

The stereoselective construction of highly functionalized small- and medium-sized carbocycles from simple substrates is an ongoing objective in organic synthesis. One approach to this goal is the use of small organic molecules that have been designed as efficient catalysts for selective cascade reactions.[1] Over the last decade, N-heterocyclic carbenes (NHCs)[2] have provided new opportunities for the development of catalytic systems based on polarity reversal or Umpolung.[3] Notably, the NHC-catalyzed generation of homoenolate equivalents from enals has emerged as a powerful tool for the synthesis of hetero- and carbocycles.[4,5]
An important discovery by Nair et al.[6] was the ability of NHCs to catalyze the addition of homoenolates to unsaturated ketones to yield 3,4-disubstituted cyclopentenes. This approach was recently extended by the addition of methanol to afford a highly substituted racemic cyclopentane with a pendent methyl ester.[5l] Although these processes expanded the reaction repertoire of carbene catalysis, the coupling partner with the enal is limited to chalcones and oxobutenoates,[7] and the products from this reaction typically have only a single alkene functional group. To advance this carbene-driven carbocycle synthesis, we envisioned that β,γ-unsaturated α-ketoesters would be a suitable class of homoenolate acceptors for the synthesis of compounds with potentially more functional groups adorning the periphery of the carbocycle framework.[8] Unfortunately, initial attempts at NHC catalysis with Lewis base activation were unsuccessful, and addition of the homoenolate intermediate to the β,γ-unsaturated α-ketoester was not observed (Scheme 1). We have been engaged recently in developing a cooperative carbene catalysis strategy by employing different Lewis acids in combination with NHCs.[9] We have shown that a MgII Lewis acid enhances the reaction rate and yield of the products in an NHC-catalyzed homoenolate addition to hydrazones.[9a] A Lewis acid in combination with carbene catalysis also completely reverses the facial selectivity of an NHC-bound homoenolate equivalent, presumably as a result of the multiple coordination sites on the metal.[9b]
Scheme 1.

NHC/Lewis acid homoenolate strategy.
Even with these advances, a major challenge in the field of carbene catalysis is to develop processes that employ new classes of electrophiles that fail as competent substrates when only NHCs are used. With this prospect in mind, we turned our attention to the activation of β,γ-unsaturated α-ketoesters with bidentate Lewis acids: a strategy that has proven successful in stereoselective Lewis acid catalyzed processes (Scheme 1).[10] Cooperative carbene catalysis might allow access to NHC-bound homoenolates in the presence of Lewis acids that coordinate and activate unsaturated α-ketoesters. Herein, we report the use of a Lewis acid as an essential component for an NHC-catalyzed annulation of enals with a new class of electrophiles. This reaction generates highly substituted cyclopentanols containing four contiguous stereogenic centers.
We began our studies by combining cinnamaldehyde (1) with (E)-methyl 2-oxo-4-phenylbut-3-enoate (2) in the presence of the azolium precatalyst A (20 mol%), DBU (40 mol%), and Ti(OiPr)4 (2 equiv). Under these conditions, cyclopentanol 3 was isolated in 69% yield as a single diastereomer (Table 1, entry 2). The excellent diastereoselectivity of this conjugate addition prompted us to develop an enantioselective version of the reaction. Use of the chiral azolium precatalysts B–D resulted in varying yields and selectivity levels (Table 1, entries 3–5). The (S,R)-aminoindanol-derived triazolium precatalyst E[9] furnished the desired cyclopentanol in 68% yield with a 7:1 d.r. and 90% ee (Table 1, entry 6).[11] The addition of 2-propanol (6 equiv) promoted a faster transesterification, and surprisingly, we observed a small increase in the diastereo- and enantioselectivity to d.r. 18:1 and 96% ee (Table 1, entry 7).[12] A decrease in the quantity of the Lewis acid used to a substoichiometric amount resulted in incomplete conversion (results not shown).[13] An increase in the amount of the Lewis acid used to 5 equivalents led to an increase in the yield to 84% (Table 1, entry 8). When this reaction was performed under our optimized conditions but in the absence of the Lewis acid, 3 was not obtained (Table 1, entries 9 and 10). This result illustrates the importance of this Lewis acid as a key component.[14]
Table 1.
