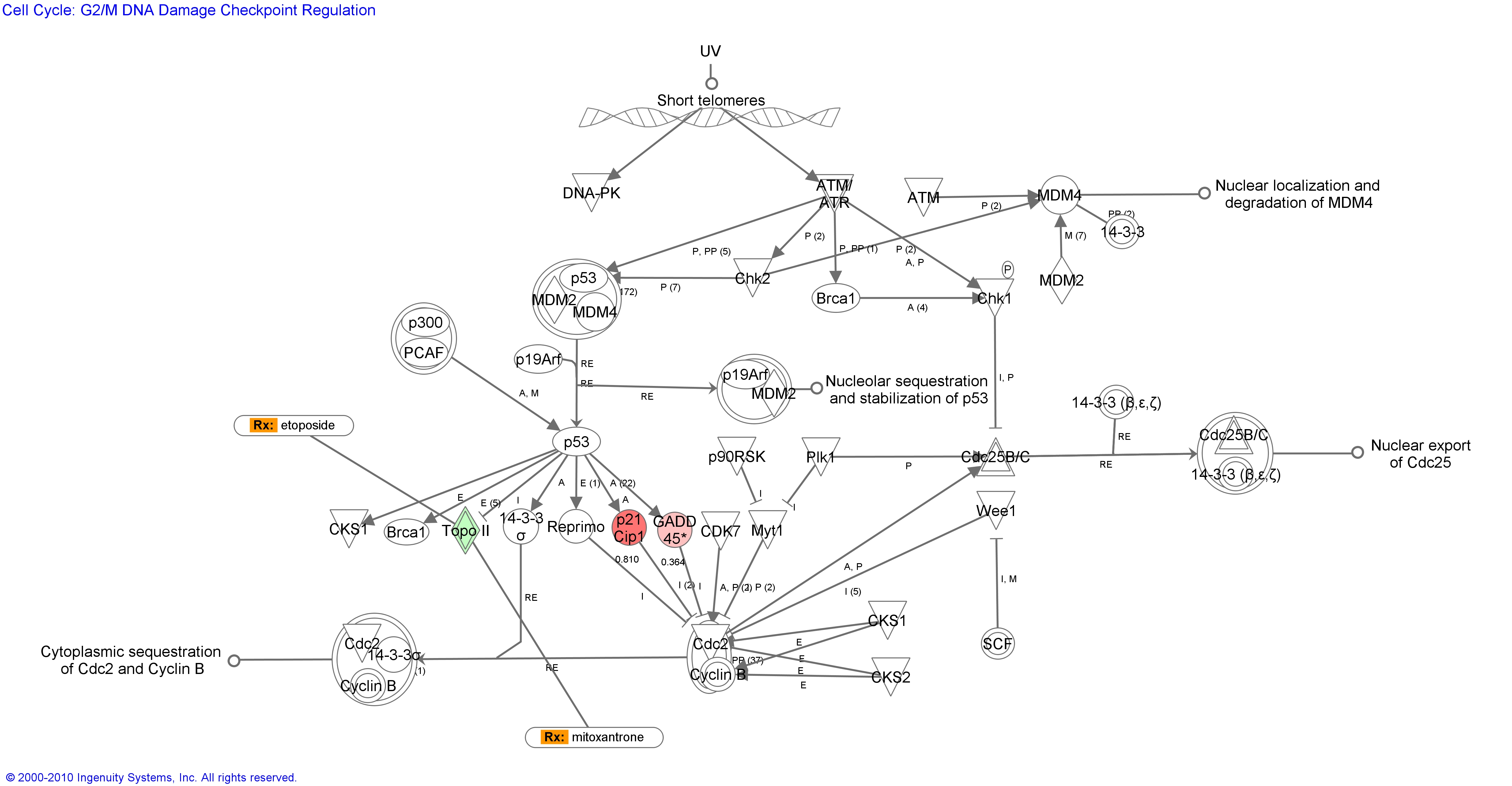
A canonical pathway (G2/M DNA damage checkpoint regulation) enriched by IPA analysis with drug targets (Topo II) of etoposide and mitoxantrone (colored in orange) indicated. The input of IPA analysis and its display is the same as in Figure S1.
(JPG)
{kind=link}
List of differentially expressed genes at 24 h post CAPE treatment. Differentially expressed gene at 24 h post CAPE treatment was shown and value of these genes at 72 h post CAPE treatment was also shown for comparison.
