

Abstract
Coupled folding-binding is central to the function of many intrinsically disordered proteins, yet not fully understood. With a continuous three-letter protein model, we explore the free-energy landscape of pairs of interacting sequences and how it is impacted by 1), variations in the binding mechanism; and 2), the addition of disordered flanks to the binding region. In particular, we focus on two sequences, one with 16 and one with 35 amino acids, which make a stable dimeric three-helix bundle at low temperatures. Three distinct binding mechanisms are realized by altering the stabilities of the individual monomers: docking, coupled folding-binding of a single α-helix, and synergistic folding and binding. Compared to docking, the free-energy barrier for binding is reduced when the single α-helix is allowed to fold upon binding, but only marginally. A greater reduction is found for synergistic folding, which in addition results in a binding transition state characterized by very few interchain contacts. Disordered flanking chain segments attached to the α-helix sequence can, despite a negligible impact on the dimer stability, lead to a downhill free-energy surface in which the barrier for binding is eliminated.
Introduction
Intrinsically disordered (ID) proteins are functional proteins that partly or wholly lack a single, stable structure under native conditions. It has become clear that intrinsic disorder plays a crucial role in a wide range of cellular processes, with particularly high occurrence in proteins involved in the regulation of transcription and translation (1,2), cell signaling (3,4), and disease (5,6). ID regions often undergo a disorder-order transition upon binding to their target molecule. Such coupled folding-binding of ID proteins typically occurs upon contact with a partner molecule that is stable, but at least one example of interacting ID regions has been characterized (7). In this case, a stable complex is formed through a mutually induced folding-binding transition (also called synergistic folding and binding). The interfaces of protein complexes formed by ID proteins have some special characteristics. For example, they exhibit a greater number of intermolecular contacts than ordered complexes, indicating especially tight fits with their partner proteins, and they rely heavily on hydrophobic-hydrophobic attraction for stability (8). Because of their peculiar properties, several functional advantages of ID proteins over globular proteins have been proposed, including the ability to bind multiple targets (9), optimize allosteric interactions (10), and decouple binding affinity and specificity (11).
It has also been suggested that the coupled folding-binding process may provide a kinetic advantage by accelerating molecular associations owing to the larger capture radius of disordered protein chains. Specifically, it was imagined that ID proteins may bind their target molecules using a fly-casting mechanism (12), where after the formation of a few initial intermolecular contacts the target is reeled in during folding. In a critical assessment of this mechanism, increased association rates were indeed seen in a native-centric Cα model for a coupled folding-binding process (13). It is interesting to note that the increased rates were due to a stabilization of the encounter complex, whereas the effect of a greater capture radius was mainly offset by a slower chain diffusion. Binding through fly-casting can be classified as a mechanism of the induced-folding type, one of the two extreme mechanistic possibilities for coupled folding-binding in which folding is initiated only after binding occurs. In the other extreme case, conformational selection, binding occurs only to the correct preformed structural element selected from the dynamic ensemble of an ID chain, meaning that folding occurs before binding. Real ID complexes are expected to form by a combination of the two mechanisms, which has also been seen in recent simulations (14,15). However, it can be mentioned that several coarse-grained and all-atom models have seen a dominant role for an induced-folding mechanism (16–20). Our group has been involved in the development of an all-atom model to capture the detailed dynamics of domain-peptide interactions (21,22), showing that coupled folding-binding can occur even for short peptide segments.
Here, we investigate and compare the fundamental features of the free-energy landscape exhibited by coupled folding-binding processes. To this end, we develop a simplified continuous sequence-based model that we initially test on the folding of a set of ideally designed sequences. This test allows us to tune the relative strengths of the effective forces of the model, primarily hydrogen bonding and hydrophobic attractions. We then apply the model to the interaction of two different sequences, making a stable dimeric α-helix bundle structure at low temperatures, and we use this interaction to address two issues in particular: the role of the intrinsic stabilities of individual protein monomers in the interaction dynamics, and the impact of disordered flanking-chain segments on the coupled folding-binding process. We find that in the limit of low monomer stabilities the two sequences associate through a mutually induced coupled folding-binding mechanism. This mode of association leads to a binding transition state characterized by very few intermolecular contacts, and it should be ideally suited for fast molecular association. Disordered chain segments flanking the primary binding region are found to be able to strongly impact the basic features of the interaction. Despite a large impact on the activation barrier, the attractive forces involving the flanks are weak enough that they do not affect the stability of the complex. This suggests that care should be taken to consider disordered flanking regions in kinetic experiments of protein interactions involving ID proteins.
Methods
Model description
The model developed is a reduced-representation, continuous protein model with three amino acid types: polar (p), hydrophobic (h), and glycine (G). All backbone atoms, N, Cα, C′, H, O, Hα1, and Hα2, are included explicitly, whereas side chains are represented by a single atom, an enlarged Cβ. Hence, h and p are geometrically related to alanine, whereas G is a normal glycine. Any N-amino-acid model conformation C is specified by the 2N dihedral angles and , in addition to the chain's overall translational and rotational orientation. All other minor degrees of freedom, such as bond lengths and angles, are fixed at standard values (see Table S1 in the Supporting Material).
The energy function can be written as , where the four terms represent excluded volume, local partial charge interaction, hydrogen bonding, and hydrophobic attraction, respectively. In the following discussion, we describe the terms in turn. The excluded-volume energy is expressed as
| (1) |
where the sum is over all atom pairs ij, rij is the ij distance, is the sum of the i and j atom radii with σi = 1.75 Å, 1.55 Å, 1.42 Å, and 1.00 Å, for C, N, O, and H atoms, respectively, and λij is a scale factor. An exception to this choice of σij is for Cβ-Cβ pairs where σij = 5.0 Å, thereby mimicking side-chain bulkiness. The scale factor λij is chosen in the following way: 1.00 for ij pairs connected by three covalent bonds, 1.25 and 1.00 for amide HH and carboxyl OO pairs, respectively, and 0.75 for all other pairs. The excluded-volume reduction (λij < 1) for global ij pairs partially alleviates the reduced flexibility of a chain with only torsional angles as degrees of freedom. The overall excluded-volume weight factor is kexvol = 0.1.
