

Abstract
Tauopathies, including Alzheimer's disease, are a group of neurodegenerative diseases characterized by abnormal tau hyperphosphorylation that leads to formation of neurofibrillary tangles. Drosophila models of tauopathy display prominent features of the human disease including compromised lifespan, impairments of learning, memory and locomotor functions and age-dependent neurodegeneration visible as vacuolization. Here, we use a Drosophila model of frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17), in order to study the neuroprotective capacity of a recently identified neuronal maintenance factor, nicotinamide mononucleotide (NAD) adenylyl transferase (NMNAT), a protein that has both NAD synthase and chaperone function. NMNAT is essential for maintaining neuronal integrity under normal conditions and has been shown to protect against several neurodegenerative conditions. However, its protective role in tauopathy has not been examined. Here, we show that overexpression of NMNAT significantly suppresses both behavioral and morphological deficits associated with tauopathy by means of reducing the levels of hyperphosphorylated tau oligomers. Importantly, the protective activity of NMNAT protein is independent of its NAD synthesis activity, indicating a role for direct protein–protein interaction. Next, we show that NMNAT interacts with phosphorylated tau in vivo and promotes the ubiquitination and clearance of toxic tau species. Consequently, apoptosis activation was significantly reduced in brains overexpressing NMNAT, and neurodegeneration was suppressed. Our report on the molecular basis of NMNAT-mediated neuroprotection in tauopathies opens future investigation of this factor in other protein foldopathies.
INTRODUCTION
Tauopathies, including Alzheimer's disease (AD) and frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17), are a group of neurodegenerative diseases characterized by the presence of abnormally hyperphosphorylated tau proteins that form predominantly paired helical filaments (PHF) (1,2). In general, tau proteins are neuronal microtubule-binding proteins promoting microtubule assembly and stabilization (1,2). In the disease state, the hyperphosphorylation of tau is about four to eight times higher than in age-matched controls, disrupting its general microtubule-binding function and facilitating the formation of neurofibrillary tangles (3–7). Investigating the role of phosphorylation states of tau towards neurotoxicity revealed that many phosphor epitopes created by proline-directed kinases (SP/TP) sites show relative specificity for disease states (8,9). In fact, blocking phosphorylation by mutating 14 of these SP/TP sites to alanine significantly reduced tau toxicity in vivo (9). Hence, tau toxicity in tauopathies can be directly correlated with its hyperphosphorylated state.
Drosophila melanogaster has been used previously as an in vivo model for tauopathy, recapitulating many of the salient features of the disease (10,11). Transgenic flies neuronally overexpressing either wild-type or mutant human tau, hTauR406W, associated with FTDP17, showed adult onset, progressive neurodegeneration with compromised lifespan and accumulation and hyperphosphorylation of tau (11). Interestingly, all these disease features are exhibited in Drosophila tauopathy models with minimal formation of insoluble tau aggregates or NFTs (11,12), which are commonly associated with the human disease pathology, thereby suggesting an essential role for the soluble, hyperphosphorylated tau oligomers in the onset and progression of degenerative phenotypes. Furthermore, in these fly models, overexpression of tau in the mushroom body neurons elicited decrements in associative olfactory learning and memory prior to the initiation of neurodegeneration, indicating that perturbations in behavioral plasticity may be one of the earliest manifestations of tauopathy (13).
In an unbiased screen in Drosophila, we identified a neuronal maintenance function of nicotinamide mononucleotide (NAD) synthase nicotinamide mononucleotide adenylyltransferase (NMNAT) (14). In addition to maintaining neuronal integrity under normal conditions, overexpression of NMNAT has been shown to protect against activity-, injury-, as well as spinocerebellar ataxia 1 (SCA1) protein, Ataxin 1, with polyglutamine expansion-induced neurodegeneration (14–16), suggesting a diverse neuroprotective function of NMNAT. Furthermore, NMNAT displays chaperone function, and NMNAT-mediated protection against neurodegeneration was achieved partly through a proteasome-mediated pathway in a manner similar to heat-shock protein 70 (Hsp70) (16,17). Several reports have implicated the therapeutic potential of chaperones in different models of tauopathy and other neurodegenerative conditions (18–21). Specifically, in a cellular model of tauopathy, an inverse relationship between aggregated tau and the levels of HSP70/90 was observed, illustrating a neuroprotective effect of overexpressing these heat shock proteins. It was shown in this study that overexpressed Hsp70 or 90 directly associated with tau aggregates and reduced hyperphosphorylated tau burden and promoted tau solubility (22).
In this study, we set out to investigate the neuroprotective capacity of NMNAT in a Drosophila model of tauopathy. We found that NMNAT suppresses the morphological vacuolization, the learning and memory decrements and the locomotor deficits associated with pan-neuronal overexpression of human wild-type or mutant tau in Drosophila. Importantly, we observed comparable neuroprotection from expressing either wild-type or enzyme-inactive NMNAT (NMNATWR) in our tauopathy model. We hypothesized that NMNAT can act like other chaperones to promote the clearance of hyperphosphorylated tau via ubiquitination. We carried out extensive biochemical analyses to test this hypothesis and found that NMNAT specifically binds human tau and disease-associated phosphorylated tau. Overexpression of either wild-type or enzyme-inactive NMNAT reduced the levels of hyperphosphorylated tau oligomers and promoted clearance of hyperphosphorylated tau oligomers through ubiquitination, in a proteasome-mediated manner. Consequently, tau-induced age-dependent neurodegeneration was suppressed by NMNAT overexpression, as indicated by reduced levels of apoptosis and activated caspase 3. Hence, we show that the neuronal maintenance factor NMNAT can offer significant protection in tauopathy, and the molecular basis for NMNAT-mediated protection is through promoting the clearance of toxic hyperphosphorylated tau oligomers.
RESULTS
NMNAT expression rescues learning and memory and locomotor deficits induced by human tau overexpression
Tauopathies, such as AD, are characterized by accumulation of abnormally phosphorylated and aggregated tau, leading to synaptic dysfunction underlying age-dependent severe dementia (11,23,24). It has been shown that overexpression of wild-type tau in the adult Drosophila mushroom body neurons, which are centers of olfactory learning and memory, is manifested in compromised associative olfactory learning and memory. This significantly precedes the onset of visible neuronal loss and neurodegeneration (13), suggesting that compromised behavioral plasticity may be the earliest detectable manifestations of tauopathies. To investigate whether neuronal maintenance factor NMNAT offers protection in tauopathy, we first assessed learning and memory functions and locomotor deficits induced by tau expression with or without NMNAT overexpression.
To model tauopathy in Drosophila CNS, we overexpressed either wild-type or Arg406→ Trp (R406W) mutant tau, an isoform associated with an early-onset familial form of dementia, with a pan-neuronal elav-GAL4 driver (11). In addition, we overexpressed either wild-type or enzyme-inactive NMNAT (NMNATWR) to assess its protective capacity in this neurodegenerative model. In NMNATWR, two of the key residues required for substrate binding are mutated, resulting in <1% enzymatic activity of the wild-type protein (14). To control for tau expression dosage in different genotypes, we used GFP expression whenever NMNAT was not overexpressed.
We have established an aversive phototaxis suppression (APS) assay to measure learning and memory function (25,26), where flies were forced to associate an aversive stimulus (bitter taste from quinine) with light and learn to avoid light and suppress phototaxis. Wild-type flies are able to learn the task after 10 training trials (25,26). Learning ability can be tested in five consecutive trials immediately after the training phase (0 h post-conditioning, PC0), whereas memory capacity can be tested in five trials 6 h after the training phase (6 h post-conditioning, PC6). A successful avoidance of light in the test trial was considered ‘pass’ and the passing rate for all trials was calculated and plotted as indexes for learning or memory abilities. We hypothesized that tau-overexpressing flies will exhibit impaired behavioral plasticity and will have compromised learning and memory function. As shown in Figure 1A and Supplementary Material, Tables S1–S4, overexpression of Tau or TauR406W in the CNS did not affect the learning (PC0) and memory (PC6) abilities of 2-day-old (2 DAE, days after eclosion) flies, but resulted in significantly reduced PC0 and PC6 in 20 DAE flies, suggesting that human Tau expression in Drosophila CNS causes learning and memory deficits in an age-dependent manner. Interestingly, loss of one copy of endogenous NMNAT (nmnat/+) significantly reduced both learning and memory abilities in 2 DAE flies overexpressing TauR406W, suggesting that a reduced endogenous NMNAT level caused an earlier onset of impairment in learning and memory and exacerbated the deficit initiated by mutant tau overexpression. Conversely, overexpressing NMNAT in the CNS significantly improved tau-induced learning and memory deficits in 20 DAE flies. Importantly, overexpression of NMNATWR was neuroprotective to a level similar to wild-type NMNAT overexpression (Fig. 1A, Supplementary Material, Tables S1–S4). These results suggest that tau expression in the CNS induced age-dependent learning and memory impairment that can be suppressed by NMNAT overexpression. The neuronal level of NMNAT is an important determinant of the age of onset as well as the severity of the deficit, as a reduced level of NMNAT lowered the age of onset of learning and memory deficits, whereas overexpression of NMNAT significantly improved learning and memory functions, in our tauopathy model. However, overexpression of either wild-type or enzyme-inactive NMNAT alone in wild-type background did not have any effect on learning and memory index in 20 DAE flies compared with controls (Supplementary Material, Fig. S1A).
Figure 1.