(XLS)
List of differentially expressed genes at 72 h post CAPE treatment. Differentially expressed gene at 72 h post CAPE treatment was shown and value of these genes at 24 h post CAPE treatment was also shown for comparison.
(XLS)
List of differentially expressed genes commonly appeared at 24 h and 72 h post CAPE treatment. Expression of genes commonly changed at both 24 h and 72 h post CAPE treatment was shown.
(XLS)
IPA gene function ontology analysis of genes whose expression are significantly changed by CAPE treatment. IPA gene function ontology analysis was shown of genes whose expression are significantly changed by CAPE treatment for 24 h and 72 h.
(XLS)
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This work was supported by CS-100-PP-12 (National Health Research Institutes) and NSC 99-2320-B-400-015-MY3 (National Science Council, NSC) in Taiwan for CPC, as well as DOH100-TD-C-111-014 (Department of Health) for SSJ and CPC. We thank support from Protein Chemistry Core Facility of Core Instrument Center in NHRI and Taiwan Bioinformatics Institute Core Facility of the National Core Facility Program for Biotechnology by NSC (NSC100-2319-B-400-001). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Bubendorf L, Schopfer A, Wagner U, Sauter G, Moch H, et al. Metastatic patterns of prostate cancer: an autopsy study of 1,589 patients. Hum Pathol. 2000;31:578–583. doi: 10.1053/hp.2000.6698. [DOI] [PubMed] [Google Scholar]
- 2.Ibrahim T, Flamini E, Mercatali L, Sacanna E, Serra P, et al. Pathogenesis of osteoblastic bone metastases from prostate cancer. Cancer. 2010;116:1406–1418. doi: 10.1002/cncr.24896. [DOI] [PubMed] [Google Scholar]
- 3.Keller ET, Zhang J, Cooper CR, Smith PC, McCauley LK, et al. Prostate carcinoma skeletal metastases: cross-talk between tumor and bone. Cancer Metastasis Rev. 2001;20:333–349. doi: 10.1023/a:1015599831232. [DOI] [PubMed] [Google Scholar]
- 4.Huggins C, Stevens R, Hodges C. Studies on prostatic cancer: II. The effects of castration on advanced carcinoma of the prostate gland. Arch Surg. 1941;43:15. [Google Scholar]
- 5.Hellerstedt BA, Pienta KJ. The current state of hormonal therapy for prostate cancer. CA Cancer J Clin. 2002;52:154–179. doi: 10.3322/canjclin.52.3.154. [DOI] [PubMed] [Google Scholar]
- 6.Chuu CP, Kokontis JM, Hiipakka RA, Fukuchi J, Lin HP, et al. Androgens as therapy for androgen receptor-positive castration-resistant prostate cancer. Journal of biomedical science. 2011;18:63. doi: 10.1186/1423-0127-18-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gilligan T, Kantoff PW. Chemotherapy for prostate cancer. Urology. 2002;60:94–100; discussion 100. doi: 10.1016/s0090-4295(02)01583-2. [DOI] [PubMed] [Google Scholar]
- 8.Pinto AC, Moreira JN, Simoes S. Liposomal imatinib-mitoxantrone combination: formulation development and therapeutic evaluation in an animal model of prostate cancer. Prostate. 2011;71:81–90. doi: 10.1002/pros.21224. [DOI] [PubMed] [Google Scholar]
- 9.Bhimani RS, Troll W, Grunberger D, Frenkel K. Inhibition of oxidative stress in HeLa cells by chemopreventive agents. Cancer Res. 1993;53:4528–4533. [PubMed] [Google Scholar]
- 10.Natarajan K, Singh S, Burke TR, Jr, Grunberger D, Aggarwal BB. Caffeic acid phenethyl ester is a potent and specific inhibitor of activation of nuclear transcription factor NF-kappa B. Proc Natl Acad Sci U S A. 1996;93:9090–9095. doi: 10.1073/pnas.93.17.9090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Watabe M, Hishikawa K, Takayanagi A, Shimizu N, Nakaki T. Caffeic acid phenethyl ester induces apoptosis by inhibition of NFkappaB and activation of Fas in human breast cancer MCF-7 cells. J Biol Chem. 2004;279:6017–6026. doi: 10.1074/jbc.M306040200. [DOI] [PubMed] [Google Scholar]