Reaction optimization.
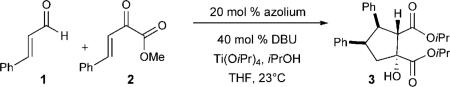
| ||||||
|---|---|---|---|---|---|---|
| Entry | Azolium | Ti(OiPr)4 [equiv] | iPrOH [equiv] | Yield [%][a] | d.r.[b] | ee [%][c] |
| 1 | A | – | 6 | 0[d] | – | – |
| 2 | A | 2 | – | 69 | > 20:1 | – |
| 3 | B | 2 | – | 76 | 12:1 | 70 |
| 4 | C | 2 | – | 44 | 10:1 | 86 |
| 5 | D | 2 | – | 69 | 20:1 | 20 |
| 6 | E | 2 | – | 68 | 7:1 | 90 |
| 7 | E | 2 | 6 | 63 | 18:1 | 96 |
| 8 | E | 5 | 6 | 84 | 20:1 | 95 |
| 9 | E | – | 6 | 0[d] | – | – |
| 10 | E | – | – | 0[d] | – | – |
Yield of the isolated product.
The diastereomeric ratio was determined by 1H NMR spectroscopy (500 MHz).
The ee value was determined by HPLC analysis using a chiral stationary phase.
Starting material was recovered after 48 h.
DBU = 1,8-diazabicyclo[5.4.0]undec-7- ene, Mes = mesityl (2,4,6-trimethylphenyl).
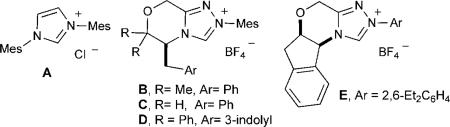
Having optimized the reaction conditions, we surveyed several β,γ-unsaturated α-ketoesters with varying substitution at the γ position (Table 2). Electron-withdrawing and electron-donating substituents on the aromatic ring were well-accommodated, with only a slight decrease in diastereoselectivity (13:1 d.r.) for the substrate with an ortho-chloro substituent (Table 2, entry 3). Heterocyclic compounds were competent substrates in the presence of TiIV; the products were obtained in moderate to good yields (52–85%) with good to excellent diastereo- and enantioselectivity (Table 2, entries 7–9). Finally, γ-cyclopropyl and γ-alkynyl substitution was well-tolerated (Table 2, entries 10 and 11), whereas only modest conversion was observed for substrates with alkyl and alkenyl groups in the γ position (results not shown).
Table 2.
Scope of the reaction with respect to the β,γ-unsaturated α-ketoester.[a]

| ||||
|---|---|---|---|---|
| Entry | R | Yield [%][b] | d.r.[c] | ee [%][d] |
| 1 | Ph | 84 (3) | 20:1 | 95 |
| 2 | 4-Cl-C6H4 | 69 (4) | 20:1 | 97 |
| 3 | 2-Cl-C6H4 | 72 (5) | 13:1 | 95 |
| 4 | 4-OMe-C6H4 | 82 (6) | 20:1 | 96 |
| 5 | 4-Me-C6H4 | 82 (7) | 20:1 | 97 |
| 6 | 3-Me-C6H4 | 68 (8) | 20:1 | 97 |
| 7 | 4-pyridyl | 52 (9) | 12:1 | 97 |
| 8 | 2-furyl | 77 (10) | 20:1 | 97 |
| 9 | 2-thienyl | 85 (11) | 20:1 | 97 |
| 10 |

|
61 (12) | 20:1 | 99 |
| 11 |
|
62 (13) | 20:1 | 94 |
See the Supporting Information for details.