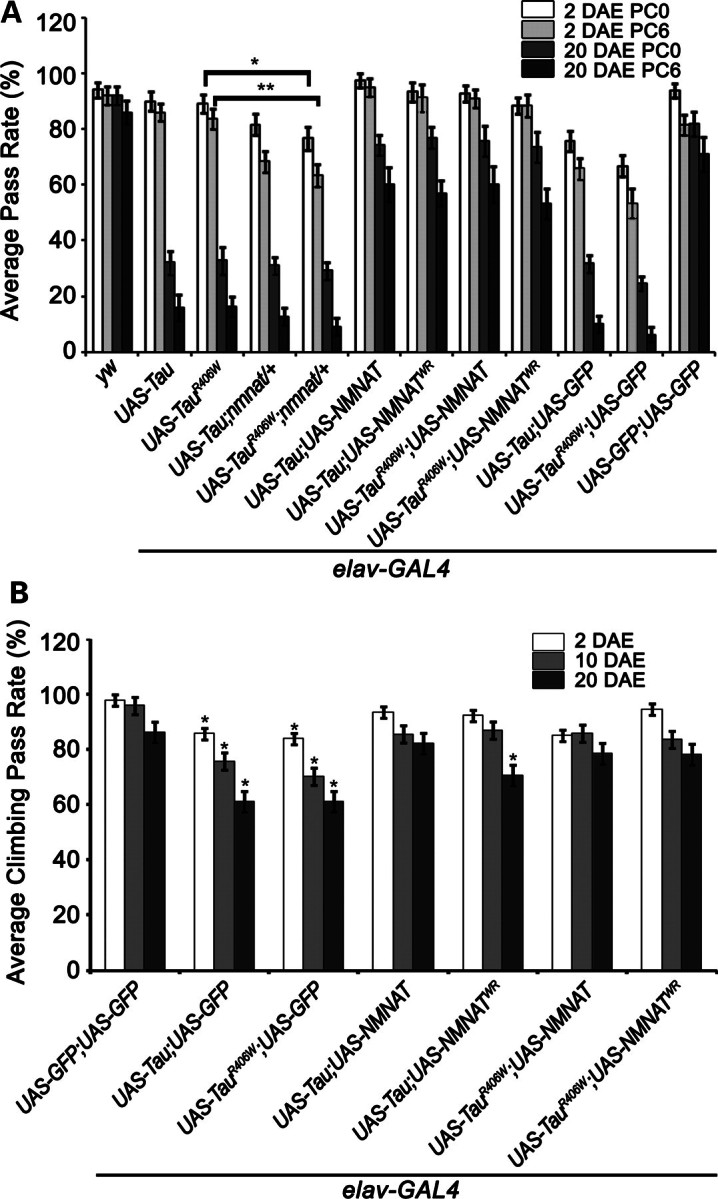
NMNAT rescues learning and memory deficits and locomotor impairments induced in tauopathy. (A) Learning and memory functions were studied in flies of specified genotypes using APS assay, where flies were conditioned to associate light with the bitter taste of quinine. PC0, 0 h post-conditioning (learning index); PC6, 6 h post-conditioning (memory index). Overexpression of tau or tauR406W causes age-dependent decrements in learning and memory functions. NMNAT or NMNATWR overexpression rescues these deficits. Error bars represent results in five consecutive trials for 20 individual flies of each age and genotype (statistical significance noted in Supplementary Material, Tables 1–4). (B) Locomotor performance was investigated using negative geotaxis assay. Overexpression of tau or tauR406W causes significant deficit in locomotor activities which are rescued by NMNAT or WR. Each bar graph is an average of 10 trials of 100 flies of each genotype and age. *P< 0.05, **P< 0.005.
In addition to learning and memory functions, we next tested locomotor activity using a negative geotaxis assay (25,27,28). As shown in Figure 1B, overexpression of either tau or tauR406W in the CNS caused an age-dependent decrease in locomotor activity as measured by climbing performance. However, overexpression of NMNAT restored the locomotor performance close to the wild-type level. Overexpressing enzyme-inactive NMNATWR restored the locomotor performance in 2 and 10 DAE flies, and an increase in locomotor performance compared with tau and GFP expressing flies, although not to the extent of the performance of GFP-only expressing flies (Fig. 1B). This observation is consistent with the level of hyperphosphorylated tau in flies overexpressing NMNATWR and Tau in 20 DAE flies (see below Fig. 4). Again, as observed with cognitive performance, overexpression of NMNAT or NMNATWR alone did not elicit any significant differences in climbing performance, compared with control flies (Supplementary Material, Fig. S1B).
Figure 4.
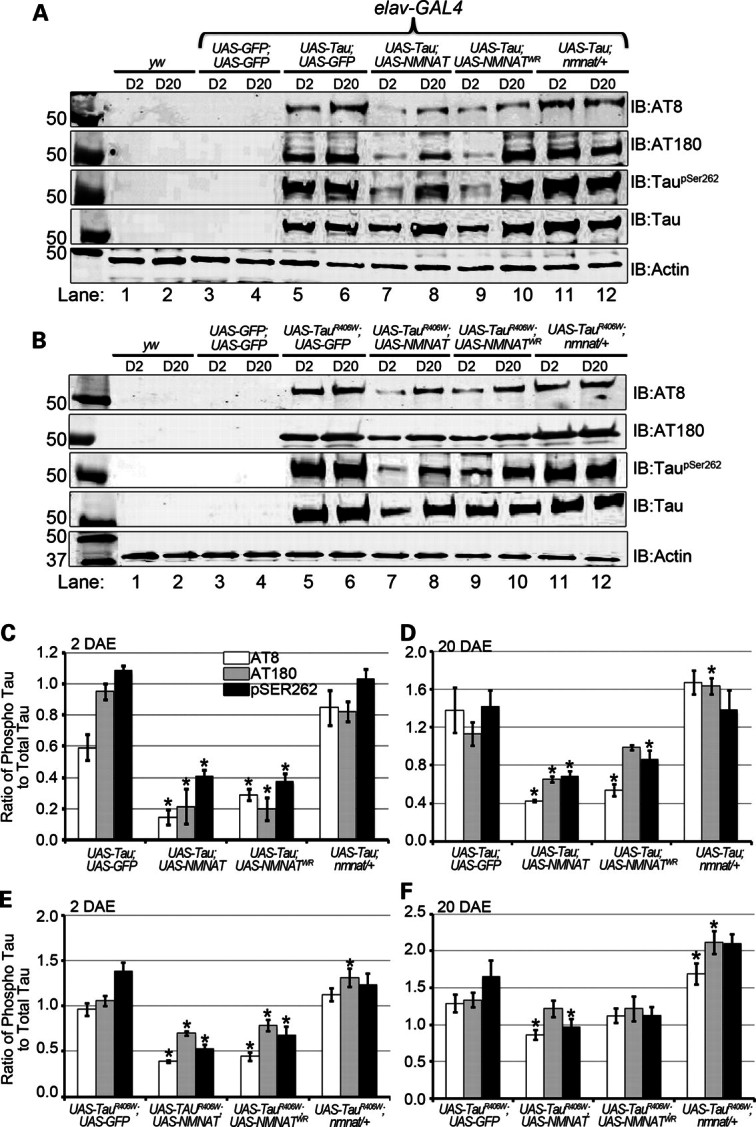
NMNAT reduces the levels of hyperphosphorylated, disease-associated tau oligomers. (A and B) Brain lysates of 2 or 20 DAE wild-type flies (lanes 1 and 2) or flies overexpressing GFP (lanes 3 and 4), hTau [WT (A) or R406W (B)] with GFP (lanes 5 and 6) or NMNAT (lanes 7 and 8) or NMNATWR (lanes 9 and 10), as well as in the nmnat/+ background (lanes 11 and 12) were probed with antibodies specific for disease-associated phospho-tau epitopes AT8, AT180 and TauSer262 as well as total hTau and actin for loading control. (C and D) Quantification of phosphorylated-tau species normalized to actin and total hTau from samples in (A) in 2 DAE (C) and 20 DAE (D) flies or in (B) in 2 DAE (E) or 20 DAE (F) flies. *P< 0.05.
The results from behavioral analyses showed that, first, overexpressing tau or tauR406W in the CNS caused an age-dependent decrease in learning and memory abilities and locomotor activity; second, the behavioral deficits were attenuated by overexpressing NMNAT; and third, the level of NMNAT is an important determinant for the age of onset and the severity of the behavioral deficits.
NMNAT rescues age-dependent vacuolization induced by tau expression
Previous work has shown that overexpression of tau in Drosophila results in an age-dependent vacuolization and degeneration of cells in the cortex as well as in neuropil structures (11). Such vacuolization is more severe in flies overexpressing mutant tauR406W. To assess whether NMNAT can rescue this tau-induced morphological change and neurodegeneration at the cellular level, we examined the morphology of the fly brain at 2 and 20 DAE with immunofluorescence labeling for NMNAT, pTauSer262 and DAPI (Fig. 2). The level and distribution of tau phosphorylation at serine 262 (pTauSer262) was examined because phosphorylation at this residue has been shown to be absolutely essential to mediate toxicity in established Drosophila models of AD (29). At 2 DAE, vacuolization was not observed in the tau-overexpressed fly brain (Fig. 2A1–4), but was present in the tauR406W overexpressed fly brain (Fig. 2B1–4, vacuole indicated by white arrowheads). At 20 DAE, multiple large vacuoles can be observed in both tau and tauR406W fly brain, indicating severe neurodegeneration (Fig. 2C1–D4). Overexpressing either wild-type (Fig. 2E1–F4) or enzyme-inactive NMNAT (Fig. 2G1–H4) significantly reduced the number and size of these age-dependent vacuolization in 20 DAE flies (Fig. 3). Interestingly, NMNAT overexpression also altered the distribution of taupSer262. In tau or tauR406W expressing brains, taupSer262 was accumulated with age and distributed in clusters or filamentous pattern (Fig. 2A2, B2, C2 and D2, black arrowheads). However, in NMNAT co-expressing brains, the level of taupSer262 was significantly reduced and no clusters were observed (Fig. 2E2, F2, G2 and H2). As a control experiment, we examined whether NMNAT or NMNATWR overexpression with GFP in the adult brain produces any morphological phenotypes. As shown in Supplementary Material, Figure S2, no morphological anomaly was observed in 20 DAE NMNAT or NMNATWR overexpressing fly brains. These results suggest that NMNAT overexpression can suppress the vacuolization phenotype caused by tau-induced neurodegeneration at the cellular level.
Figure 2.