- 12.McEleny K, Coffey R, Morrissey C, Fitzpatrick JM, Watson RW. Caffeic acid phenethyl ester-induced PC-3 cell apoptosis is caspase-dependent and mediated through the loss of inhibitors of apoptosis proteins. BJU Int. 2004;94:402–406. doi: 10.1111/j.1464-410X.2004.04936.x. [DOI] [PubMed] [Google Scholar]
- 13.Chen YJ, Shiao MS, Hsu ML, Tsai TH, Wang SY. Effect of caffeic acid phenethyl ester, an antioxidant from propolis, on inducing apoptosis in human leukemic HL-60 cells. J Agric Food Chem. 2001;49:5615–5619. doi: 10.1021/jf0107252. [DOI] [PubMed] [Google Scholar]
- 14.Kuo HC, Kuo WH, Lee YJ, Lin WL, Chou FP, et al. Inhibitory effect of caffeic acid phenethyl ester on the growth of C6 glioma cells in vitro and in vivo. Cancer Lett. 2006;234:199–208. doi: 10.1016/j.canlet.2005.03.046. [DOI] [PubMed] [Google Scholar]
- 15.He YJ, Liu BH, Xiang DB, Qiao ZY, Fu T, et al. Inhibitory effect of caffeic acid phenethyl ester on the growth of SW480 colorectal tumor cells involves beta-catenin associated signaling pathway down-regulation. World J Gastroenterol. 2006;12:4981–4985. doi: 10.3748/wjg.v12.i31.4981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chuu CP, Kokontis JM, Hiipakka RA, Liao S. Modulation of liver X receptor signaling as novel therapy for prostate cancer. J Biomed Sci. 2007;14:543–553. doi: 10.1007/s11373-007-9160-8. [DOI] [PubMed] [Google Scholar]
- 17.Lebedeva I, Rando R, Ojwang J, Cossum P, Stein CA. Bcl-xL in prostate cancer cells: effects of overexpression and down-regulation on chemosensitivity. Cancer Res. 2000;60:6052–6060. [PubMed] [Google Scholar]
- 18.Liu QY, Stein CA. Taxol and estramustine-induced modulation of human prostate cancer cell apoptosis via alteration in bcl-xL and bak expression. Clin Cancer Res. 1997;3:2039–2046. [PubMed] [Google Scholar]
- 19.Williams JF, Muenchen HJ, Kamradt JM, Korenchuk S, Pienta KJ. Treatment of androgen-independent prostate cancer using antimicrotubule agents docetaxel and estramustine in combination: an experimental study. Prostate. 2000;44:275–278. doi: 10.1002/1097-0045(20000901)44:4<275::aid-pros3>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 20.Pienta KJ, Lehr JE. Inhibition of prostate cancer growth by estramustine and etoposide: evidence for interaction at the nuclear matrix. J Urol. 1993;149:1622–1625. doi: 10.1016/s0022-5347(17)36463-7. [DOI] [PubMed] [Google Scholar]
- 21.Shankar S, Chen X, Srivastava RK. Effects of sequential treatments with chemotherapeutic drugs followed by TRAIL on prostate cancer in vitro and in vivo. Prostate. 2005;62:165–186. doi: 10.1002/pros.20126. [DOI] [PubMed] [Google Scholar]
- 22.Polin L, Valeriote F, White K, Panchapor C, Pugh S, et al. Treatment of human prostate tumors PC-3 and TSU-PR1 with standard and investigational agents in SCID mice. Invest New Drugs. 1997;15:99–108. doi: 10.1023/a:1005856605726. [DOI] [PubMed] [Google Scholar]
- 23.Celli N, Dragani LK, Murzilli S, Pagliani T, Poggi A. In vitro and in vivo stability of caffeic acid phenethyl ester, a bioactive compound of propolis. J Agric Food Chem. 2007;55:3398–3407. doi: 10.1021/jf063477o. [DOI] [PubMed] [Google Scholar]
- 24.Majumder PK, Sellers WR. Akt-regulated pathways in prostate cancer. Oncogene. 2005;24:7465–7474. doi: 10.1038/sj.onc.1209096. [DOI] [PubMed] [Google Scholar]
- 25.Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 26.Gartel AL, Radhakrishnan SK. Lost in transcription: p21 repression, mechanisms, and consequences. Cancer Res. 2005;65:3980–3985. doi: 10.1158/0008-5472.CAN-04-3995. [DOI] [PubMed] [Google Scholar]
- 27.Frescas D, Pagano M. Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: tipping the scales of cancer. Nat Rev Cancer. 2008;8:438–449. doi: 10.1038/nrc2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chellappan SP, Hiebert S, Mudryj M, Horowitz JM, Nevins JR. The E2F transcription factor is a cellular target for the RB protein. Cell. 1991;65:1053–1061. doi: 10.1016/0092-8674(91)90557-f. [DOI] [PubMed] [Google Scholar]