The next term takes into account locally the interactions between partial charges on the backbone,
| (2) |
where the ij sum is over all pairs of N, H, C′, and O atoms in the amino acid I with partial charges qi = –0.2, +0.2, +0.42, and –0.42, respectively, and the I sum is over all amino acids. The strength is klocal = 50.
The hydrogen-bond term can be written as
| (3) |
where i and j sum over all NH and CO groups, respectively, but ij pairs must be separated by at least two NH (or CO) groups to be included. The quantities αij and βij are the N-H-O and H-O-C′ angles, respectively (the contribution of any ij with either αij < 90° or βij < 90° is set to zero), and the parameter σhb = 2.0 Å. The hydrogen-bonding strength is khbond = 3.1, but it can be reduced through the sequence-dependent scale factor γij. For hh, hp, and pp hydrogen bonds, γij = 1, and for GG, Gh, and Gp pairs, γij = 0.75. We added the factor γij < 1 for G mainly to enhance its propensity to break secondary structure in the model. This reduction may not be physically unreasonable, however, given glycine's lack of side chain, which may allow it to more easily interact with surrounding water molecules and therefore lead to effectively weaker intra chain hydrogen bonds compared to other amino acids (23).
The hydrophobicity energy, Ehp, is given by
| (4) |
where the sum runs over all hydrophobic Cβ atoms, excluding nearest and next-nearest amino acid neighbors. The optimal Cβ-Cβ distance for a hydrophobic contact is σhp = 5.0 Å, which is set to coincide with the excluded-volume parameter σij = 5.0 Å for Cβ-Cβ pairs. In this way, the excluded-volume repulsion (Eq. 1) and hydrophobic attraction (Eq. 4) combine into a smooth Lennard-Jones-like interaction (24,25). The hydrophobicity strength is khp = 0.805.
Monte Carlo simulation
In all simulations, the protein chains are kept in a (100 Å)3 box with periodic boundary conditions. To find the thermodynamic behavior, as determined by the amino acid sequence and the energy function E(C), we use simulated tempering (ST) (26–28), which is an expanded-ensemble Monte Carlo (MC) method. We perform three independent ST simulations for each system studied, and these are used to estimate statistical errors. Each run is at least 109 elementary MC updates. For the single-chain simulations, two different MC update types are used: a pivot move, where a single or angle is selected and assigned a new, random value, and a biased gaussian step (BGS) (29), where eight consecutive , angles are turned in a coordinated way to yield approximately local chain deformations. The pivot and BGS moves are efficient at evolving chains at high and low temperatures, respectively. For the two-chain systems, we use, in addition, single-chain rigid body translation and rotation updates. The fixed A1(S) and A2(S) conformations were picked from an A1+A2 test simulation at low E, i.e., where the three-helix bundle dimer was fully formed. In simulations with A1(S) or A2(S), only rigid-body updates are used for the (S)-chains, such that their internal conformations remain entirely fixed. The Monte Carlo kinetics runs differ from the thermodynamics runs in two ways. First, the unphysical, global pivot move is turned off. Second, simulations are performed at a single, fixed temperature. The A1_Lh5 and A1_Lp5 chains are initiated from random, high-T conformations and 500 independent runs are performed for each system.
Order parameters for folding and binding
As measures of binding progress we use two different order parameters, RCM, the distance between the centers of mass (CM) of the 2 chains, and Nhh, the number of interchain hh contacts. Two h amino acids kl are considered in contact if , where is the kl contribution to the total hydrophobicity energy, Ehp. The CM is calculated over all Cα atoms in the A1 or A2 chains. All flanking chain segments are ignored in the CM calculation. Progress in folding is measured using the root mean-square deviation (RMSD), calculated over all Cα atoms, and the fractions of amino acids in an α or a β state. An amino acid i is considered to be in an α-helical state if and , and in a β-sheet state if and .
Results
Continuous hydrophobic/polar model for protein folding and binding
To investigate and compare the basic features of various protein-protein association modes, we developed a simplified protein model with three amino acid types, h, p, and G. The model is described in detail in Methods. The two main molecular driving forces in the model are pairwise hh attraction and hydrogen bonding. To determine optimal model parameters, we applied the model to the six sequences A1, A2, A3, B1, B2, and B3, which can be found in Table 1. The A sequences were designed according to the strategy of Regan and DeGrado (30). In A1, the 16 h and p amino acids are distributed along the sequence such that when the chain makes an α-helix, all the h amino acids appear on the same side. In A2 and A3, two and three A1 sequences, respectively, are joined by flexible GGG segments, allowing the formation of α-helical bundles stabilized in part by hydrophobic forces. The B1 sequence was inspired by the de novo design of the three-stranded antiparallel β-sheet Betanova (31). Its six h amino acids are distributed such that they can cluster on the same side of the β-sheet. B2 and B3 are similar to B1 but have slightly longer strands.
Table 1.
Three-letter amino acid sequences studied
| Protein | N | Sequence |
|---|---|---|
| A1 | 16 | pphpphhpphpphhpp |
| A2 | 35 | A1-GGG-A1 |
| A3 | 54 | A1-GGG-A1-GGG-A1 |
| B1 | 18 | phphpGGphphGGhphph |
| B2 | 21 | phphphGGhphphGGhphphp |
| B3 | 24 | phphphpGGphphphGGhphphph |
| A1_Lhk | 18 + 6k | (GhG)kG-A1-G(GhG)k |
| A1_Lpk | 18 + 6k | (GpG)kG-A1-G(GpG)k |
The three amino acids types are p (polar), h (hydrophobic), and G (glycine); N is the number of amino acids; and k = 1, 2, 3, 4, and 5.