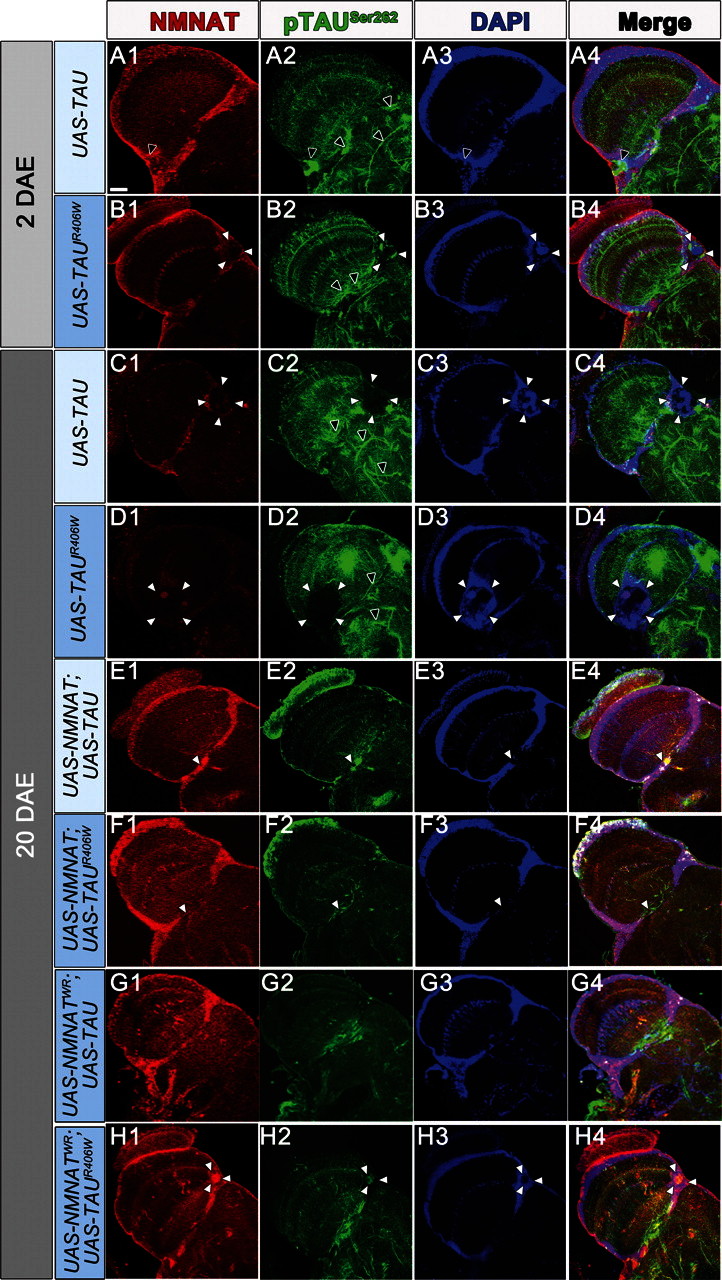
NMNAT suppresses tau-induced morphological vacuolization. Tau or tauR406W overexpression causes age-dependent vacuolization (A–D) demarcated by white arrowheads. Brains were stained for pTauSer262 to detect hyperphosphorylated tau and also NMNAT levels (red) and DAPI to mark cell bodies. Overexpression of wild-type NMNAT (E and F) or enzyme-inactive NMNATWR (G and H) significantly reduced the size and occurrence of vacuoles in 20-day-old flies. Scale bar = 20 μm. Black arrows: filamentous tau; white arrows demarcate vacuole boundary.
Figure 3.
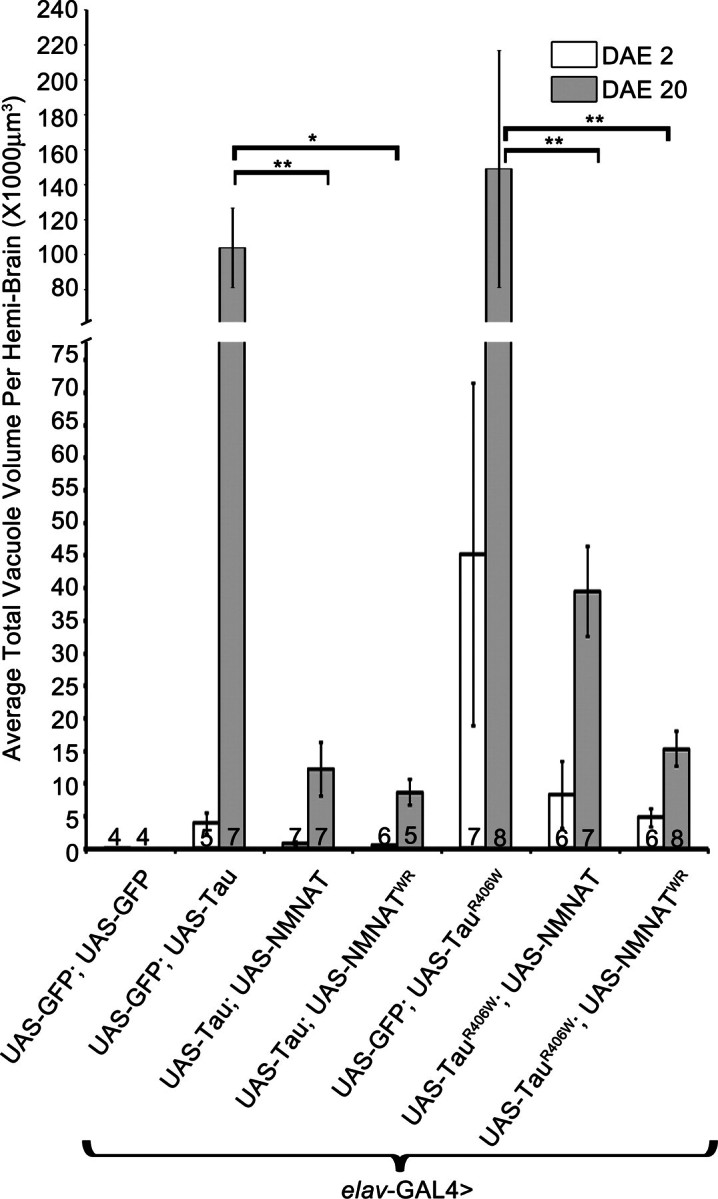
NMNAT reduces the total vacuole volume per brain induced by hTau overexpression. Overexpression of both wild-type and enzyme-deficient NMNAT reduces total vacuole volume per brain. Vacuole volume was quantified by using the FluoView10-ASW (Olympus) software, assuming each vacuole to be spherical in form. Numbers at the bottom of the bars indicate the number of brains quantified per age per genotype. *P< 0.001, **P< 0.05.
NMNAT reduces the level of tau hyperphosphorylation
The significant rescue of tau-induced behavioral and morphological impairments by either wild-type or enzyme-inactive NMNAT suggested that the mode of protection availed by NMNAT might possibly have an enzyme-independent mechanism. Studies using tauopathy models have suggested the main cause of neurodegeneration to be tau hyperphosphorylation and oligomerization that interfere with proper neuronal functions and lead to cell death (9,11,29–31). In order to reveal the mechanism underlying the protective effects of NMNAT, we next determined the impact of NMNAT expression on the levels of hyperphosphorylated tau.
To determine the conformation and phosphorylation state of tau protein, we used the following specific antibodies that are commonly used to detect tau abnormalities in disease state: AT8 and AT180 antibodies to preferentially recognize tau phosphorylation characteristic of AD state (11,32,33), taupSer262 antibody to probe for serine 262-specific phosphorylation of tau, as phosphorylation at this residue has been shown to be essential for neurotoxicity associated with a different model of AD (29), and human tau antibody to probe the total level of tau protein expressed. The specificity of these antibodies was indicated by lack of immunoreactivity in the control flies that were not expressing human tau (Fig. 4A and B, lanes 1–4). In tau- or tauR406W-expressing flies, a significant level of phosphorylated tau was detected in the CNS by all three phospho-specific antibodies (Fig. 4A and B, lanes 5–6). When NMNAT or NMNATWR was co-overexpressed, a significant decrease in phospho-tau was detected by AT8, AT180 and pSer262 antibodies in 2 DAE flies (Fig. 4A and B, lanes 7 and 9, quantifications in 4C and E). The level of phospho-tau was higher in tauR406W-expressing flies than that in tau flies, consistent with a more aggressive phenotype produced by tauR406W overexpression (11). Phospho-tau accumulated with age as the levels were substantially elevated in 20 DAE tau- or tauR406W-expressing flies, and overexpressing NMNAT provided significant reduction in the level of phospho-tau (Fig. 4A and B, lane 8, and quantifications in 4D and F). More interestingly, removing one copy of endogenous NMNAT significantly increased the level of phospho-tau in tauR406W-expressing flies (Fig 4A and B, lanes 11 and 12, and quantifications in 4C–F), consistent with the observation that removing one copy of endogenous NMNAT exacerbated the learning and memory defect caused by tauR406W overexpression (Fig. 1). These results suggest that the level of phospho-tau is correlated with the severity of the neurodegenerative phenotypes and that NMNAT-mediated suppression of tau-induced behavioral and morphological deficits is the result of a reduced level of hyperphosphorylated tau, hence mitigating toxicity caused by such modifications.