- 29.Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- 30.Elledge SJ, Harper JW. Cdk inhibitors: on the threshold of checkpoints and development. Curr Opin Cell Biol. 1994;6:847–852. doi: 10.1016/0955-0674(94)90055-8. [DOI] [PubMed] [Google Scholar]
- 31.Kreisberg JI, Malik SN, Prihoda TJ, Bedolla RG, Troyer DA, et al. Phosphorylation of Akt (Ser473) is an excellent predictor of poor clinical outcome in prostate cancer. Cancer Res. 2004;64:5232–5236. doi: 10.1158/0008-5472.CAN-04-0272. [DOI] [PubMed] [Google Scholar]
- 32.Coffer PJ, Jin J, Woodgett JR. Protein kinase B (c-Akt): a multifunctional mediator of phosphatidylinositol 3-kinase activation. Biochem J. 1998;355(Pt 1):1–13. doi: 10.1042/bj3350001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gonzalez E, McGraw TE. The Akt kinases: isoform specificity in metabolism and cancer. Cell Cycle. 2009;8:2502–2508. doi: 10.4161/cc.8.16.9335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakatani K, Thompson DA, Barthel A, Sakaue H, Liu W, et al. Up-regulation of Akt3 in estrogen receptor-deficient breast cancers and androgen-independent prostate cancer lines. J Biol Chem. 1999;274:21528–21532. doi: 10.1074/jbc.274.31.21528. [DOI] [PubMed] [Google Scholar]
- 35.Sasaki T, Nakashiro K, Tanaka H, Azuma K, Goda H, et al. Knockdown of Akt isoforms by RNA silencing suppresses the growth of human prostate cancer cells in vitro and in vivo. Biochem Biophys Res Commun. 2010;399:79–83. doi: 10.1016/j.bbrc.2010.07.045. [DOI] [PubMed] [Google Scholar]
- 36.Wang LC, Chu KH, Liang YC, Lin YL, Chiang BL. Caffeic acid phenethyl ester inhibits nuclear factor-kappaB and protein kinase B signalling pathways and induces caspase-3 expression in primary human CD4+ T cells. Clin Exp Immunol. 2010;160:223–232. doi: 10.1111/j.1365-2249.2009.04067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ho HC, Hsu SL, Ting CT, Kuo CY, Yang VC. Caffeic acid phenethyl ester inhibits arterial smooth muscle cell proliferation and migration in vitro and in vivo using a local delivery system. Cell Mol Biol (Noisy-le-grand) 2009;55(Suppl):OL1161–1167. [PubMed] [Google Scholar]
- 38.Shigeoka Y, Igishi T, Matsumoto S, Nakanishi H, Kodani M, et al. Sulindac sulfide and caffeic acid phenethyl ester suppress the motility of lung adenocarcinoma cells promoted by transforming growth factor-beta through Akt inhibition. J Cancer Res Clin Oncol. 2004;130:146–152. doi: 10.1007/s00432-003-0520-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci U S A. 1999;96:4240–4245. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 41.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 42.Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, et al. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 43.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 44.Liang J, Slingerland JM. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle. 2003;2:339–345. [PubMed] [Google Scholar]
- 45.Alao JP. The regulation of cyclin D1 degradation: roles in cancer development and the potential for therapeutic invention. Molecular cancer. 2007;6:24. doi: 10.1186/1476-4598-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu R, Wang L, Chen G, Katoh H, Chen C, et al. FOXP3 up-regulates p21 expression by site-specific inhibition of histone deacetylase 2/histone deacetylase 4 association to the locus. Cancer Res. 2009;69:2252–2259. doi: 10.1158/0008-5472.CAN-08-3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen X, Johns DC, Geiman DE, Marban E, Dang DT, et al. Kruppel-like factor 4 (gut-enriched Kruppel-like factor) inhibits cell proliferation by blocking G1/S progression of the cell cycle. J Biol Chem. 2001;276:30423–30428. doi: 10.1074/jbc.M101194200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang W, Geiman DE, Shields JM, Dang DT, Mahatan CS, et al. The gut-enriched Kruppel-like factor (Kruppel-like factor 4) mediates the transactivating effect of p53 on the p21WAF1/Cip1 promoter. J Biol Chem. 2000;275:18391–18398. doi: 10.1074/jbc.C000062200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Katsuki Y, Sakamoto K, Minamizato T, Makino H, Umezawa A, et al. Inhibitory effect of CT domain of CCN3/NOV on proliferation and differentiation of osteogenic mesenchymal stem cells, Kusa-A1. Biochem Biophys Res Commun. 