Yield of the isolated product.
The diastereomeric ratio was determined by 1H NMR spectroscopy (500 MHz) of the unpurified reaction mixture.
The ee value was determined by HPLC analysis using a chiral stationary phase.
Modification of the aldehyde component was also explored (Table 3). Electron-withdrawing groups were well-tolerated at all positions of the aromatic ring, although a slight decrease in diastereo- and enantioselectivity was observed in some cases (Table 3, entries 3 and 5). The presence of electron-donating groups led to moderate yields and good enantioselectivities. However, the diastereoselectivity dropped for methoxy-substituted aryl enals (Table 3, entries 7 and 8). Finally, naphthyl-derived enals furnished the desired cyclopentanols in good yields with good enantioselectivity but with slightly decreased diastereoselectivity (Table 3, entries 9 and 10).[15]
Table 3.
Scope of the reaction with respect to the aldehyde substrate.[a]
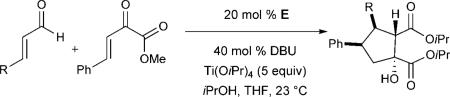
| ||||
|---|---|---|---|---|
| Entry | R | Yield [%][b] | d.r.[c] | ee [%][d] |
| 1 | 4-Cl-C6H4 | 74 (14) | 20:1 | 97 |
| 2 | 3-Cl-C6H4 | 73 (15) | 20:1 | 97 |
| 3 | 2-Cl-C6H4 | 68 (16) | 10:1 | 98 |
| 4 | 4-Br-C6H4 | 75 (17) | 20:1 | 96 |
| 5[e,f] | 4-CO2Me-C6H4 | 82 (18) | 18:1 | 91 |
| 6 | 4-Me-C6H4 | 68 (19) | 20:1 | 97 |
| 7 | 4-MeO-C6H4 | 56 (20) | 17:1 | 96 |
| 8 | 2-MeO-C6H4 | 62 (21) | 5:1 | 97 |
| 9 | 1-napthyl | 78 (22) | 12:1 | 97 |
| 10 | 2-napthyl | 77 (23) | 16:1 | 96 |
| 11[g] | 4-Cl-C6H4 | 78 (24) | 20:1 | 97 |
See the Supporting Information for details.
Yield of the isolated product.
The diastereomeric ratio was determined by 1H NMR spectroscopy (500 MHz).
The ee value was determined by HPLC analysis using a chiral stationary phase.
The reaction was carried out with 6 equivalents of Ti(OiPr)4.
Transesterification to 4-CO2iPr-C6H4 was observed.
(E)-Methyl 4-(4-methoxyphenyl)-2-oxobut-3-enoate was used.
Our proposed pathway for this reaction is illustrated in Scheme 2. Initial coordination of the α,β-unsaturated alde-hyde to the titanium(IV) Lewis acid, followed by the addition of the NHC, induces the formation of the extended Breslow intermediate I, presumably coordinated to the oxophilic titanium center. The Lewis acid concurrently coordinates to the β,γ-unsaturated α-ketoester to give II, thereby activating the α-ketoester and promoting the conjugate addition.[16] Following C—C bond formation, the bisenolate III undergoes protonation, tautomerization, and an intramolecular aldol reaction to afford intermediate IV. Subsequent acylation and catalyst turnover gives the mixed ester V, which then undergoes transesterification to furnish 3.[17] Surprisingly, neither the β-lactone nor the cyclopentene is observed, even though the metal alkoxide and the acyl azolium moiety are cis in intermediate IV: an arrangement that could lead to an intramolecular acylation. Our current proposal is that the titanium Lewis acid prevents intramolecular acylation of IV as a result of the stability of the various titanium–oxygen interactions/ligations, which undergo hydrolysis upon workup and release of the product.
Scheme 2.

Proposed reaction pathway.