There could be two possible modes of action of how NMNAT reduces phosphorylated tau oligomers. First, NMNAT could affect the activity of some of the kinases involved in hyperphosphorylating Tau and therefore reduce the level of phosphorylation of tau. Second, NMNAT could act as a chaperone to interact with these hyperphosphorylated oligomers and promote clearance in a proteasome-mediated manner. To test the first hypothesis, we overexpressed NMNAT in Cos7 cells and examined the expression level of a list of kinases previously implicated in tau phosphorylation (Supplementary Material, Fig. S3). Two types of phosphorylation motifs are present within tau protein: proline-directed serine/threonine sites and the KXGS motifs within the four microtubule-binding repeat domains (serine residues at 262, 293, 324 and 356) (34,35). We examined the following six major kinases that have been implicated in hyperphosphorylation of tau. First, we examined p38 microtubule-associated protein (MAP) kinase and its activated form phosphorylated at Thr180/pTyr182 as MAPK/ERK-P was found in a subset of neurons and glial cells bearing abnormal tau deposition, and strong p38-P immunoreactivity was observed in ∼50–70% of neurons with neurofibrillary tangles and in dystrophic neurites of senile plaques in AD (36). Second, we investigated stress-activated protein kinase JNK and its activated form phosphorylated at Thr183/Tyr185 (37). Third, we checked the levels of GSK3β and its activated form phosphorylated at Ser9 as it has been shown that in AD, hyperphosphorylated tau plays a very important role in neurodegeneration and tau is hyperphosphorylated on residues that overlap with GSK3β targets; in animal models, tau hyperphosphorylation by GSK3β accelerates neurodegeneration in the context of both wild-type and mutant tau (38–41). Fourth, we investigated ERK and its active form phosphorylated at Thr202/Tyr204 (42). Fifth, we examined MAP/microtubule affinity-regulating kinases family (MARK1–4) of serine/threonine kinases that can phosphorylate MAPs including tau at KXGS sites such as Ser262 (34), as recent evidence supports a role for the prior phosphorylation of tau by MARK to initiate the pathogenic cascade of hyperphosphorylation by other kinases leading to the formation of tau lesions (43). Finally, we investigated whether NMNAT levels affected PP2A, one of the major protein phosphatases involved in tau dephosphorylation in vivo (44). As shown in Supplementary Material, Figure S3, we did not observe any changes in the expression or activation for p38MAPK, ERK, JNK, GSK3β, MARK, as detected by a phospho-MARK family (Activation Loop) antibody, and PP2A, from NMNAT overexpression (Supplementary Material, Fig. S3). These results suggest that NMNAT does not affect the levels of either total or phosphorylated forms of the major kinases implicated in tau phosphorylation. Therefore, it is unlikely that NMNAT reduces the level of phosphorylation of tau by affecting the activity of these known major kinases involved in hyperphosphorylating Tau.
NMNAT interacts with tau oligomers
We next examined the second possibility that NMNAT could act as a chaperone to interact with hyperphosphorylated tau oligomers and promote their clearance in a proteasome-mediated manner. Our previous work on the biochemical properties of NMNAT has uncovered an enzyme-independent chaperone function that contributes to its neuroprotective effects in models of SCA1 (16). NMNAT was shown to be recruited with chaperone heat shock protein 70 (Hsp70) into the protein aggregates of human Ataxin1 with 82 poly-glutamin expansion (hATX[82Q]) and to reduce aggregation load partly through a proteasome-mediated pathway (16). To test the hypothesis that the chaperone function of NMNAT may contribute to the clearance of toxic phosphor tau oligomers, we first examined whether NMNAT interacts directly with overexpressed human tau proteins by immunoprecipitation analysis using brain lysates from tau- or tauR406W-expressing flies. As shown in Figure 5A, endogenous NMNAT specifically interacted with tau and phospho-tau proteins in tau- or tauR406W-expressing fly brains. These phospho-tau epitopes included AT8 and AT180, corresponding to tau conformations found in disease. Furthermore, we observed interaction between NMNAT and tau phosphorylated at Ser262, a KXGS (MARK) site, normally phosphorylated by MARK affecting microtubule stability and integrity (45). Moreover, inhibition of PP2A by okadaic acid, which mimics tau pathology in vitro, was sufficient to induce hyperphosphorylation of tau at Ser262 via hyperactivation of GSK3β. Therefore, the current consensus is that even though phosphorylation at Ser-262 in normal conditions might regulate the function of tau (35), the abnormal hyperphosphorylation at this residue is only observed in disease state (46,47). Using a specific antibody recognizing tau with pSER262, we observed a strong 64 kDa band and an additional lower band corresponding to 55–60 kDa in the brain lysate. Previous work has shown that pathologically hyperphosphorylated (including pSer262) human Tau band runs at 64 kDa, whereas the normally phosphorylated Tau at Ser262 runs in the range of 55–60 kDa (48). Normally phosphorylated hTau is known to play an important role in the regulation of Tau function and microtubule dynamics (45). Interestingly, we observed that only the pTauSer262 at 64 kDa was detected in the NMNAT immunoprecipitated fraction but not the 55–60 kDa band (Fig. 5A). These results suggest that NMNAT directly interacts with toxic hyperphosphorylated Tau species.
Figure 5.

Endogenous NMNAT interacts with hTau oligomers to promote proteasome-mediated clearance of ubiquitinated tau. (A) Immunoprecipitated whole-brain lysates with NMNAT antibody were probed with total hTau or disease-associated phospho-tau-specific antibodies AT8 and AT180. Lack of tau bands in the negative wild-type and GFP-overexpressing brains reveal that the NMNAT–tau interaction is specific to overexpressed hTau. (B) Total hTau was immunoprecipitated from brains of 2 DAE flies overexpressing wild-type hTau, hTauR406W, hTauR406W +NMNAT, hTauR406W + NMNATWR and probed for ubiquitin. Stacking gel shows HMW tau oligomers and lower gel shows LMW oligomers (note the arrowheads) that can be mono- or polyubiquitinated. Note in NMNAT-overexpressing flies, the band immunoprecipitated at 75 kDa probably corresponds to ubiquitinated hyperphosphorylated tau that can be directly cleared by the proteasome. (C) Blot in (B) probed with a human Tau antibody; stacking gel shows HMW tau oligomers, and lower gel shows LMW oligomers at similar size to those detected by ubiquitin antibody in (B). (D) Total NMNAT was immunoprecipitated from brains of 2 DAE flies overexpressing wild-type hTau, hTauR406W, hTauR406W +NMNAT or hTauR406W + NMNATWR and probed for Tau.
Next, to uncover the mechanism of NMNAT-mediated tau clearance, we examined the state of tau ubiquitination, as several studies have reported the involvement of the ubiquitin-proteasome in tau degradation (30,49–57). Moreover, tau extracted from AD brains but not from control brains was shown to be heavily ubiquitinated by the E3 ubiquitin ligase CHIP in vitro (5). We therefore investigated whether NMNAT overexpression can promote ubiquitination of tau oligomers. To test this, we overexpressed either wild-type or enzyme-inactive NMNAT in the tauR406W-expressing flies and immunoprecipitated total human tau from the brain lysates. As shown in Figure 5B, western analysis with a ubiquitin-specific antibody showed that overexpression of NMNAT or NMNATWR promoted the clearance of lower-molecular-weight (≤250 kDa)-ubiquitinated tau oligomers (Fig. 5B arrowheads) while increasing the levels of ubiquitinated tau monomer (Fig. 5B, ∼75 kDa bands indicated by an arrow). These results indicate that NMNAT overexpression reduces mutant human tau-induced toxicity by promoting clearance of the lower-molecular-weight-ubiquitinated tau oligomers via the ubiquitin-proteasome pathway. In addition, higher-molecular-weight (>250 kDa)-ubiquitinated tau species observed in the stacking gel in tau- or tauR406W-expressing flies (Fig. 5B, lanes 3 and 6) were significantly increased with NMNAT overexpression (Fig. 5B, lanes 9 and 12). This suggests that NMNAT may be able to promote the formation of higher molecular weight tau oligomers that are far less toxic than low molecular weight tau oligomers. To confirm that these ubiquitin-positive bands that immunoprecipitated with tau antibody were indeed ubiquitinated tau oligomers, we probed the same blot with anti-tau antibody (Fig. 5C). Here, we detected protein bands with the same molecular weight as those previously probed by ubiquitin antibody, and further observed that NMNAT expression promoted the formation of higher molecular weight tau oligomers (stacking gel, Fig.5C) and reduced the level of ubiquitinated tau monomer and low molecular weight oligomers (Fig. 5C). Furthermore, we also probed the same blot with an antibody specific for pTauSer262 and confirmed that some of these high molecular weight tau oligomers are also positive for phosphorylation at Ser262 residue (Supplementary Material, Fig. S4). To further examine the direct interaction between NMNAT and ubiquitinated tau oligomers, we immunoprecipitated the fly brain lysates with an anti-NMNAT antibody and probed for total tau. As shown in Figure 5D, we observed that indeed NMNAT interacted with ubiquitinated tau oligomers, and NMNAT overexpression increased the binding between NMNAT and ubiquitin-tau monomer and high molecular weight oligomers. These results suggest that NMNAT directly interacts with tau proteins and promotes the clearance and breakdown of toxic oligomeric tau species of low molecular weight through the ubiquitin-proteasome pathway, and also facilitates the formation of less toxic high molecular weight tau aggregates, thereby reducing the neuronal load of highly toxic oligomeric tau species.
NMNAT protects against tau-induced apoptosis
Neuronal overexpression of hTau in Drosophila is shown to induce apoptosis resulting in age-dependent vacuolization (11). So far, we have shown that overexpression of NMNAT suppresses human tau- induced neurodegeneration, and reduces hyperphosphorylated tau levels by promoting ubiquitination and clearance. Next, we wanted to test whether NMNAT expression affects the level of apoptosis induced by tau overexpression. As shown in Figure 6, overexpression of either tau or tauR406W causes significant increases in the levels of cleaved caspase 3 (Fig. 6A and B, lanes 5 and 6), consistent with previous observations (58,59). However, overexpressing NMNAT or NMNATWR significantly reduced caspase 3 activation via cleavage in both D2 and D20 flies (Fig. 6A and B, lanes 7–10, quantifications in 6C–D). In contrast, loss of an endogenous copy of NMNAT (nmnat/+) caused a significant increase in cleaved caspase 3 levels (Fig. 6A and B, lanes 11–12, quantifications in 6C–D). Furthermore, we also stained 10 DAE adult brains overexpressing tauR406W with GFP, NMNAT or WR for cleaved-caspase 3 immunoreactivity, to estimate the number of cells about to undergo apoptosis (Fig. 7). We observed more number of cleaved-caspase 3-positive cells in brains overexpressing tauR406W alone (10.6 ± 1.21), compared with tauR406W brains overexpressing either NMNAT (3 ± 0.45, P < 0.005) or NMNATWR (4.4 ± 0.51, P< 0.005). These results suggest that human tau overexpression induces caspase activation and apoptosis, and higher levels of NMNAT expression significantly reduced the level of caspase 3 activation and the resultant apoptosis, whereas a reduced level of NMNAT expression exacerbated apoptosis induced by tau expression.
Figure 6.