2008;368:808–814. doi: 10.1016/j.bbrc.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 50.Zimmers TA, Jin X, Gutierrez JC, Acosta C, McKillop IH, et al. Effect of in vivo loss of GDF-15 on hepatocellular carcinogenesis. J Cancer Res Clin Oncol. 2008;134:753–759. doi: 10.1007/s00432-007-0336-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schutyser E, Struyf S, Menten P, Lenaerts JP, Conings R, et al. Regulated production and molecular diversity of human liver and activation-regulated chemokine/macrophage inflammatory protein-3 alpha from normal and transformed cells. J Immunol. 2000;165:4470–4477. doi: 10.4049/jimmunol.165.8.4470. [DOI] [PubMed] [Google Scholar]
- 52.Wolpe SD, Sherry B, Juers D, Davatelis G, Yurt RW, et al. Identification and characterization of macrophage inflammatory protein 2. Proc Natl Acad Sci U S A. 1989;86:612–616. doi: 10.1073/pnas.86.2.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Duchene J, Lecomte F, Ahmed S, Cayla C, Pesquero J, et al. A novel inflammatory pathway involved in leukocyte recruitment: role for the kinin B1 receptor and the chemokine CXCL5. J Immunol. 2007;179:4849–4856. doi: 10.4049/jimmunol.179.7.4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bektic J, Pfeil K, Berger AP, Ramoner R, Pelzer A, et al. Small G-protein RhoE is underexpressed in prostate cancer and induces cell cycle arrest and apoptosis. Prostate. 2005;64:332–340. doi: 10.1002/pros.20243. [DOI] [PubMed] [Google Scholar]
- 55.Narla G, Heath KE, Reeves HL, Li D, Giono LE, et al. KLF6, a candidate tumor suppressor gene mutated in prostate cancer. Science. 2001;294:2563–2566. doi: 10.1126/science.1066326. [DOI] [PubMed] [Google Scholar]
- 56.Malhotra S, Lapointe J, Salari K, Higgins JP, Ferrari M, et al. A tri-marker proliferation index predicts biochemical recurrence after surgery for prostate cancer. PLoS One. 2011;6:e20293. doi: 10.1371/journal.pone.0020293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhao H, Jin S, Antinore MJ, Lung FD, Fan F, et al. The central region of Gadd45 is required for its interaction with p21/WAF1. Exp Cell Res. 2000;258:92–100. doi: 10.1006/excr.2000.4906. [DOI] [PubMed] [Google Scholar]
- 58.Jin S, Antinore MJ, Lung FD, Dong X, Zhao H, et al. The GADD45 inhibition of Cdc2 kinase correlates with GADD45-mediated growth suppression. J Biol Chem. 2000;275:16602–16608. doi: 10.1074/jbc.M000284200. [DOI] [PubMed] [Google Scholar]
- 59.Gao H, Jin S, Song Y, Fu M, Wang M, et al. B23 regulates GADD45a nuclear translocation and contributes to GADD45a-induced cell cycle G2-M arrest. J Biol Chem. 2005;280:10988–10996. doi: 10.1074/jbc.M412720200. [DOI] [PubMed] [Google Scholar]
- 60.Yagi A, Hasegawa Y, Xiao H, Haneda M, Kojima E, et al. GADD34 induces p53 phosphorylation and p21/WAF1 transcription. J Cell Biochem. 2003;90:1242–1249. doi: 10.1002/jcb.10711. [DOI] [PubMed] [Google Scholar]
- 61.Ding H, Duan W, Zhu WG, Ju R, Srinivasan K, et al. P21 response to DNA damage induced by genistein and etoposide in human lung cancer cells. Biochem Biophys Res Commun. 2003;305:950–956. doi: 10.1016/s0006-291x(03)00873-8. [DOI] [PubMed] [Google Scholar]
- 62.Horiguchi-Yamada J, Fukumi S, Saito S, Nakayama R, Iwase S, et al. DNA topoisomerase II inhibitor, etoposide, induces p21WAF1/CIP1 through down-regulation of c-Myc in K562 cells. Anticancer Res. 2002;22:3827–3832. [PubMed] [Google Scholar]