The thermodynamic behavior of the six sequences is shown in Fig. 1. At high temperatures, T, all sequences exhibit a similarly low propensity for ordered structures. As T is lowered, however, the A and B sequences behave differently and form predominantly α- and β-structure, respectively. In fact, all sequences fold into distinct stable native states that are well represented by the minimum-energy conformations, shown in Fig. 2. This can be seen from the E-RMSD free-energy surfaces in Fig. 3, where E is the total energy and the RMSD is with respect to the corresponding min-E structures, which show global minima at low-E/low-RMSD values. The temperature dependence of the specific heat capacity, Cv, exhibits single-peak behavior for all sequences, indicating that folding is a cooperative transition from the unfolded to the native state (see Fig. 1, C and D). We identify the Cv peaks as the folding temperature, Tf, where the folded and unfolded ensembles are roughly equally populated. We note that the Cv peaks generally become more pronounced with chain length N and that the Tf values for the helical A sequences are generally higher than those for the B sequences, indicating lower thermal stabilities for the β-sheets compared to the α-helix bundles. Although we cannot directly compare the results for our model sequences with experimental thermodynamics data, we note that Betanova (31) and its variants (32) are relatively unstable, whereas three-helix bundle proteins can be quite thermostable (33,34).
Figure 1.

Folding thermodynamics of the A1–A3 and B1–B3 sequences. (A and B) The α-helix (blue lines) and β-sheet (red lines) contents, denoted and , where angled brackets indicate the thermodynamic average, as a function of the temperature (T). (C and D) The specific heat capacity, Cv, calculated using , where E is the total energy, N the number of amino acids, and kB the Boltzmann constant, as a function of T. For reference, we note that α = 0.83 – 0.88 for the structures in Fig. 2, A–C, and β = 0.67 – 0.71 for the structures in Fig. 2, D–F.
Figure 2.
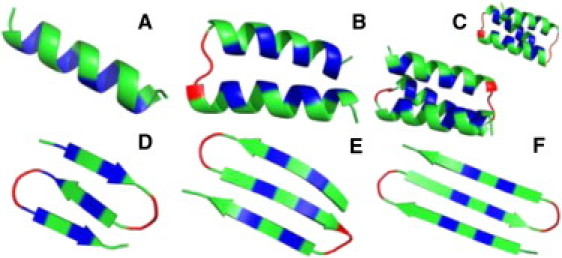
Minimum-energy conformations found across all MC simulations for the sequences A1, A2, A3, B1, B2, and B3 (A–F, respectively). A compact three-helix bundle has two distinct topological states corresponding to the two different ways of wrapping the α-helices, either clockwise or counterclockwise. Coarse-grained models of the type used in this study do not distinguish these two possibilities (54), and as a result, the native state of A3 exhibits a twofold topological degeneracy. For A3, we show the min-E conformations obtained within each topological state. Colors indicate h (blue), p (green), and G (red) amino acid types.
Figure 3.

Free-energy surfaces F(E, RMSD) taken at the folding temperature, Tf, for the sequences A1, A2, A3, B1, B2, and B3 (A–F, respectively), where E is the total energy and RMSD the root mean-square deviation measured with respect to the corresponding conformations in Fig. 2. For A3, we use, for each generated conformation, the smallest RMSD value obtained when comparing to both the clockwise and counterclockwise structures (see Fig. 2C). Free energies are calculated using F(E, RMSD) = –kBT ln P(E, RMSD), where P is the equilibrium probability distribution at T = Tf.
Models with a similar level of coarse-graining have been developed previously, and applied to both protein folding (35–38) and misfolding and aggregation (39,40). An advantage of our approach is that by initially applying the model to the folding of a set of sequences, the relative strengths of the molecular driving forces can be adjusted to reasonable values (see Fig. S1 and Text S1 in the Supporting Material). Because this test is performed on sequences making both α- and β-structure, an unphysical bias toward one type of secondary structure is avoided. Moreover, nonnative interactions, which are likely particularly important for ID proteins, are naturally included in our model. In Gō-type or native-centric modeling approaches, by contrast, nonnative forces must be added separately, and calibrating their strength is not straightforward (14,24,41). All results presented in this work are obtained for the single set of model parameters, given in Methods.
Comparing binding mechanisms: docking, coupled folding-binding, and mutually induced folding-binding
Having seen that our model is able to capture the folding of the ideal A and B sequences in Table 1, we turn now to the interaction of two of the sequences, A1 and A2. Our aim is to investigate how various features of the free-energy landscape of binding depend on the binding mechanism. To this end, we enclose A1 and A2 in a periodic box and find the thermodynamic behavior using MC simulations, as before, but with additional single-chain rigid-body updates (see Methods). We denote this system by A1 + A2. The A1 and A2 chains are at low temperatures found predominantly in a stable bound state in which the two chains are folded into a heterodimeric three-helix bundle (see Fig. 4 A, inset). To understand the formation of this complex, we use two order parameters, Nhh and RCM, which measure the number of interchain hh contacts and the CM distance between the two chains, respectively. Nhh and RCM are thus independent of the folding process, allowing progress in the two processes to be delineated.
Figure 4.
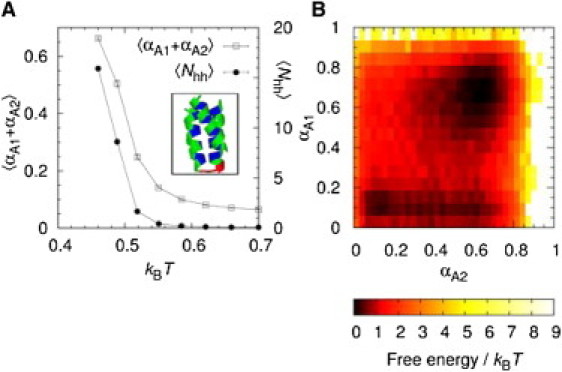
Mutually induced folding upon binding exhibited by A1 + A2, i.e., the interaction of the A1 and A2 sequences. (A) The total α-helix content, , and the number of interchain hh contacts, , as a function of T. (Inset) The A1 + A2 min-E conformation. (B) The free-energy surface, F(αA1, αA2) = –kBT ln P(αA1, αA2), where P is the probability distribution taken at the binding temperature, Tb.