NMNAT-mediated protection of morphological defects in tauopathy is partly through reduction in apoptosis. (A and B) Levels of apoptosis were measured in 2 and 20 DAE wild-type flies (yw, lanes 1 and 2) and flies overexpressing GFP (lanes 3 and 4); wild-type hTau (A) or hTauR406W (B) with GFP (lanes 5 and 6) or NMNAT (lanes 7 and 8) or NMNATWR (lanes 9 and 10) or in the nmnat/+ background (lanes 11 and 12) by probing for cleaved caspase 3, total caspase 3 and actin as a loading control. (C and D) Quantification of cleaved caspase 3 levels in panel A (C) or B (D). The level of cleaved or total caspase 3 were normalized to that of actin. The ratio of cleaved caspase 3 to total caspase 3 was displayed. *P< 0.005.
Figure 7.
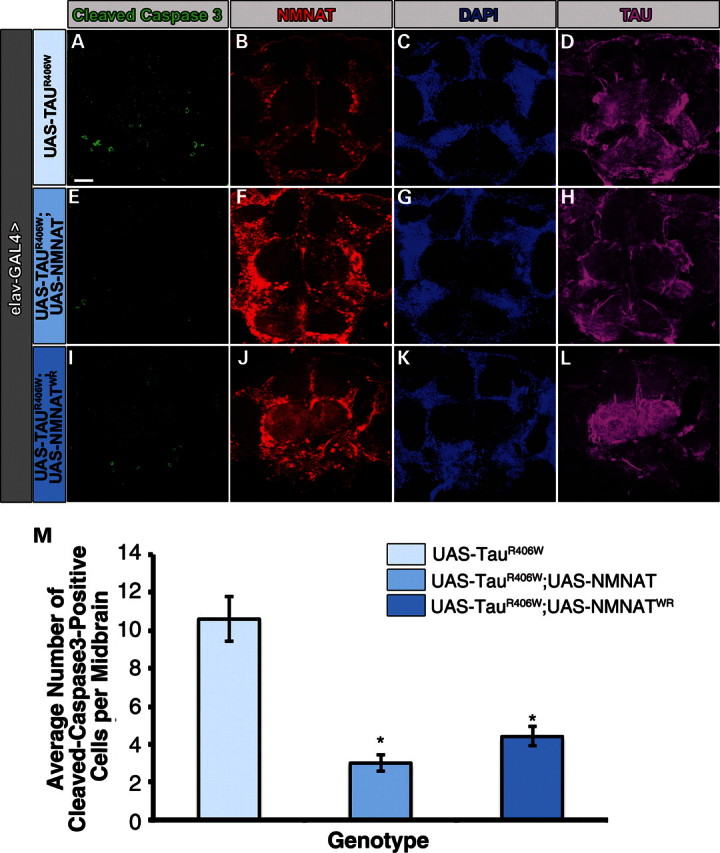
Overexpression of NMNAT or NMNATWR reduces the total number of cleaved caspase 3-positive cells in TauR406W brains. Whole-mount adult brains overexpressing either TauR406W with GFP, or NMNAT or NMNATWR were stained for cleaved caspase 3 (green, A, E and I), NMNAT (red, B, F and J), DAPI (blue, C, G and K) and hTau (magenta, D, H and L). (M) The total number of cleaved caspase 3-positive cells per mid-brain were averaged per genotype and plotted. N= 8, *P< 0.005.
DISCUSSION
Drosophila models of neuronal tauopathy exhibit key features of the human disorders including adult onset, age-dependent increase in neurodegeneration, accumulation of abnormal tau and compromised lifespan (11). Interestingly, neuronal tauopathy in Drosophila exhibits all these pathological hallmarks of the disease without forming neurofibrillary tangles, showing that the toxic species compromising behavior and lifespan seem to be the soluble phosphorylated tau oligomers (11). Neuronal maintenance factor NMNAT has been shown to protect against polyglutamine-expansion-induced neurodegeneration in a Drosophila model of SCA1, partly through a ubiqutitin-proteosome-mediated pathway (14,16). The chaperone function of NMNAT predicted a possibility of a role of NMNAT in regulating protein homeostasis and aggregation in neurons with toxic tau overload. We tested this possibility directly in this study by showing that expressing either wild-type or mutant human tau in the CNS induced age-dependent neurodegenerative phenotypes including vacuolization in the brain, impaired learning and memory functions and behavioral deficits in locomotor capabilities in Drosophila, all of which can be significantly suppressed by overexpression of NMNAT proteins. Importantly, we found that the severity of neurodegenerative phenotypes was correlated with the level of hyperphosphorylated tau oligomers, and NMNAT overexpression specifically reduced hyperphosphorylated tau oligomers, although increased the level of tau monomers and high molecular weight aggregates. In addition, we further show that NMNAT protein directly interacted with the tau oligomers to promote ubiquitylation and clearance of the toxic tau oligomeric species. The reduction of tau oligomers was correlated with reduced caspase 3 activation, suggesting that NMNAT significantly reduces tau-induced apoptosis and hence rescues tau-induced neurodegenerative phenotypes.
NMNAT rescues morphological and behavioral defects in tauopathy
Previous work has shown that overexpression of wild-type or mutant tau causes abnormal vacuolization in the cortex and neuropil structures in flies in an age-dependent manner (11). Such vacuolization is commonly observed in other neurodegeneration models in Drosophila and is correlated with the relative speed and severity of degeneration in flies (60–62). However, such degeneration in flies is not coupled with the formation of insoluble neurofibrillary tangles (11). It was shown that cholinergic neurons were significantly affected by tau overexpression (11); however, in our studies, we observed vacuole formation throughout various parts of the brain when tau was expressed in the CNS. Therefore, other neuronal populations may also be susceptible to tau-induced neurodegeneration. In addition to vacuole formation, tau expression in the CNS resulted in a high level of pTauSer262 accumulation, a phosphorylated form of tau that is linked with several neurodegenerative conditions (29) throughout the brain. Overexpression of NMNAT proteins (wild-type or enzyme-inactive) significantly reduced the size and occurrence of vacuolization throughout the brain. Interestingly, NMNAT expression also affected the levels and distribution of pTauSer262 proteins. Specifically, increased NMNAT levels decreased the overall levels of pTauSer262 and reduced the filamentous accumulation of pTauSer262. These observations indicate that NMNAT protein can regulate the clearance of misfolded tau oligomers. The ability of NMNAT protein to regulate misfolded protein aggregates has previously been observed in a Drosophila model of SCA1, where NMNAT was recruited together with chaperone Hsp70 to the hATX1[82Q] protein aggregates and affected the levels of aggregation (16). Therefore, a similar neuroprotective mechanism may be at play in the current tauopathy model, where the chaperone function of NMNAT contributes to the regulation of misfolded protein load in the brain.
Learning and memory deficits associated with tauopathies in Drosophila were first reported when tau was overexpressed in mushroom body neurons, which are known as the centers for olfactory learning and memory (13). In this case, selective tau overexpression in these neurons affected associative olfactory learning and memory before the onset of apparent neuronal loss. In our study, we expressed tau proteins in all neurons in the CNS and observed an age-dependent decline of learning and memory indices. In previous studies, such a decline in cognitive functions seems to precede the morphological defects, which only become apparent with age (13). Interestingly, removing one copy of endogenous NMNAT significantly exacerbated the learning and memory deficits induced by tauR406W expression, suggesting that the level of neuronal NMNAT protein is a critical determinant of the onset and severity of the degeneration induced by mutant tau. In contrast, higher levels of NMNAT expression rescued the learning and memory and locomotor deficits induced by either wild-type or mutant tau, further suggesting the importance of the neuronal NMNAT levels and a likely protein–protein interaction-mediated neuroprotective mechanism afforded by NMNAT.
Mechanism of tau-induced degeneration and NMNAT-mediated protection in tauopathy
Using a Drosophila model of tauopathy, we were able to analyze a plausible mechanism of NMNAT's neuroprotective capacity in ameliorating tau-induced neurodegeneration. We found that the severity of morphological and behavioral deficits associated with tau overexpression is directly correlated with the level of hyperphosphorylated tau species associated with various diseases (Fig. 4). Importantly, NMNAT expression reduced tau-induced degenerative phenotypes by reducing the level of hyperphosphorylated tau and promoting the ubiquitination and clearance of toxic tau oligomers (Fig. 5).
The key mediator of toxicity in tauopathy is the hyperphosphorylation of tau. Several studies in animal models with altered kinase or phosphatase activity supported the role of tau hyperphosphorylation in promoting neurotoxicity (8–10,43,48,63–67). For example, when 14 key phospho-epitopes created by proline-directed kinase SP/TP sites in tau were mutated to alanine, the toxicity associated with tau overexpression was significantly reduced, identifying a positive correlation between increased phosphorylation at disease-associated sites and neurotoxicity (8,9). In our study, increased NMNAT levels effectively reduced tau hyperphosphorylation specifically at sites that are known to be phosphorylated in disease states, including those recognized by AT8 and AT180, and consequently reduced the level of apoptosis and neurodegeneration. One important finding from our study is the specific reduction of ubiquitinated tau oligomeric species in the range of 150–250 kDa and increase of ubiquitinated monomeric tau and ubiquitinated higher molecular weight oligomeric tau >250 kDa in NMNAT-overexpressing brains. As NMNAT expression suppressed tau-induced degeneration, this finding suggests that the mid-range (150–250 kDa) tau oligomers are likely the species mediating neuronal toxicity, whereas the monomers and high-range (>250 kDa) tau oligomers are less toxic. This finding is consistent with the emerging notion that high molecular weight (tau) protein aggregates may be protective when the formation of aggregates can reduce the neuronal load of oligomeric species (39,68,69). We showed for the first time that a neuroprotective protein NMNAT exerts its protective effects by reducing specifically the toxic tau oligomers while increasing the less toxic and maybe even protective higher molecular weight tau species.
