

Abstract
Common variation in over 100 genes has been implicated in the risk of developing asthma, but the contribution of rare variants to asthma susceptibility remains largely unexplored. We selected nine genes that showed the strongest signatures of weak purifying selection from among 53 candidate asthma-associated genes, and we sequenced the coding exons and flanking noncoding regions in 450 asthmatic cases and 515 nonasthmatic controls. We observed an overall excess of p values <0.05 (p = 0.02), and rare variants in four genes (AGT, DPP10, IKBKAP, and IL12RB1) contributed to asthma susceptibility among African Americans. Rare variants in IL12RB1 were also associated with asthma susceptibility among European Americans, despite the fact that the majority of rare variants in IL12RB1 were specific to either one of the populations. The combined evidence of association with rare noncoding variants in IL12RB1 remained significant (p = 3.7 × 10−4) after correcting for multiple testing. Overall, the contribution of rare variants to asthma susceptibility was predominantly due to noncoding variants in sequences flanking the exons, although nonsynonymous rare variants in DPP10 and in IL12RB1 were associated with asthma in African Americans and European Americans, respectively. This study provides evidence that rare variants contribute to asthma susceptibility. Additional studies are required for testing whether prioritizing genes for resequencing on the basis of signatures of purifying selection is an efficient means of identifying novel rare variants that contribute to complex disease.
Introduction
Despite the many successes of genome-wide association studies (GWASs), only a small fraction of the heritability of common diseases is accounted for by the risk alleles identified through these studies.1–10 One possible explanation for this “missing heritability” is that the genotyping platforms typically used for GWASs include mostly common variants (those with a minor allele frequency [MAF] >0.05) selected for their “tagging” of larger haplotype blocks. This strategy is unlikely to tag most of the rare variants in the genome. In fact, theoretical modeling favors a scenario in which most of the genetic risk for common diseases is due to mildly deleterious mutations that are maintained at low frequency in the population by weak purifying (negative) selection.11 Such low-frequency or rare variants are likely to have larger effects on disease risk than the common variants detected by GWASs. To date, however, the relative contributions of alleles with MAF <5% to the heritability of common diseases have not been comprehensively surveyed.
A limitation to studies of rare variants is that they require resequencing of genes in cases and controls (or individuals selected from the tails of a distribution of quantitative phenotypes). Until recently, such studies were not feasible in large samples. However, reductions in sequencing costs and advances in sequencing technologies have now made it possible to directly evaluate the contribution of rare variants to the risk of common diseases. A second challenge is in selecting candidate genes for resequencing or for prioritizing the selection of genes identified from genome-wide studies. One approach has been to select genes underlying Mendelian forms of common diseases. For example, Cohen and colleagues resequenced genes containing mutations that result in Mendelian forms of lipidemias in subjects sampled from the upper and lower 5% of the distribution of cholesterol levels.12 In a series of studies of relatively small samples, rare nonsynonymous polymorphisms influencing normal variation in cholesterol levels were identified in many of these genes.13–15 Although this strategy might help identify genes harboring rare variants that increase the risk of disease, either Mendelian subforms do not exist for many common diseases, or the causal genetic mutations are unknown. Another approach is to focus on candidate genes and regions identified through GWASs. Johansen and colleagues used this approach to implicate rare variants in four genes identified through a GWAS of hypertriglyceridemia.16 However, genes with rare variants that contribute to disease might not necessarily also harbor common variants with sufficient effect sizes to be detected through GWASs. Thus, additional strategies are required for selecting genes for resequencing studies so that the genes harboring rare variants that contribute to common disease risk are more likely to be identified.
Asthma (MIM 600807) represents an excellent example of a common disease for which GWASs have identified common variants that collectively account for a very small proportion of the total genetic risk1–10 and for which Mendelian subforms are unknown. Collectively, common variation in well over 100 genes has been associated with asthma—either through GWASs, linkage and positional-cloning studies, or candidate-gene studies—with varying degrees of replication.17–19 A theoretical framework developed by Pritchard suggested that genes with molecular signatures of weak purifying selection are more likely to harbor an excess of rare or low-frequency variants involved in a complex disease.11 Weak purifying selection prevents mildly deleterious mutations from reaching appreciable frequencies in a population as compared to neutral or adaptive mutations. For example, genes involved in Mendelian diseases have proportionally more rare variation20 and show stronger signatures of purifying selection at nonsynonymous sites.21
We selected nine candidate asthma-associated genes for resequencing on the basis of evidence of weak purifying selection in a genome-wide scan for natural selection in both European and African Americans.22 Our results suggest that rare variation contributes to asthma susceptibility in both coding exons and their flanking noncoding regions. Discovering whether prioritizing genes with evidence of purifying selection is an efficient way to identify genes with rare variation that contributes to the etiology of common diseases will require additional studies.