Fig. 4 A shows Nhh and the total α-helix content, αA1 + αA2, as functions of the temperature. The two quantities are closely linked, meaning that for A1 + A2, folding of the two chains is concurrent with binding. An important factor controlling how a protein chain interacts with its partner is the stability of the complex as quantified by, e.g., the binding temperature, Tb, relative to the stabilities of the individual monomer native states. In analogy with the definition of Tf, we determine Tb using the maximum in the Cv curve for A1 + A2 (data not shown). We find that , which is higher than but lower than . Hence, at Tb, both chains have some residual structure on their own, a situation common for many real ID segments (42). The impact of this residual structure can be seen from the αA1, αA2 free-energy surface in Fig. 4 B, which displays two major free-energy minima corresponding to low-αA1 and high-αA1/high-αA2 states, respectively. This indicates a slight preference for A2 folding before A1. However, the free-energy landscape is rather flat, such that many different pathways are possible between the unbound, unfolded and the bound, folded states.
How is the A1 + A2 binding process influenced by an increase or decrease in the stability of the individual monomers? To address this question, we study two additional systems, A1 + A2(S) and A1(S) + A2(S). Here, (S) indicates that the chain's native state has been made artificially entirely stable, i.e., A1(S) is fixed in a single α-helix conformation and A2(S) in a two-helix bundle conformation (see Methods for details). By construction, association in A1(S) + A2(S) will occur via a docking mechanism. For A1 + A2(S), we find that kBTb = 0.579, i.e., the binding temperature is significantly higher than the folding temperature for A1, and binding can therefore occur under conditions where A1 is disordered. As it turns out, binding of A1 to A2(S) triggers the folding of A1. This is clear from the increase in the average A1 α-helix content, , from 0.10 for an isolated A1 to 0.68 for A1 + A2(S), at T = Tb. The coupled folding-binding process of A1 is also evident from the rather dramatically different free-energy profiles, F(αA1), taken separately when the molecules are spatially separate (Nhh = 0) and in contact (Nhh ≥ 1), as shown in Fig. 5. We conclude that the presence of A2(S) induces the folding of A1, starting from a highly unstructured chain. It is interesting to note that a small activation barrier in the folding of A1 persists even in the presence of A2(S).
Figure 5.

Coupled folding-binding of the A1 sequence upon interaction with A2(S), a stable two-helix-bundle A2 conformation. (A) The free-energy surface F(αA1, Nhh), where αA1 is the α-helix content of A1 and Nhh is the number of interchain hh contacts, taken at T = Tb. (B) Free-energy profile, F (αA1), taken separately for Nhh ≥ 1 (A1 and A2(S) in contact) and Nhh = 0 (no A1-A2(S) contact formed), at Tb.
Fig. 6 shows the free-energy profiles in the binding order parameters, Nhh and RCM. All three systems exhibit a single local maximum (the transition state (TS)) between the unbound (U) and bound (B) states. The activation energy for binding (ΔFTS–U) is highest for A1(S) + A2(S), i.e., when A1 and A2 associate via a pure docking mechanism. By comparison, for A1 + A2(S), where binding involves the coupled folding-binding of A1, the barrier height as measured by both order parameters is decreased. In Nhh, we find and (5.9 ± 0.2)kBT for A1(S) + A2(S) and A1 + A2(S), respectively. Another difference is a small shift in the TS toward larger RCM values, such that for A1(S) + A2(S) and for A1 + A2(S). This signals a greater capture radius for a disordered A1 chain compared to the folded A1(S), as predicted by the fly-casting mechanism and seen in previous simulations (13,43,44).
Figure 6.

Comparing the binding-free-energy profiles for different association mechanisms: A1(S) + A2(S) (docking), A1 + A2(S) (coupled folding-binding of A1), and A1 + A2 (mutually induced folding upon binding). Shown is the free energy, F, as a function of (A) the number of interchain hh contacts, Nhh, and (B) the center-of-mass distance, RCM, taken at the systems' respective binding temperatures, Tb.
The increase in and decrease in ΔFTS–U in response to the coupled folding-binding of A1 are rather modest changes, however. A greater impact is obtained when both A1 and A2 are mutually induced to fold upon binding, as in the A1 + A2 system. For this case, we obtain . The A1 + A2 binding-free-energy landscape also exhibits a different shape such that a bias toward the bound state can be accessed after only one to two initial interchain hh contacts are formed (see Fig. 6 A). By contrast, both A1(S) + A2(S) and A1 + A2(S) require the formation of additional hh contacts before the TS is reached, involving a further climb in free energy. Hence, in the mutual folding-upon-binding scenario, as seen in A1 + A2, the binding TS is reached extremely easily upon chain-chain contact, a situation that should be highly suitable for fast binding kinetics.
Impact of flanking chain segments on coupled folding-binding
Many protein regions undergoing coupled folding-binding are found within longer regions of intrinsic disorder (45,46). To fully understand the properties of these interactions in vivo, it is therefore important to investigate the role played by flanking chain segments. Previously, it has been suggested that disordered flanks adjacent to short linear binding motifs may be important to suppress aggregation of ID proteins (47). Here, we investigate the impact of flanks on the coupled folding-binding process. To this end, we use our coarse-grained approach and model disordered flanking chain segments as repetitions of the sequence triplets, GhG or GpG, and attach them to both ends of A1 to give the long A1 sequences A1_Lhk and A1_Lpk, respectively, where k indicates the number of triplets attached at each end (see Table 1). For the A1_Lhk and A1_Lpk sequences with k = 1, 2, 3, 4, and 5, the flanking regions remain entirely disordered even under conditions where the A1 α-helix is fully formed (see Fig. S2). We study the interaction of A1_Lhk and A1_Lpk with A2(S) for various k and ask in particular how the attached p- and h-flanks impact 1), the stability of the complex and 2), the binding-free-energy landscape.