Molecular chaperones and their indispensible role in protein folding have been implicated in many neurodegenerative diseases, including Parkinson's disease, AD and Huntington's disease (18–21). These stress-regulated proteins prevent improper folding and aggregation of proteins and facilitate formation of proper conformation by recognizing and binding to exposed hydrophobic residues of unfolded proteins (70–72). The molecular chaperones Hsp70 and Hsp90 were shown to prevent aggregation in a cellular model of tauopathy (22). In fact, in AD brains, the levels of Hsp90 and Hsp70 are shown to be inversely related to the expression of aggregated tau, raising a good possibility that molecular chaperones may protect against tau aggregation and NFT formation (22). We have previously shown that NMNAT, in addition to its role as a housekeeping enzyme in NAD synthesis, also functions as a stress-responsive chaperone (16,73). Similar to Hsp70 and Hsp90 (22), NMNAT is also capable of binding to tau oligomers in our study, and reduce tau toxic load by promoting ubiquitination and clearance of tau oligomers. Our studies also found that a reduced endogenous level of NMNAT significantly exacerbated the tau-induced degeneration, further indicating the critical role of NMNAT in maintaining neuronal integrity and regulating the neuronal misfolded protein load. Interestingly, gene-array studies found that the level of human neuronal NMNAT isoform NMNAT2 was reduced in brain specimens derived from AD patients (https://www.nextbio.com), providing a causative link between reduced NMNAT levels and AD.
In summary, we report here that overexpression of human wild-type of mutant tau-induced neurodegeneration can be suppressed by NMNAT expression. NMNAT interacts directly with tau and modulates neuroprotection in tauopathy in an enzyme-independent manner. We further report a molecular mechanism of tau-induced degeneration and NMNAT-mediated neuroprotection. Our analysis places NMNAT in the category of neuronal chaperones that are critical in regulating neuronal protein homeostasis, modulating misfolded protein load and maintaining neuronal integrity. Future work should elucidate the mechanism of NMNAT down-regulation in these protein-folding diseases. Understanding the regulation of NMNAT would be interesting and crucial to further explain its role in maintaining proper neuronal health, and would open novel therapeutic windows for this protein in neurodegenerative disease treatment.
MATERIALS AND METHODS
Fly stocks and genetics
Flies were reared at room temperature in ambient light under a normal 12 h light/dark cycle. Human wild-type tau (UAS-Tau) or Arg406→ Trp (R406W) mutant tau containing no N-terminal inserts and four microtubule-binding domains (0N, 4R) (UAS-TauR406W) were generously provided by Dr Mel Feany (Harvard Medical School) (11). For our tauopathy model, human wild-type tau or Arg406→ Trp (R406W) mutant tau was expressed with a pan-neuronal driver elav-GAL4. Fly strains used in this experiment included yw (WT), UAS-CD8GFP, w;;UAS-NMNAT, w;;UAS-NMNATWR and w;;nmnat/TM6B.
Immunocytochemistry
Adult brains were fixed in phosphate-buffered saline (PBS) with 4% formaldehyde for 20 min and washed in PBS with 0.4% Triton X-100. Antibody dilutions used were anti-NMNAT 1:800; anti-tau 1:200 (DAKO); anti-taupSer262 1:200 (Millipore, Billerica, MA, USA). Secondary antibodies conjugated to Alexa-488, -555 and -647, Cy3 or Cy5 (Jackson ImmunoResearch, West Grove, PA, USA; and Molecular Probes) were used at 1:250. DAPI staining was performed after the washes post-secondary antibody staining, using 1:1000 dilution of the original stock solution (5 mg/ml in ddH2O; Invitrogen, Eugene, OR, USA) in PBS–0.1% Triton for 10min at room temperature. DAPI staining was followed by three 10 min washes in PBS–0.1%Triton. All primary antibody incubations were performed at 4°C overnight or secondary for 2 h at room temperature, in the presence of 5% normal goat serum.
Image acquisition and processing
Images from fluorescently labeled specimens were taken on an Olympus IX81 confocal microscope and processed using FluoView10-ASW (Olympus) and Adobe Photoshop CS4 (Adobe Systems, San Jose, CA, USA).
Protein extraction, immunoprecipitation and western blotting
Twenty female fly heads of each genotype were collected at specific ages and immediately flash-frozen before extraction. For protein extraction, brains were homogenized in lysis buffer consisting of 100 mm KCl, 20 mm HEPES, 10 mm EDTA, 1 mm DTT, 5% glycerol, 0.1% Triton-X100, protease inhibitor cocktail (Sigma) and a phosphatase inhibitor cocktail (Roche). After homogenizing, the samples were centrifuged briefly at 500g to remove debris. Ten microliters of total protein lysate per sample (equivalent to 1 head) was loaded onto a polyacrylamide gel (PAGE) and resolved by SDS–PAGE and transferred onto nitrocellulose membrane. For immunoprecipitation experiments, 40 2-day-old female fly heads were homogenized in lysis buffer, precleared with Protein-G beads for 1h and incubated with Protein-G beads conjugated with 10 µg of antibody per sample overnight at 4°C. The bead pellets were collected and washed four times with lysis buffer before eluting out the bead fraction with 4× Lamelli buffer, heating the samples at 95°C and centrifuging for 5 min to collect the bead-eluted fraction. Lysates were probed with anti-NMNAT (1:8000), anti-tau (1:1000, DAKO, Carpinteria, CA, USA), anti-phosphotauSer262 (1:1000; Millipore); AT8 for PHF-TauSer202/Thr205 (1:1000; ThermoScientific, Rockford, IL, USA); AT180 for PHF-TauThr231 (1:1000; ThermoScientific); anti-caspase 3 (1:1000, Cell Signaling, Beverly, MA, USA); anti-cleaved caspase 3 Asp175 (1:1000, Cell Signaling), anti-p38MAPK (1:1000, Cell Signaling), anti-phospho-p38MAPK (pThr180/pTyr182) (1:1000, Cell Signaling), anti-JNK (1:1000, Cell Signaling), anti-phosphoJNK (pThr 183/Tyr 185) (1:1000, Cell Signaling), anti-ERK (1:1000, Cell Signaling), anti-phosphoERK (pThr 202/pTyr 204) (1:1000, Cell Signaling), anti GSK3β (1:1000, Cell Signaling), anti phosphor-GSK3β (Ser9) (1:1000, Cell Signaling), anti-MARK1 (1:1000, Cell Signaling), anti phospho-MARK family (activation loop) (1:1000, Cell Signaling), anti-dsRED (1:1000, Clontech, Mountain View, CA, USA) and anti-actin antibodies (1:4000; Sigma, St Louis, MO, USA). Western blot analysis was performed with infrared dye-conjugated secondary antibodies, IR700 and IR800 (LI-COR Biosciences); blots were imaged and processed on an Odyssey Infrared Imaging System. The wide linear range of the Odyssey system made it easier to detect bands and analyze changes in protein levels quantitatively.
Behavior assays
Negative geotaxis assay
Briefly, 10 age-matched female flies per genotype were placed in a vial marked with a line drawn horizontally 8 cm above the surface. The flies were tapped to the surface and given 10 s to demonstrate climbing activity as a negative geotaxic response. After 10 s, the number of flies that successfully climbed above the 8 cm line was recorded. This assay was repeated 10 times and the averaged data (SEMs) represented as percentages where the number of flies above the 8 cm mark was divided by the total number of flies tested within each group.
APS assay
The APS assay was performed as described by Le Bourg and Buecher (25,26). In this assay, each female fly per genotype was individually conditioned to associate the bitter taste of quinine with light and was then tested in a T-maze and allowed to choose between a dark and a lighted vial. Each positively phototaxic fly was trained in 10 trials to move into the lighted vial, which contains the quinine-soaked filter paper. Repetitive training reinforced them to make the aversive association. Immediately after training, the fly was tested in a series of five trials to see whether it learned to make this association (PC0; learning function). These flies were again tested 6 h later to see whether they remembered to make this negative association of bitter taste with light (PC6; memory function). The pass rate for each fly was averaged out of five trials for PC0 and PC6. Score differences between control and experimental groups were assessed using ANOVA and Tukey's test (significance noted at P< 0.05).
Densitometry, quantification and statistical analyses
Densitometric analysis of blots was performed using the ImageJ software (NIH). Data are represented as means + SE. Statistical analysis was performed using ANOVA Tukey's test and Scheffe's test. Values of P< 0.05 were considered statistically significant.