The synthetic utility of this annulation reaction was initially demonstrated by further elaboration of the product cyclopentanols. The treatment of bisester 22 with lithium aluminum hydride followed by silica-gel-supported sodium periodate resulted in the formation of β-hydroxyketone 26 (Scheme 3).[18] Additionally, reduction of the bisester 22 with sodium borohydride in a THF/methanol mixture at 0 °C was regioselective (> 20:1) in favor of the 1,2-diol (the 1,3-diol was not observed), which was isolated in 71% yield. Subsequent oxidative cleavage under the aforementioned conditions, followed by decarboxylation in DMSO/H2O at 130 °C, afforded the 3,4-cis-disubstituted cyclopentanone 28.[19] Overall, these transformations demonstrate the utility of the carbonyl units that remain during this novel process promoted by an NHC and a Lewis acid. These reactions also enable efficient differentiation of the two esters as well as the formation of compounds that are challenging to access otherwise, such as 3,4-cis-substituted cyclopentanones.
Scheme 3.

Synthetic transformations: a) LiAlH4, THF, 0–25 °C; b) NaIO4·SiO2, CH2Cl2, 25 °C; c) NaBH4, THF/MeOH (2:1), 0 °C. d) NaIO4·SiO2, CH2Cl2, 25 °C; e) DMSO/H2O, 130 °C. DMSO = di-methyl sulfoxide.
In conclusion, we have developed the first NHC-catalyzed addition of homoenolates to β,γ-unsaturated α-ketoesters. The use of Ti(OiPr)4 as a mild Lewis acid compatible with NHC catalysis is essential for activation of the electrophile and promotion of the conjugate addition. This powerful NHC–Lewis acid combination enables the rapid assembly of highly substituted and functionalizable cyclopentanols from simple substrates with excellent levels of diastereo- and enantioselectivity. Furthermore, derivatization of the products provides enantiomerically enriched cyclopentanones. The two esters in the products can be differentiated by directed reduction. The powerful strategy combining Lewis basic NHC catalysis with Lewis acid activation can provide innovative ways of incorporating new reaction components and continues to be a promising area of research. New directions related to this strategy are under way and will be reported in due course.
Experimental Section
The azolium precatalyst E (0.2 equiv) and the γ-aryl (E)-α-oxobutenoic ester (3.0 equiv) were placed in an oven-dried screw-capped vial equipped with a magnetic stir bar. The vial was capped with a septum cap, removed from the dry box, and put under positive N2 pressure. Cinnamaldehyde (32.2 mg, 0.244 mmol), THF (0.5 m), Ti(OiPr)4 (5.0 equiv), iPrOH (6.0 equiv), and DBU (0.4 equiv) were added successively to the vial with a syringe, and the reaction mixture was stirred at room temperature under a static nitrogen atmosphere. Upon consumption of the aldehyde and transesterification (all reactions were complete within 48 h), the reaction mixture was filtered through a short plug of SiO2 and washed with EtOAc. The solution was concentrated under reduced pressure and purified by flash chromatography (silica gel, 9% EtOAc/hexanes) to afford the corresponding cyclopentanol. Analytical data for 3: IR (film): ν̃ = 3502, 3058, 3030, 2981, 2920, 2851, 1737, 1679, 1604, 1498, 1455, 1375, 1321, 1263, 1241, 1182, 1107, 1067, 911, 742, 699 cm–1; 1H NMR (500 MHz, CDCl3): δ = 7.06–6.97 (m, 6 H), 6.97–6.90 (m, 4 H), 5.26 (sept, J = 6.3 Hz, 1 H), 4.97 (sept, J = 6.2 Hz, 1 H), 4.26 (dd, J = 9.5, 9.5 Hz, 1 H), 4.05 (ddd, J = 9.8, 7.3, 7.3 Hz, 1 H), 3.99 (s, 1 H), 3.85 (d, J = 9.2 Hz, 1 H), 2.80 (dd, J = 13.4, 10.1 Hz, 1 H), 2.36 (dd, J = 13.5, 7.2 Hz, 1 H), 1.45 (d, J = 6.3 Hz, 3 H), 1.43 (d, J = 6.3 Hz, 3 H), 1.17 (d, J = 6.2 Hz, 3 H), 1.10 ppm (d, J = 6.3 Hz, 3 H); 13C NMR (125 mhz, CDCl3): δ = 174.8, 170.2, 140.9, 140.7, 128.7 (2 C), 128.5 (2 C), 127.8 (2 C), 127.7 (2 C), 126.1, 126.0, 81.5, 70.7, 68.5, 59.0, 50.1, 47.9, 44.3, 22.0 (2 C), 21.9 ppm (2 C); MS (ESI): m/z calcd for C25H31O5: 411 [M+H]+; found: 411. The enantiomeric ratio was measured by chiral-phase HPLC (Chiralcel OD-H, 5% IPA/hexanes, 0.50 mL min–1, 210 nm): Rt (major) = 13.2 min, Rt (minor) = 18.7 min; 95% ee.