Fig. 7 A shows that the activation free energy of binding, , decreases sharply with the flank length for A1_Lhk + A2(S), whereas for A1_Lpk + A2(S), there is an almost constant trend in k. This suggests that hh interactions between the h-flanks and the binding target A2(S) act to lower the free-energy barrier between the unbound and bound states. As it turns out, the impact of h-flanks is especially large in the RCM order parameter where the shape of the free-energy curves changes, as can be seen from Fig. 8. For A1_Lh5 + A2(S), the free-energy profile in RCM exhibits a plateaulike behavior over a relatively large RCM range, ≈ 17 – 37Å, rather than a pronounced single peak. This indicates that long disordered h-flanks attached to A1 might be effective at increasing the A1 capture radius, reminiscent of the fly-casting mechanism (12,44), although our flanks remain disordered in both the bound and unbound states.
Figure 7.

Impact of disordered flanking chain segments on the coupled folding-binding process. (A) The activation free energy of binding, ΔFTS–U, as a function of flank length, k, for A1_Lhk + A2(S) and A1_Lpk + A2(S), where k is the number of GhG (A1_Lhk) or GpG (A1_Lpk) triplets attached to both ends of A1 (see Table 1). ΔFTS–Udecreases with k for the h-flanks while remaining more or less constant for the p-flanks. (B) The binding temperature, Tb, as a function of k.
Figure 8.
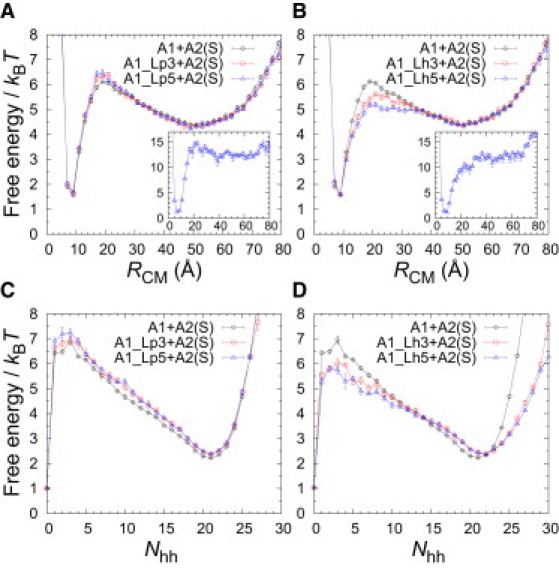
Impact of disordered flanks on the shape of binding-free-energy profiles. Free energy, F, as a function of RCM (A and B) and Nhh (C and D) for different sequences interacting with A2(S): A1 with long flanks (A1_Lh5 and A1_Lp5), A1 with intermediate flanks (A1_Lh3 and A1_Lp3), and, for comparison, A1 with no flanks attached. The free energies are calculated at the A1 + A2(S) binding temperature, Tb. (Insets) F(RCM) for A1_Lh5 and A1_Lp5, respectively, at kBT = 0.48, corresponding to T = 0.83Tb.
To test this effect more directly, we perform a large number of small-step Monte Carlo kinetic runs for A1_Lh5 + A2(S) and A1_Lp5 + A2(S). All these runs are initiated with the two chains at the largest possible separation within our periodic box ( Å), and relaxation toward equilibrium is thereafter observed, as shown in Fig. 9. Overall, the folding of the long A1 sequences, as measured by , where t is the number of MC steps, and their binding with A2(S), as measured by , follow a similar trend. This is a direct observation of coupled folding-binding in our model. Despite the general similarity between the two systems, there are interesting differences in the details, especially at small t, where decreases significantly more rapidly for A1_Lh5 than for A1_Lp5 (see Fig. 9 A). This difference can be quantified by the mean time of formation of the first interchain hh contact. We find this capture time to be tcap ≈ 6.3 × 105 and ≈ 8.6 × 105 MC steps for A1_Lh5 and A1_Lp5, respectively. Hence, on average, A1 with h-flanks captures its target faster than A1 with p-flanks, reflecting the different shapes of their free-energy profiles (see Fig. 8, A and B). However, in total, the relaxation process is not faster for A1_Lh5. In fact, after t MC steps, helix formation is slightly slower for A1_Lh5 than for A1_Lp5 (see Fig. 9 B). A possibility is therefore that although attractive flanks may aid in capturing the target, they may also result in kinetic traps, leading to slower subsequent folding.
Figure 9.

Impact of disordered flanks on coupled folding-binding kinetics. A large number of independent small-step MC relaxation runs are performed in which A2(S) binds either the A1_Lh5 sequence (A1 with attached h-flanks) or the A1_Lp5 sequence (A1 with attached p-flanks). Shown are (A) and (B) , where indicates the average over all runs at different MC time steps t. The runs are performed at kBT = 0.579, corresponding to the A1 + A2(S) binding temperature.
Another interesting difference between the p- and h-flank systems emerges at lower temperatures. At T = 0.83Tb, e.g., a small but clear free-energy barrier in RCM remains for A1_Lp5, whereas the barrier is absent for A1_Lh5 (see Fig. 8, A and B, insets), suggesting a different type of process. A fairly large free-energy barrier remains in Nhh for both systems, even at low T (data not shown), but this measure takes into account only a subset of formed hh contacts. A more revealing parameter by which to follow the combined folding-binding process would be , counting both inter- and intrachain hh contacts. For , the situation is analogous to RCM, i.e., A1_Lp5 displays a distinct free-energy barrier, whereas the free-energy profile for A1_Lh5 is barrierless at low T (see Fig. S3). The observed differences thus indicate that flanking chain segments, if attracted to the target molecule, may lead to a binding scenario that is downhill in free energy. Finally, let us stress that while the h-flanks can be highly significant for the folding-binding process, as we have seen, their effect on the overall thermal stability of the bound state is negligible (see Fig. 7 B). In our model, therefore, attractive flanks transiently stabilize the binding TS during folding-binding thereby speeding up target capture while at the same time being weak enough that they do not affect the overall stability of the final complex.