SUPPLEMENTARY MATERIAL
FUNDING
This work is supported by the American Heart Association Predoctoral Fellowship (to Y.O.A.), the National Institute for Neurological Disorders and Stroke Grant R01NS64269 (to R.G.Z.) and the Pew Charitable Trust (to R.G.Z.).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr M.B. Feany for kindly sharing the reagents and Dr B.D. Watson for critical reading of the manuscript.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Iqbal K., Alonso Adel C., Chen S., Chohan M.O., El-Akkad E., Gong C.X., Khatoon S., Li B., Liu F., Rahman A., et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim. Biophys. Acta. 2005;1739:198–210. doi: 10.1016/j.bbadis.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 2.Sergeant N., Delacourte A., Buee L. Tau protein as a differential biomarker of tauopathies. Biochim. Biophys. Acta. 2005;1739:179–197. doi: 10.1016/j.bbadis.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 3.Alonso A.C., Grundke-Iqbal I., Iqbal K. Alzheimer's disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat. Med. 1996;2:783–787. doi: 10.1038/nm0796-783. [DOI] [PubMed] [Google Scholar]
- 4.Chatterjee S., Sang T.K., Lawless G.M., Jackson G.R. Dissociation of tau toxicity and phosphorylation: role of GSK-3beta, MARK and Cdk5 in a Drosophila model. Hum. Mol. Genet. 2009;18:164–177. doi: 10.1093/hmg/ddn326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khatoon S., Grundke-Iqbal I., Iqbal K. Brain levels of microtubule-associated protein tau are elevated in Alzheimer's disease: a radioimmuno-slot-blot assay for nanograms of the protein. J. Neurochem. 1992;59:750–753. doi: 10.1111/j.1471-4159.1992.tb09432.x. [DOI] [PubMed] [Google Scholar]
- 6.Ledesma M.D., Medina M., Avila J. The in vitro formation of recombinant tau polymers: effect of phosphorylation and glycation. Mol. Chem. Neuropathol. 1996;27:249–258. doi: 10.1007/BF02815107. [DOI] [PubMed] [Google Scholar]
- 7.Lovestone S., Davis D.R., Webster M.T., Kaech S., Brion J.P., Matus A., Anderton B.H. Lithium reduces tau phosphorylation: effects in living cells and in neurons at therapeutic concentrations. Biol. Psychiatry. 1999;45:995–1003. doi: 10.1016/s0006-3223(98)00183-8. [DOI] [PubMed] [Google Scholar]
- 8.Steinhilb M.L., Dias-Santagata D., Fulga T.A., Felch D.L., Feany M.B. Tau phosphorylation sites work in concert to promote neurotoxicity in vivo. Mol. Biol. Cell. 2007;18:5060–5068. doi: 10.1091/mbc.E07-04-0327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Steinhilb M.L., Dias-Santagata D., Mulkearns E.E., Shulman J.M., Biernat J., Mandelkow E.M., Feany M.B. S/P and T/P phosphorylation is critical for tau neurotoxicity in Drosophila. J. Neurosci. Res. 2007;85:1271–1278. doi: 10.1002/jnr.21232. [DOI] [PubMed] [Google Scholar]
- 10.Jackson G.R., Wiedau-Pazos M., Sang T.K., Wagle N., Brown C.A., Massachi S., Geschwind D.H. Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron. 2002;34:509–519. doi: 10.1016/s0896-6273(02)00706-7. [DOI] [PubMed] [Google Scholar]
- 11.Wittmann C.W., Wszolek M.F., Shulman J.M., Salvaterra P.M., Lewis J., Hutton M., Feany M.B. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science. 2001;293:711–714. doi: 10.1126/science.1062382. [DOI] [PubMed] [Google Scholar]
- 12.Williams D.W., Tyrer M., Shepherd D. Tau and tau reporters disrupt central projections of sensory neurons in Drosophila. J. Comp. Neurol. 2000;428:630–640. doi: 10.1002/1096-9861(20001225)428:4<630::aid-cne4>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 13.Mershin A., Pavlopoulos E., Fitch O., Braden B.C., Nanopoulos D.V., Skoulakis E.M. Learning and memory deficits upon TAU accumulation in Drosophila mushroom body neurons. Learn Mem. 2004;11:277–287. doi: 10.1101/lm.70804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhai R.G., Cao Y., Hiesinger P.R., Zhou Y., Mehta S.Q., Schulze K.L., Verstreken P., Bellen H.J. Drosophila NMNAT maintains neural integrity independent of its NAD synthesis activity. PLoS Biol. 2006;4:e416. doi: 10.1371/journal.pbio.0040416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.MacDonald J.M., Beach M.G., Porpiglia E., Sheehan A.E., Watts R.J., Freeman M.R. The Drosophila cell corpse engulfment receptor Draper mediates glial clearance of severed axons. Neuron. 2006;50:869–881. doi: 10.1016/j.neuron.2006.04.028. [DOI] [PubMed] [Google Scholar]
- 16.Zhai R.G., Zhang F., Hiesinger P.R., Cao Y., Haueter C.M., Bellen H.J. NAD synthase NMNAT acts as a chaperone to protect against neurodegeneration. Nature. 2008;452:887–891. doi: 10.1038/nature06721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhai R.G., Rizzi M., Garavaglia S. Nicotinamide/nicotinic acid mononucleotide adenylyltransferase, new insights into an ancient enzyme. Cell. Mol. Life Sci. 2009;66:2805–2818. doi: 10.1007/s00018-009-0047-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carmichael J., Chatellier J., Woolfson A., Milstein C., Fersht A.R., Rubinsztein D.C. Bacterial and yeast chaperones reduce both aggregate formation and cell death in mammalian cell models of Huntington's disease. Proc. Natl Acad. Sci. USA. 2000;97:9701–9705. doi: 10.1073/pnas.170280697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ostrerova N., Petrucelli L., Farrer M., Mehta N., Choi P., Hardy J., Wolozin B. alpha-Synuclein shares physical and functional homology with 14–3–3 proteins. J. Neurosci. 1999;19:5782–5791. doi: 10.1523/JNEUROSCI.19-14-05782.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sittler A., Lurz R., Lueder G., Priller J., Lehrach H., Hayer-Hartl M.K., Hartl F.U., Wanker E.E. Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington's disease. Hum. Mol. Genet. 2001;10:1307–1315. doi: 10.1093/hmg/10.12.1307. [DOI] [PubMed] [Google Scholar]
- 21.Warrick J.M., Chan H.Y., Gray-Board G.L., Chai Y., Paulson H.L., Bonini N.M. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat. Genet. 1999;23:425–428. doi: 10.1038/70532. [DOI] [PubMed] [Google Scholar]
- 22.Dou F., Netzer W.J., Tanemura K., Li F., Hartl F.U., Takashima A., Gouras G.K., Greengard P., Xu H. Chaperones increase association of tau protein with microtubules. Proc. Natl Acad. Sci. USA. 2003;100:721–726. doi: 10.1073/pnas.242720499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lewis J., McGowan E., Rockwood J., Melrose H., Nacharaju P., Van Slegtenhorst M., Gwinn-Hardy K., Paul Murphy M., Baker M., Yu X., et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat. Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- 24.Hutton M., Lewis J., Dickson D., Yen S.H., McGowan E. Analysis of tauopathies with transgenic mice. Trends Mol. Med. 2001;7:467–470. doi: 10.1016/s1471-4914(01)02123-2. [DOI] [PubMed] [Google Scholar]
- 25.Ali Y.O., Escala W., Ruan K., Zhai R.G. Assaying locomotor and learning and memory deficits in Drosophila models of neurodegeneration. J. Vis. Exp. 2011;49 doi: 10.3791/2504. doi:10.3791/2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Le Bourg E., Buecher C. Learned suppression of photopositive tendencies in Drosophila melanogaster. Anim. Learn Behav. 2002;30:330–341. doi: 10.3758/bf03195958. [DOI] [PubMed] [Google Scholar]
- 27.Benzer S. Behavioral mutants of Drosophila isolated by countercurrent distribution. Proc. Natl Acad. Sci. USA. 1967;58:1112–1119. doi: 10.1073/pnas.58.3.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Greenspan R.J., Finn J.A., Jr., Hall J.C. Acetylcholinesterase mutants in Drosophila and their effects on the structure and function of the central nervous system. J. Comp. Neurol. 1980;189:741–774. doi: 10.1002/cne.901890409. [DOI] [PubMed] [Google Scholar]
- 29.Iijima K., Gatt A., Iijima-Ando K. Tau Ser262 phosphorylation is critical for Abeta42-induced tau toxicity in a transgenic Drosophila model of Alzheimer's disease. Hum. Mol. Genet. 2010;19:2947–2957. doi: 10.1093/hmg/ddq200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blard O., Frebourg T., Campion D., Lecourtois M. Inhibition of proteasome and Shaggy/Glycogen synthase kinase-3beta kinase prevents clearance of phosphorylated tau in Drosophila. J. Neurosci. Res. 2006;84:1107–1115. doi: 10.1002/jnr.21006. [DOI] [PubMed] [Google Scholar]
- 31.Dickey C.A., Kamal A., Lundgren K., Klosak N., Bailey R.M., Dunmore J., Ash P., Shoraka S., Zlatkovic J., Eckman C.B., et al. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J. Clin. Invest. 2007;117:648–658. doi: 10.1172/JCI29715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jicha G.A., Bowser R., Kazam I.G., Davies P. Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J. Neurosci. Res. 1997;48:128–132. doi: 10.1002/(sici)1097-4547(19970415)48:2<128::aid-jnr5>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 33.Jicha G.A., Lane E., Vincent I., Otvos L., Jr., Hoffmann R., Davies P. A conformation- and phosphorylation-dependent antibody recognizing the paired helical filaments of Alzheimer's disease. J. Neurochem. 1997;69:2087–2095. doi: 10.1046/j.1471-4159.1997.69052087.x. [DOI] [PubMed] [Google Scholar]
- 34.Drewes G., Ebneth A., Preuss U., Mandelkow E.M., Mandelkow E. MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell. 1997;89:297–308. doi: 10.1016/s0092-8674(00)80208-1. [DOI] [PubMed] [Google Scholar]
- 35.Dickey C.A., Dunmore J., Lu B., Wang J.W., Lee W.C., Kamal A., Burrows F., Eckman C., Hutton M., Petrucelli L. HSP induction mediates selective clearance of tau phosphorylated at proline-directed Ser/Thr sites but not KXGS (MARK) sites. FASEB J. 2006;20:753–755. doi: 10.1096/fj.05-5343fje. [DOI] [PubMed] [Google Scholar]
- 36.Ferrer I., Blanco R., Carmona M., Puig B. Phosphorylated mitogen-activated protein kinase (MAPK/ERK-P), protein kinase of 38 kDa (p38-P), stress-activated protein kinase (SAPK/JNK-P), and calcium/calmodulin-dependent kinase II (CaM kinase II) are differentially expressed in tau deposits in neurons and glial cells in tauopathies. J. Neural. Transm. 2001;108:1397–1415. doi: 10.1007/s007020100016. [DOI] [PubMed] [Google Scholar]