Supplementary Material
Footnotes
Financial support was generously provided by the NIH (NIGMS RO1 GM73072), Amgen, AstraZeneca, GlaxoSmithKline, and a GAANN (Graduate Assistance in Areas of Nation Need) fellowship to D.T.C. FQRNT (Fonds Québécois de la Recherche sur la Nature et les Technologies) is also gratefully acknowledged for a postdoctoral fellowship to B.C.-D. We thank John M. Roberts (NU) for assistance with X-ray crystallography.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201005908.
References
- 1.Enders D, Grondal C, Hüttl MRM. Angew. Chem. 2007;119:1590. doi: 10.1002/anie.200603129.; Angew. Chem. Int. Ed. 2007;46:1570.; Almasi D, Alonso DA, Nájera C. Tetrahedron: Asymmetry. 2007;18:299.; Erkkilä A, Majander I, Pihko PM. Chem. Rev. 2007;107:5416. doi: 10.1021/cr068388p.; Grondal C, Jeanty M, Enders D. Nat. Chem. 2010;2:167. doi: 10.1038/nchem.539.; for selected examples, see: Rendler S, MacMillan DWC. J. Am. Chem. Soc. 2010;132:5027. doi: 10.1021/ja100185p.; Jones SB, Simmons B, MacMillan DWC. J. Am. Chem. Soc. 2009;131:13606. doi: 10.1021/ja906472m.
- 2.a Enders D, Balensiefer T. Acc. Chem. Res. 2004;37:534. doi: 10.1021/ar030050j. [DOI] [PubMed] [Google Scholar]; b Nair V, Bindu S, Sreekumar V. Angew. Chem. 2004;116:5240. doi: 10.1002/anie.200301714. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2004;43:5130. [Google Scholar]; c Zeitler K. Angew. Chem. 2005;117:7674. [Google Scholar]; Angew. Chem. Int. Ed. 2005;44:7506. [Google Scholar]; d Marion N, Diez-González S, Nolan SP. Angew. Chem. 2007;119:3046. doi: 10.1002/anie.200603380. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2007;46:2988. [Google Scholar]; e Enders D, Niemeier O, Henseler A. Chem. Rev. 2007;107:5606. doi: 10.1021/cr068372z. [DOI] [PubMed] [Google Scholar]; f Zeitler K. In: Organocatalysis. Reetz MT, List B, Jaroch S, Weinmann H, editors. Springer; Heidelberg: 2008. pp. 183–206. [Google Scholar]; g Glorius F, Hirano K. In: Organocatalysis. Reetz MT, List B, Jaroch S, Weinmann H, editors. Springer; Heidelberg: 2008. pp. 183–206. [Google Scholar]; h Phillips EM, Chan A, Scheidt KA. Aldrichimica Acta. 2009;42:55. [PMC free article] [PubMed] [Google Scholar]
- 3.Seebach D. Angew. Chem. 1979;91:259. [Google Scholar]; Angew. Chem. Int. Ed. Engl. 1979;18:239. [Google Scholar]
- 4.For a recent review of methodology for the generation of homoenolates with NHCs, see: Nair V, Vellalath S, Babu BP. Chem. Soc. Rev. 2008;37:2691. doi: 10.1039/b719083m.