Discussion
ID regions undergoing coupled folding-binding are common in protein-interaction networks and involve the formation of all types of secondary and irregular structures (48). One of the suggested biological advantages of coupled folding-binding is that it may result in fast associating/dissociating, weak-affinity complexes and therefore make ID proteins especially well suited for regulatory and cell-signaling processes. Although some ID regions fold into entire globular domains upon binding, most coupled folding-binding events involve short amphiphilic segments within longer regions of intrinsic disorder (11).
In our coarse-grained model, the amphiphilic A1 sequence undergoes a near-complete binding-induced folding transition upon interaction with the stable two-helix bundle structure A2(S) (see Fig. 5). By comparing the binding free-energy landscape of this interaction with the docking case of the same sequences, we find that the coupled folding-binding process both increases the capture radius of the A1 chain and reduces the height of the free-energy barrier for binding. This suggests an accelerated molecular association rate. Previous simulation studies (13,43) have shown qualitatively similar trends. In particular, Huang and Liu (13) showed that a key factor in controlling the binding kinetics is the height of the free-energy barrier for binding and that a greater capture radius does not automatically produce faster association kinetics, because it also leads to a slower chain diffusion (13). It is interesting, therefore, that the coupled folding-binding of our A1 sequence, with a length typical of many coupled folding-binding segments (11), reduces the binding barrier with only ≈ 0.5kBT (see Fig. 6, A and B), a rather marginal effect compared to the overall barrier height, which is ≈10-fold larger.
This raises the possibility that other mechanisms may be used to further speed up the association/dissociation of short binding segments. Our simple model suggests two possibilities. The first is an association mode in which both chains bind and fold simultaneously, as exhibited by our A1 + A2 system (see Fig. 4). We find that such a mutually induced folding-binding mechanism reduces the binding barrier more substantially, with ≈ 1.5kBT. In addition, A1 + A2 displays a binding TS characterized by the formation of only one or two attractive interchain contacts, which should further facilitate molecular association. Second, our results indicate that the shape of the binding free-energy surface can be strongly impacted by disordered flanking segments adjacent to the primary binding region. Evidence for the concerted evolution between short linear protein motifs and their flanks has been found (49), suggesting that they are often functional, and the conformational ensemble of a small α-helical peptide was found to be affected by its flanking residues (50).
The addition of flanks to our A1 sequence reveals that attractive intermolecular forces involving the flanks can be highly efficient at stabilizing the TS in a coupled folding-binding scenario. This stabilization occurs in our model despite the fact that the attached h-flanks have little impact on the bound-state stability, meaning that these nonnative intermolecular forces are relatively weak. Let us stress that nonattractive disordered flanks by themselves are not expected to impact the stability of a complex if they remain fully disordered in the bound state, as we see in the case of p-flanks attached to the A1 sequence.
In two recent restrained molecular dynamics simulations of the barnase-barstar complex (51,52), a downhill binding-free-energy surface was found, despite the fact that both proteins are stable on their own. This apparent disagreement with our results points to the potentially large role that can be played by electrostatic interactions in steering molecular associations. The stability of the barnase-barstar complex is, however, particularly strongly dependent on electrostatic interactions (53), with an association driven in part by a large net charge difference between the two molecules.
Conclusion
A simple, sequence-based protein model was developed and tested on the folding of a set of ideal sequences, allowing the relative strength of the molecular driving forces to be carefully tuned. It was thereafter used to investigate the general features of the binding-free-energy landscape of coupled folding-binding processes. Within the context of a helical system, our results indicate that coupled folding-binding, as compared to a docking scenario, provides a mechanism to lower the binding activation barrier, in line with previous investigations, and suggests that increased molecular association rates can be obtained. This reduction is, however, relatively small for a single α-helix with 16 amino acids which is a typical length for many functional coupled folding-binding events. We find that an association in which both chains are mutually induced to fold and bind concurrently provides a greater reduction of the activation free energy. Such synergistic folding-binding also leads to a binding TS that is characterized by the formation of very few intermolecular contacts, which could further accelerate binding. Disordered chain segments flanking a coupled folding-binding region are found to have a profound impact on the binding characteristics. In particular, attractive nonspecific interactions involving flanking chain segments may lead to an elimination of the activation barrier and a downhill free-energy surface. This can occur even when the addition of flanking sequences does not affect the overall stability of the complex. The model developed captures key elements of protein folding and binding, including secondary-structure formation and hydrophobic attraction, yet it is computationally manageable. It would therefore be interesting to apply it to coupled folding-binding processes of longer chains or those leading to the formation of β-sheet structure.
Acknowledgments
This work was supported by the Swedish Research Council (S.W.) and the Royal Swedish Physiographic Society (A.B.).