- 63.Zhao H, Halicka HD, Traganos F, Jorgensen E, Darzynkiewicz Z. New biomarkers probing depth of cell senescence assessed by laser scanning cytometry. Cytometry A. 2010;77:999–1007. doi: 10.1002/cyto.a.20983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kolomeichuk SN, Bene A, Upreti M, Dennis RA, Lyle CS, et al. Induction of apoptosis by vinblastine via c-Jun autoamplification and p53-independent down-regulation of p21WAF1/CIP1. Mol Pharmacol. 2008;73:128–136. doi: 10.1124/mol.108.039750. [DOI] [PubMed] [Google Scholar]
- 65.Heliez C, Baricault L, Barboule N, Valette A. Paclitaxel increases p21 synthesis and accumulation of its AKT-phosphorylated form in the cytoplasm of cancer cells. Oncogene. 2003;22:3260–3268. doi: 10.1038/sj.onc.1206409. [DOI] [PubMed] [Google Scholar]
- 66.Chuu CP, Chen RY, Hiipakka RA, Kokontis JM, Warner KV, et al. The liver X receptor agonist T0901317 acts as androgen receptor antagonist in human prostate cancer cells. Biochem Biophys Res Commun. 2007;357:341–346. doi: 10.1016/j.bbrc.2007.03.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chuu CP, Chen RY, Kokontis JM, Hiipakka RA, Liao S. Suppression of androgen receptor signaling and prostate specific antigen expression by (-)-epigallocatechin-3-gallate in different progression stages of LNCaP prostate cancer cells. Cancer Lett. 2009;275:86–92. doi: 10.1016/j.canlet.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chuu CP, Lin HP. Antiproliferative effect of LXR agonists T0901317 and 22(R)-hydroxycholesterol on multiple human cancer cell lines. Anticancer Res. 2010;30:3643–3648. [PubMed] [Google Scholar]
- 69.Chuu CP, Kokontis JM, Hiipakka RA, Fukuchi J, Lin HP, et al. Androgen suppresses proliferation of castration-resistant LNCaP 104-R2 prostate cancer cells through androgen receptor, Skp2, and c-Myc. Cancer science. 2011;102:2022–2028. doi: 10.1111/j.1349-7006.2011.02043.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jiang SS, Fang WT, Hou YH, Huang SF, Yen BL, et al. Upregulation of SOX9 in lung adenocarcinoma and its involvement in the regulation of cell growth and tumorigenicity. Clin Cancer Res. 2010;16:4363–4373. doi: 10.1158/1078-0432.CCR-10-0138. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A network enriched by IPA analysis with drug targets (TUBA) of docetaxel and vinblastine (colored in orange) indicated. The union of differentially expressed genes (DEGs) at 24 h and 72 h post CAPE treatment was input to IPA. Upregulated genes are colored in red, and downregulated genes in green. Values of log ratio of expression change were also shown in the bottom of DEGs.
(JPG)
A network enriched by IPA analysis with drug targets (beta tublin) of docetaxel and vinblastine (colored in orange) indicated. The input of IPA analysis and its display is the same as in Figure S1.
(JPG)
A canonical pathway (G2/M DNA damage checkpoint regulation) enriched by IPA analysis with drug targets (Topo II) of etoposide and mitoxantrone (colored in orange) indicated. The input of IPA analysis and its display is the same as in Figure S1.
(JPG)
List of differentially expressed genes at 24 h post CAPE treatment. Differentially expressed gene at 24 h post CAPE treatment was shown and value of these genes at 72 h post CAPE treatment was also shown for comparison.
(XLS)
List of differentially expressed genes at 72 h post CAPE treatment. Differentially expressed gene at 72 h post CAPE treatment was shown and value of these genes at 24 h post CAPE treatment was also shown for comparison.
(XLS)
List of differentially expressed genes commonly appeared at 24 h and 72 h post CAPE treatment. Expression of genes commonly changed at both 24 h and 72 h post CAPE treatment was shown.
(XLS)
IPA gene function ontology analysis of genes whose expression are significantly changed by CAPE treatment. IPA gene function ontology analysis was shown of genes whose expression are significantly changed by CAPE treatment for 24 h and 72 h.
(XLS)