- 5.Burstein C, Glorius F. Angew. Chem. 2004;116:6331. doi: 10.1002/anie.200461572.; Angew. Chem. Int. Ed. 2004;43:6205.; Sohn SS, Rosen EL, Bode JW. J. Am. Chem. Soc. 2004;126:14370. doi: 10.1021/ja044714b.; Chan A, Scheidt KA. Org. Lett. 2005;7:905. doi: 10.1021/ol050100f.; He M, Bode JW. Org. Lett. 2005;7:3131. doi: 10.1021/ol051234w.; Burstein C, Tschan S, Xie XL, Glorius F. Synthesis. 2006:2418.; Phillips EM, Wadamoto M, Chan A, Scheidt KA. Angew. Chem. 2007;119:3167. doi: 10.1002/anie.200605235.; Angew. Chem. Int. Ed. 2007;46:3107.; Chan A, Scheidt KA. J. Am. Chem. Soc. 2007;129:5334. doi: 10.1021/ja0709167.; Wadamoto M, Phillips EM, Reynolds TE, Scheidt KA. J. Am. Chem. Soc. 2007;129:10098. doi: 10.1021/ja073987e.; Chan A, Scheidt KA. J. Am. Chem. Soc. 2008;130:2740. doi: 10.1021/ja711130p.; Phillips EM, Reynolds TE, Scheidt KA. J. Am. Chem. Soc. 2008;130:2416. doi: 10.1021/ja710521m.; Maki BE, Chan A, Scheidt KA. Synthesis. 2008:1306. doi: 10.1055/s-2008-1072516.; Nair V, Babu BP, Vellalath S, Varghese V, Raveendran AE, Suresh E. Org. Lett. 2009;11:2507. doi: 10.1021/ol900571x.; for a recent report on the use of NHCs as Brønsted base catalysts, see: Phillips EM, Riedrich M, Scheidt KA. J. Am. Chem. Soc. 2010;132:13179. doi: 10.1021/ja1061196.
- 6.Nair V, Vellalath S, Poonoth M, Suresh E. J. Am. Chem. Soc. 2006;128:8736. doi: 10.1021/ja0625677. [DOI] [PubMed] [Google Scholar]
- 7.For an enantioselective variant of the NHC reaction described by Nair et al., see: Chiang P-C, Kaeobamrung J, Bode JW. J. Am. Chem. Soc. 2007;129:3520. doi: 10.1021/ja0705543.
- 8.For NHC–homoenolate additions to 1,2-dicarbonyl compounds, see: Nair V, Vellalath S, Poonoth M, Mohan R, Suresh E. Org. Lett. 2006;8:507. doi: 10.1021/ol052926n.
- 9.Raup DEA, Cardinal-David B, Holte D, Scheidt KA. Nat. Chem. 2010;2:766. doi: 10.1038/nchem.727.; Cardinal-David B, Raup DEA, Scheidt KA. J. Am. Chem. Soc. 2010;132:5345. doi: 10.1021/ja910666n.; for other examples of NHC–metal cooperative catalysis, see: Nemoto T, Fukuda T, Hamada Y. Tetrahedron Lett. 2006;47:4365.; Lebeuf R, Hirano K, Glorius F. Org. Lett. 2008;10:4243. doi: 10.1021/ol801644f.; He J, Tang S, Liu J, Sun Y, Pan X, She X. Tetrahedron Lett. 2009;50:430.; Chen Z, Yu X, Wu J. Chem. Commun. 2010;46:6356. doi: 10.1039/c0cc01207f.