Supporting Material
References
- 1.Dyson H.J., Wright P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 2.Fuxreiter M., Tompa P., Asturias F.J. Malleable machines take shape in eukaryotic transcriptional regulation. Nat. Chem. Biol. 2008;4:728–737. doi: 10.1038/nchembio.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dyson H.J., Wright P.E. Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol. 2002;12:54–60. doi: 10.1016/s0959-440x(02)00289-0. [DOI] [PubMed] [Google Scholar]
- 4.Wright P.E., Dyson H.J. Linking folding and binding. Curr. Opin. Struct. Biol. 2009;19:31–38. doi: 10.1016/j.sbi.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iakoucheva L.M., Brown C.J., Dunker A.K. Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 2002;323:573–584. doi: 10.1016/s0022-2836(02)00969-5. [DOI] [PubMed] [Google Scholar]
- 6.Uversky V.N., Oldfield C.J., Dunker A.K. Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu. Rev. Biophys. 2008;37:215–246. doi: 10.1146/annurev.biophys.37.032807.125924. [DOI] [PubMed] [Google Scholar]
- 7.Demarest S.J., Martinez-Yamout M., Wright P.E. Mutual synergistic folding in recruitment of CBP/p300 by p160 nuclear receptor coactivators. Nature. 2002;415:549–553. doi: 10.1038/415549a. [DOI] [PubMed] [Google Scholar]
- 8.Mészáros B., Tompa P., Dosztányi Z. Molecular principles of the interactions of disordered proteins. J. Mol. Biol. 2007;372:549–561. doi: 10.1016/j.jmb.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 9.Oldfield C.J., Meng J., Dunker A.K. Flexible nets: disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genomics. 2008;9(Suppl 1):S1. doi: 10.1186/1471-2164-9-S1-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hilser V.J., Thompson E.B. Intrinsic disorder as a mechanism to optimize allosteric coupling in proteins. Proc. Natl. Acad. Sci. USA. 2007;104:8311–8315. doi: 10.1073/pnas.0700329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oldfield C.J., Cheng Y., Dunker A.K. Coupled folding and binding with α-helix-forming molecular recognition elements. Biochemistry. 2005;44:12454–12470. doi: 10.1021/bi050736e. [DOI] [PubMed] [Google Scholar]
- 12.Shoemaker B.A., Portman J.J., Wolynes P.G. Speeding molecular recognition by using the folding funnel: the fly-casting mechanism. Proc. Natl. Acad. Sci. USA. 2000;97:8868–8873. doi: 10.1073/pnas.160259697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang Y., Liu Z. Kinetic advantage of intrinsically disordered proteins in coupled folding-binding process: a critical assessment of the “fly-casting” mechanism. J. Mol. Biol. 2009;393:1143–1159. doi: 10.1016/j.jmb.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 14.Ganguly D., Zhang W., Chen J. Synergistic folding of two intrinsically disordered proteins: searching for conformational selection. Mol. Biosyst. 2011;8:198–209. doi: 10.1039/c1mb05156c. [DOI] [PubMed] [Google Scholar]
- 15.Higo J., Nishimura Y., Nakamura H. A free-energy landscape for coupled folding and binding of an intrinsically disordered protein in explicit solvent from detailed all-atom computations. J. Am. Chem. Soc. 2011;133:10448–10458. doi: 10.1021/ja110338e. [DOI] [PubMed] [Google Scholar]
- 16.Chen H.F., Luo R. Binding induced folding in p53-MDM2 complex. J. Am. Chem. Soc. 2007;129:2930–2937. doi: 10.1021/ja0678774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Turjanski A.G., Gutkind J.S., Hummer G. Binding-induced folding of a natively unstructured transcription factor. PLoS Comput. Biol. 2008;4:e1000060. doi: 10.1371/journal.pcbi.1000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen J. Intrinsically disordered p53 extreme C-terminus binds to S100B(ββ) through “fly-casting”. J. Am. Chem. Soc. 2009;131:2088–2089. doi: 10.1021/ja809547p. [DOI] [PubMed] [Google Scholar]
- 19.Wang J., Wang Y., Wang E. Multi-scaled explorations of binding-induced folding of intrinsically disordered protein inhibitor IA3 to its target enzyme. PLoS Comput. Biol. 2011;7:e1001118. doi: 10.1371/journal.pcbi.1001118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ganguly D., Chen J. Topology-based modeling of intrinsically disordered proteins: balancing intrinsic folding and intermolecular interactions. Proteins. 2011;79:1251–1266. doi: 10.1002/prot.22960. [DOI] [PubMed] [Google Scholar]
- 21.Staneva I., Wallin S. All-atom Monte Carlo approach to protein-peptide binding. J. Mol. Biol. 2009;393:1118–1128. doi: 10.1016/j.jmb.2009.08.063. [DOI] [PubMed] [Google Scholar]
- 22.Staneva I., Wallin S. Binding free energy landscape of domain-peptide interactions. PLoS Comput. Biol. 2011;7:e1002131. doi: 10.1371/journal.pcbi.1002131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Merkel J.S., Regan L. Aromatic rescue of glycine in beta sheets. Fold. Des. 1998;3:449–455. doi: 10.1016/s1359-0278(98)00062-5. [DOI] [PubMed] [Google Scholar]
- 24.Zarrine-Afsar A., Wallin S., Chan H.S. Theoretical and experimental demonstration of the importance of specific nonnative interactions in protein folding. Proc. Natl. Acad. Sci. USA. 2008;105:9999–10004. doi: 10.1073/pnas.0801874105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wallin S., Zeldovich K.B., Shakhnovich E.I. The folding mechanics of a knotted protein. J. Mol. Biol. 2007;368:884–893. doi: 10.1016/j.jmb.2007.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marinari E., Parisi G. Simulated tempering: a new Monte Carlo scheme. Europhys. Lett. 1992;19:451–458. [Google Scholar]
- 27.Lyubartsev A.P., Martsinovski A.A., Vorontsov-Velyaminov P.N. New approach to Monte Carlo calculation of the free energy: method of expanded ensembles. J. Chem. Phys. 1992;96:1776–1783. [Google Scholar]
- 28.Irbäck A., Potthast F. Studies of an off-lattice model for protein folding: sequence dependence and improved sampling at finite temperature. J. Chem. Phys. 1995;103:10298–10305. [Google Scholar]