- 37.Vogel J., Anand V.S., Ludwig B., Nawoschik S., Dunlop J., Braithwaite S.P. The JNK pathway amplifies and drives subcellular changes in tau phosphorylation. Neuropharmacology. 2009;57:539–550. doi: 10.1016/j.neuropharm.2009.07.021. [DOI] [PubMed] [Google Scholar]
- 38.Schaffer B.A., Bertram L., Miller B.L., Mullin K., Weintraub S., Johnson N., Bigio E.H., Mesulam M., Wiedau-Pazos M., Jackson G.R., et al. Association of GSK3B with Alzheimer disease and frontotemporal dementia. Arch. Neurol. 2008;65:1368–1374. doi: 10.1001/archneur.65.10.1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takashima A. Hyperphosphorylated tau is a cause of neuronal dysfunction in tauopathy. J. Alzheimers Dis. 2008;14:371–375. doi: 10.3233/jad-2008-14403. [DOI] [PubMed] [Google Scholar]
- 40.Kim H.S., Kim E.M., Lee J.P., Park C.H., Kim S., Seo J.H., Chang K.A., Yu E., Jeong S.J., Chong Y.H., et al. C-terminal fragments of amyloid precursor protein exert neurotoxicity by inducing glycogen synthase kinase-3beta expression. FASEB J. 2003;17:1951–1953. doi: 10.1096/fj.03-0106fje. [DOI] [PubMed] [Google Scholar]
- 41.Ishizawa T., Mattila P., Davies P., Wang D., Dickson D.W. Colocalization of tau and alpha-synuclein epitopes in Lewy bodies. J. Neuropathol. Exp. Neurol. 2003;62:389–397. doi: 10.1093/jnen/62.4.389. [DOI] [PubMed] [Google Scholar]
- 42.Guise S., Braguer D., Carles G., Delacourte A., Briand C. Hyperphosphorylation of tau is mediated by ERK activation during anticancer drug-induced apoptosis in neuroblastoma cells. J. Neurosci. Res. 2001;63:257–267. doi: 10.1002/1097-4547(20010201)63:3<257::AID-JNR1019>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 43.Nishimura I., Yang Y., Lu B. PAR-1 kinase plays an initiator role in a temporally ordered phosphorylation process that confers tau toxicity in Drosophila. Cell. 2004;116:671–682. doi: 10.1016/s0092-8674(04)00170-9. [DOI] [PubMed] [Google Scholar]
- 44.Sontag E., Nunbhakdi-Craig V., Lee G., Bloom G.S., Mumby M.C. Regulation of the phosphorylation state and microtubule-binding activity of Tau by protein phosphatase 2A. Neuron. 1996;17:1201–1207. doi: 10.1016/s0896-6273(00)80250-0. [DOI] [PubMed] [Google Scholar]
- 45.Stoothoff W.H., Johnson G.V. Tau phosphorylation: physiological and pathological consequences. Biochim. Biophys. Acta. 2005;1739:280–297. doi: 10.1016/j.bbadis.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 46.Martin L., Magnaudeix A., Wilson C.M., Yardin C., Terro F. The new indirubin derivative inhibitors of glycogen synthase kinase-3, 6-BIDECO and 6-BIMYEO, prevent tau phosphorylation and apoptosis induced by the inhibition of protein phosphatase-2A by okadaic acid in cultured neurons. J. Neurosci. Res. 2011;89:1802–1811. doi: 10.1002/jnr.22723. [DOI] [PubMed] [Google Scholar]
- 47.Martin L., Page G., Terro F. Tau phosphorylation and neuronal apoptosis induced by the blockade of PP2A preferentially involve GSK3beta. Neurochem. Int. 2011;59:235–250. doi: 10.1016/j.neuint.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 48.Le Corre S., Klafki H.W., Plesnila N., Hubinger G., Obermeier A., Sahagun H., Monse B., Seneci P., Lewis J., Eriksen J., et al. An inhibitor of tau hyperphosphorylation prevents severe motor impairments in tau transgenic mice. Proc. Natl Acad. Sci. USA. 2006;103:9673–9678. doi: 10.1073/pnas.0602913103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cardozo C., Michaud C. Proteasome-mediated degradation of tau proteins occurs independently of the chymotrypsin-like activity by a nonprocessive pathway. Arch. Biochem. Biophys. 2002;408:103–110. doi: 10.1016/s0003-9861(02)00493-9. [DOI] [PubMed] [Google Scholar]
- 50.David D.C., Layfield R., Serpell L., Narain Y., Goedert M., Spillantini M.G. Proteasomal degradation of tau protein. J. Neurochem. 2002;83:176–185. doi: 10.1046/j.1471-4159.2002.01137.x. [DOI] [PubMed] [Google Scholar]
- 51.Goldbaum O., Oppermann M., Handschuh M., Dabir D., Zhang B., Forman M.S., Trojanowski J.Q., Lee V.M., Richter-Landsberg C. Proteasome inhibition stabilizes tau inclusions in oligodendroglial cells that occur after treatment with okadaic acid. J. Neurosci. 2003;23:8872–8880. doi: 10.1523/JNEUROSCI.23-26-08872.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hatakeyama S., Matsumoto M., Kamura T., Murayama M., Chui D.H., Planel E., Takahashi R., Nakayama K.I., Takashima A. U-box protein carboxyl terminus of Hsc70-interacting protein (CHIP) mediates poly-ubiquitylation preferentially on four-repeat Tau and is involved in neurodegeneration of tauopathy. J. Neurochem. 2004;91:299–307. doi: 10.1111/j.1471-4159.2004.02713.x. [DOI] [PubMed] [Google Scholar]
- 53.Petrucelli L., Dawson T.M. Mechanism of neurodegenerative disease: role of the ubiquitin proteasome system. Ann. Med. 2004;36:315–320. doi: 10.1080/07853890410031948. [DOI] [PubMed] [Google Scholar]
- 54.Petrucelli L., Dickson D., Kehoe K., Taylor J., Snyder H., Grover A., De Lucia M., McGowan E., Lewis J., Prihar G., et al. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum. Mol. Genet. 2004;13:703–714. doi: 10.1093/hmg/ddh083. [DOI] [PubMed] [Google Scholar]
- 55.Shimura H., Schwartz D., Gygi S.P., Kosik K.S. CHIP-Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival. J. Biol. Chem. 2004;279:4869–4876. doi: 10.1074/jbc.M305838200. [DOI] [PubMed] [Google Scholar]
- 56.Zhang J.Y., Liu S.J., Li H.L., Wang J.Z. Microtubule-associated protein tau is a substrate of ATP/Mg(2+)-dependent proteasome protease system. J. Neural Transm. 2005;112:547–555. doi: 10.1007/s00702-004-0196-x. [DOI] [PubMed] [Google Scholar]
- 57.Babu J.R., Geetha T., Wooten M.W. Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation. J. Neurochem. 2005;94:192–203. doi: 10.1111/j.1471-4159.2005.03181.x. [DOI] [PubMed] [Google Scholar]
- 58.Fan Y., Bergmann A. The cleaved-Caspase-3 antibody is a marker of Caspase-9-like DRONC activity in Drosophila. Cell Death Differ. 2010;17:534–539. doi: 10.1038/cdd.2009.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Khurana V., Lu Y., Steinhilb M.L., Oldham S., Shulman J.M., Feany M.B. TOR-mediated cell-cycle activation causes neurodegeneration in a Drosophila tauopathy model. Curr. Biol. 2006;16:230–241. doi: 10.1016/j.cub.2005.12.042. [DOI] [PubMed] [Google Scholar]
- 60.Coombe P.E., Heisenberg M. The structural brain mutant vacuolar medulla of Drosophila melanogaster with specific behavioral defects and cell degeneration in the adult. J. Neurogenet. 1986;3:135–158. doi: 10.3109/01677068609106845. [DOI] [PubMed] [Google Scholar]
- 61.Buchanan R.L., Benzer S. Defective glia in the Drosophila brain degeneration mutant drop-dead. Neuron. 1993;10:839–850. doi: 10.1016/0896-6273(93)90200-b. [DOI] [PubMed] [Google Scholar]
- 62.Kretzschmar D., Hasan G., Sharma S., Heisenberg M., Benzer S. The Swiss cheese mutant causes glial hyperwrapping and brain degeneration in Drosophila. J. Neurosci. 1997;17:7425–7432. doi: 10.1523/JNEUROSCI.17-19-07425.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ahlijanian M.K., Barrezueta N.X., Williams R.D., Jakowski A., Kowsz K.P., McCarthy S., Coskran T., Carlo A., Seymour P.A., Burkhardt J.E., et al. Hyperphosphorylated tau and neurofilament and cytoskeletal disruptions in mice overexpressing human p25, an activator of cdk5. Proc. Natl Acad. Sci. USA. 2000;97:2910–2915. doi: 10.1073/pnas.040577797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lucas J.J., Hernandez F., Gomez-Ramos P., Moran M.A., Hen R., Avila J. Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J. 2001;20:27–39. doi: 10.1093/emboj/20.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cruz J.C., Tseng H.C., Goldman J.A., Shih H., Tsai L.H. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron. 2003;40:471–483. doi: 10.1016/s0896-6273(03)00627-5. [DOI] [PubMed] [Google Scholar]
- 66.Noble W., Planel E., Zehr C., Olm V., Meyerson J., Suleman F., Gaynor K., Wang L., LaFrancois J., Feinstein B., et al. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc. Natl Acad. Sci. USA. 2005;102:6990–6995. doi: 10.1073/pnas.0500466102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Noble W., Olm V., Takata K., Casey E., Mary O., Meyerson J., Gaynor K., LaFrancois J., Wang L., Kondo T., et al. Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron. 2003;38:555–565. doi: 10.1016/s0896-6273(03)00259-9. [DOI] [PubMed] [Google Scholar]
- 68.Brunden K.R., Trojanowski J.Q., Lee V.M. Evidence that non-fibrillar tau causes pathology linked to neurodegeneration and behavioral impairments. J. Alzheimers Dis. 2008;14:393–399. doi: 10.3233/jad-2008-14406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Iqbal K., Alonso Adel C., Grundke-Iqbal I. Cytosolic abnormally hyperphosphorylated tau but not paired helical filaments sequester normal MAPs and inhibit microtubule assembly. J. Alzheimers Dis. 2008;14:365–370. doi: 10.3233/jad-2008-14402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hartl F.U. Molecular chaperones in cellular protein folding. Nature. 1996;381:571–579. doi: 10.1038/381571a0. [DOI] [PubMed] [Google Scholar]
- 71.Slavotinek A.M., Biesecker L.G. Unfolding the role of chaperones and chaperonins in human disease. Trends Genet. 2001;17:528–535. doi: 10.1016/s0168-9525(01)02413-1. [DOI] [PubMed] [Google Scholar]
- 72.Ali Y.O., Kitay B.M., Zhai R.G. Dealing with misfolded proteins: examining the neuroprotective role of molecular chaperones in neurodegeneration. Molecules. 2010;15:6859–6887. doi: 10.3390/molecules15106859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ali Y.O., McCormack R., Darr A., Zhai R.G. Nicotinamide mononucleotide adenylyltransferase is a stress response protein regulated by the heat shock factor/hypoxia-inducible factor 1{alpha} pathway. J. Biol. Chem. 2011;286:19089–19099. doi: 10.1074/jbc.M111.219295. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.