- 10.Dujardin G, Molato S, Brown B. Tetrahedron: Asymmetry. 1993;4:193.; Tietze LF, Schneider C, Motenbruck A. Angew. Chem. 1994;106:1031.; Angew. Chem. Int. Ed. Engl. 1994;33:980.; Ciufolini MA, Roschangar F. J. Am. Chem. Soc. 1996;118:12082.; Johnson SC. Chem. Commun. 1998:1019.; Thorhauge J, Johannsen M, Jørgensen KA. Angew. Chem. 1998;110:2543.; Angew. Chem. Int. Ed. 1998;37:2404.; Evans DA, Johnson JS, Olhava EJ. J. Am. Chem. Soc. 2000;122:1635.; Wada E, Koga H, Kumaran G. Tetrahedron Lett. 2002;43:9397.; Koga H, Wada E. Tetrahedron Lett. 2003;44:715.; Kurosu M, Porter JR, Foley MA. Tetrahedron Lett. 2004;45:145.; Ünaleroglu C, Temelli B, Demir AS. Synthesis. 2004:2574.; Hughes KD, Nguyen T-LN, Dyckman D, Dulay D, Boyko WJ, Giuliano RM. Tetrahedron: Asymmetry. 2005;16:273.; for an application of unsaturated α-ketoesters as substrates in total synthesis, see: Evans DA, Dunn TB, Kvœrnø L, Beauchemin A, Raymer B, Olhava EJ, Mulder JA, Juhl M, Kagechika K, Favor DA. Angew. Chem. 2007;119:4782. doi: 10.1002/anie.200701515.; Angew. Chem. Int. Ed. 2007;46:4698.
- 11.The absolute and relative configuration were determined by X-ray crystallography for compound 4 (see Table 2, entry 2) and the others were assigned by analogy (see the Supporting Information). The relative configuration of the minor diastereomer 29 was determined by X-ray crystallography and others were assigned by analogy (see the Supporting Information). CCDC 793381 (4) and 793382 (29) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- 12.The minor positive influence that 2-propanol has on the stereoselectivity is currently under investigation.
- 13.A decrease in the catalyst loading (to 5 and 10 mol%) resulted in incomplete conversion (50 and 80%, respectively) after 48 h.
- 14.In NMR spectroscopic experiments (1H 500 MHz, 13C 125 MHz) in which the azolium precatalyst E, the base DBU, and α-ketoester 2 were combined in [D8]THF, no detectable differences were observed in the signals relative to those in the spectra of the starting material. Efforts are currently under way to understand the full role of the Lewis acid in these cooperative carbene catalytic processes.
- 15.At present, the use of achiral carbene A and chiral TiIV complexes does not provide the desired cyclopentane products. Further investigations in this area are ongoing.
- 16.For a recent computational study that supports a conjugate-addition pathway over a benzoin/oxy-Cope process, see: Domingo LR, Zaragozá RJ, Arnó M. Org. Biomol. Chem. 2010;8:4884. doi: 10.1039/c0ob00088d.
- 17.The transesterification reaction can be monitored by thin-layer chromatography or by 1H NMR spectroscopy (500 MHz). The mixed ester can be isolated by flash chromatography.
- 18.Zhong Y-L, Shing TKM. J. Org. Chem. 1997;62:2622. doi: 10.1021/jo9621581. [DOI] [PubMed] [Google Scholar]
- 19.a Krapcho AP, Weimaster JF, Eldridge JM, Jahngen EGE, Jr., Lovey AJ, Stephens WP. J. Org. Chem. 1978;43:138. [Google Scholar]; b Sibi MP, Asano Y. J. Am. Chem. Soc. 2001;123:9708. doi: 10.1021/ja016492c. [DOI] [PubMed] [Google Scholar]; c Morris WJ, Custar DW, Scheidt KA. Org. Lett. 2005;7:1113. doi: 10.1021/ol050093v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.