- 29.Favrin G., Irbäck A., Sjunnesson F. Monte Carlo update for chain molecules: biased Gaussian steps in torsional space. J. Chem. Phys. 2001;114:8154–8158. [Google Scholar]
- 30.Regan L., DeGrado W.F. Characterization of a helical protein designed from first principles. Science. 1988;241:976–978. doi: 10.1126/science.3043666. [DOI] [PubMed] [Google Scholar]
- 31.Kortemme T., Ramírez-Alvarado M., Serrano L. Design of a 20-amino acid, three-stranded β-sheet protein. Science. 1998;281:253–256. doi: 10.1126/science.281.5374.253. [DOI] [PubMed] [Google Scholar]
- 32.López de la Paz M., Lacroix E., Serrano L., López de la M. Computer-aided design of β-sheet peptides. J. Mol. Biol. 2001;312:229–246. doi: 10.1006/jmbi.2001.4918. [DOI] [PubMed] [Google Scholar]
- 33.Johansson J., Gibney B., Dutton P. A native-like three-α-helix bundle protein from structure-based redesign: a novel maquette scaffold. J. Am. Chem. Soc. 1998;120:3881–3886. [Google Scholar]
- 34.Dimitriadis G., Drysdale A., Smith D.A. Microsecond folding dynamics of the F13W G29A mutant of the B domain of staphylococcal protein A by laser-induced temperature jump. Proc. Natl. Acad. Sci. USA. 2004;101:3809–3814. doi: 10.1073/pnas.0306433101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takada S., Luthey-Schulten Z., Wolynes P.G. Folding dynamics with nonadditive forces: a simulation study of a designed helical protein and a random heteropolymer. J. Chem. Phys. 1999;110:11616–11629. [Google Scholar]
- 36.Irbäck A., Sjunnesson F., Wallin S. Three-helix-bundle protein in a Ramachandran model. Proc. Natl. Acad. Sci. USA. 2000;97:13614–13618. doi: 10.1073/pnas.240245297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Knott M., Chan H.S. Exploring the effects of hydrogen bonding and hydrophobic interactions on the foldability and cooperativity of helical proteins using a simplified atomic model. Chem. Phys. 2004;307:187–199. [Google Scholar]
- 38.Hoang T.X., Trovato A., Maritan A. Geometry and symmetry presculpt the free-energy landscape of proteins. Proc. Natl. Acad. Sci. USA. 2004;101:7960–7964. doi: 10.1073/pnas.0402525101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nguyen H.D., Hall C.K. Kinetics of fibril formation by polyalanine peptides. J. Biol. Chem. 2005;280:9074–9082. doi: 10.1074/jbc.M407338200. [DOI] [PubMed] [Google Scholar]
- 40.Małolepsza E., Boniecki M., Piela L. Theoretical model of prion propagation: a misfolded protein induces misfolding. Proc. Natl. Acad. Sci. USA. 2005;102:7835–7840. doi: 10.1073/pnas.0409389102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang Y., Liu Z. Nonnative interactions in coupled folding and binding processes of intrinsically disordered proteins. PLoS ONE. 2010;5:e15375. doi: 10.1371/journal.pone.0015375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Choi U.B., McCann J.J., Bowen M.E. Beyond the random coil: stochastic conformational switching in intrinsically disordered proteins. Structure. 2011;19:566–576. doi: 10.1016/j.str.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gupta N., Irbäck A. Coupled folding-binding versus docking: a lattice model study. J. Chem. Phys. 2004;120:3983–3989. doi: 10.1063/1.1643900. [DOI] [PubMed] [Google Scholar]
- 44.Levy Y., Onuchic J.N., Wolynes P.G. Fly-casting in protein-DNA binding: frustration between protein folding and electrostatics facilitates target recognition. J. Am. Chem. Soc. 2007;129:738–739. doi: 10.1021/ja065531n. [DOI] [PubMed] [Google Scholar]
- 45.Vacic V., Oldfield C.J., Dunker A.K. Characterization of molecular recognition features, MoRFs, and their binding partners. J. Proteome Res. 2007;6:2351–2366. doi: 10.1021/pr0701411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fuxreiter M., Tompa P., Simon I. Local structural disorder imparts plasticity on linear motifs. Bioinformatics. 2007;23:950–956. doi: 10.1093/bioinformatics/btm035. [DOI] [PubMed] [Google Scholar]
- 47.Abeln S., Frenkel D. Disordered flanks prevent peptide aggregation. PLoS Comput. Biol. 2008;4:e1000241. doi: 10.1371/journal.pcbi.1000241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vanhee P., Stricher F., Schymkowitz J. Protein-peptide interactions adopt the same structural motifs as monomeric protein folds. Structure. 2009;17:1128–1136. doi: 10.1016/j.str.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 49.Chica C., Diella F., Gibson T.J. Evidence for the concerted evolution between short linear protein motifs and their flanking regions. PLoS ONE. 2009;4:e6052. doi: 10.1371/journal.pone.0006052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jaskierny A.J., Panahi A., Feig M. Effect of flanking residues on the conformational sampling of the internal fusion peptide from Ebola virus. Proteins. 2011;79:1109–1117. doi: 10.1002/prot.22947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang L., Siu S.W., Helms V. Downhill binding energy surface of the barnase-barstar complex. Biopolymers. 2010;93:977–985. doi: 10.1002/bip.21507. [DOI] [PubMed] [Google Scholar]
- 52.Hoefling M., Gottschalk K.E. Barnase-Barstar: from first encounter to final complex. J. Struct. Biol. 2010;171:52–63. doi: 10.1016/j.jsb.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 53.Frisch C., Fersht A.R., Schreiber G. Experimental assignment of the structure of the transition state for the association of barnase and barstar. J. Mol. Biol. 2001;308:69–77. doi: 10.1006/jmbi.2001.4577. [DOI] [PubMed] [Google Scholar]
- 54.Wallin S., Farwer J., Bastolla U. Testing similarity measures with continuous and discrete protein models. Proteins. 2003;50:144–157. doi: 10.1002/prot.10271. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.