Subjects and Methods
Study Subjects
Sequencing of coding exons and their corresponding flanking regions for the nine genes was performed in a total of 510 asthmatic cases and 515 nonasthmatic controls. After excluding 60 European American cases whose samples did not meet quality standards, we ended up with a final study population of 108 cases and 248 controls of European American descent and 342 cases and 267 controls of African American descent. Subjects included 58 European American children who have mild to moderate asthma and who participated in the Childhood Asthma Research and Education (CARE) Network,23 150 African American children recruited from Johns Hopkins University as part of the Genomic Research on Asthma in the African Diaspora (GRAAD)4 and from the University of Chicago as part of the National Heart, Lung, and Blood Institute (NHLBI)-funded Collaborative Studies on the Genetics of Asthma (CSGA),24 and 50 European American and 192 African American asthmatic adults recruited from Wake Forest University and the University of Chicago as part of the CSGA and the Severe Asthma Research Program (SARP; also funded by the NHLBI).25 Two hundred forty eight European American and 267 African American controls were recruited from Johns Hopkins University, Wake Forest University, and the University of Chicago. Controls were 18 years old or older, nonasthmatic, and without any first-degree relatives with asthma.
CARE included European American asthmatic cases; detailed descriptions of each trial are available online (see Web Resources).
SARP and CSGA included both African American and European American cases and controls. Subjects with mild to severe asthma were recruited from SARP centers and the CSGA, and they met the American Thoracic Society (ATS) definition of severe persistent asthma.26 All subjects were characterized according to asthma severity.25,27 Controls were recruited from the same medical centers and had no personal or first-degree-relative family history of asthma.
The GRAAD study consisted of self-reported African American cases and controls from the Baltimore-Washington, D.C. metropolitan area. To determine the asthma status of all affected individuals, we had a clinical coordinator administer a standardized questionnaire based on the asthma criteria set by either the American Thoracic Society28 or the International Study of Asthma and Allergy in Childhood (ISAAC).29 Asthma was defined as both a self-reported history of asthma and as a documented history of physician-diagnosed asthma (past or current). Controls were likewise administered a standardized questionnaire and were determined to be negative for a history of asthma.
Study protocols were approved by the institutional review boards at Harvard University, Johns Hopkins University, Wake Forest University, the University of Arizona, and the University of Chicago.
Selection of Genes
Because we do not know how to identify genes that harbor rare risk alleles, we decided to cast a wide net and include all genes that had been even marginally (or weakly) implicated in asthma risk. We compiled a list of 120 candidate asthma-associated genes, 118 from a literature review17 and an additional two, ORMDL3 and CHI3L1, that GWASs had recently revealed to be associated with asthma and an asthma-like quantitative phenotype, respectively, at the time this study was designed6,30. For sequencing, we ultimately selected nine genes that showed the strongest evidence of purifying selection at nonsynonymous sites in both European Americans and African Americans from a genome-wide scan for natural selection22 (Tables 1 and S1, available online). In brief, we obtained signatures of natural selection at nonsynonymous sites by comparing levels of human polymorphism (within-species variation) to chimpanzee sequence divergence (between-species variation) to estimate the population-scaled selection coefficient (γ = 2Nes) for each gene. We used an extension of the McDonald-Kreitman test and the MKPRF program31to identify loci showing the strongest signatures of purifying selection, as indicated by a higher proportion of polymorphism within species versus divergence between species. We ranked genes on the basis of the probability that γ was <−0.5; 53 of the asthma-associated genes had at least one SNP or fixed difference at nonsynonymous sites in the 20 European Americans and 15 African Americans22 (sequence data were available for 91 of the 120 genes, and 53 of these genes matched this criterion and were included in the study) (Table S1). From these, we selected the top nine genes with the strongest evidence of purifying selection in both populations for our resequencing study.
Table 1.
Estimates of the Probability of Negative Selection and the Population-Scaled Selection Coefficient for Nonsynonymous Sites in Nine Candidate Asthma-Associated Genes in African Americans and European Americans
| Gene | Symbol | MIM Number | Pr[γ < −0.5]a(γ)bAA | Pr[γ < −0.5] (γ) EA |
|---|---|---|---|---|
| Adrenergic, beta-2-, receptor, surface | ADRB2 | 109690 | 0.88 (−2.5) | 0.88 (−2.6) |
| Angiotensinogen | AGT | 106150 | 0.89 (−1.3) | 0.87 (−2.8) |
| Cystic fibrosis transmembrane Conductance regulator | CFTR | 602421 | 0.84 (−1.9) | 0.96 (−3.5) |
| Chitinase, acidic | CHIA | 606080 | 0.89 (−2.2) | 0.92 (−2.7) |
| Dipeptidyl-peptidase 10 | DPP10 | 608209 | 0.80 (−1.6) | 0.89 (−2.5) |
| Inhibitor of kappa light polypeptide gene enhancer in B cells | IKBKAP | 603722 | 0.96 (−2.6) | 0.81 (−1.2) |
| Interleukin 12 receptor, beta 1 | IL12RB1 | 601604 | 0.84 (−2.1) | 0.91 (−2.9) |
| Phospholipase A2, group VII | PLA2G7 | 601690 | 0.88 (−2.3) | 0.90 (−2.6) |
| Transforming growth factor, beta 1 | TGFB1 | 190180 | 0.83 (−2.4) | 0.95 (−4.0) |
The estimates of the probability of selection and the selection coefficient are available as supplemental data in Torgerson et al.22 Estimates for the complete list of genes are in Table S1.The following abbreviations are used: EA, European Americans; AA, African Americans.
Probability of negative selection.
Population-scaled selection coefficient (γ = 2Nes).
The nine genes selected for sequencing included one gene (CFTR [MIM 602421]) in which severe mutations cause a Mendelian lung disease and in which common variants have been associated with asthma32 and another gene (DPP10 [MIM 608209]) that was initially discovered in a positional cloning study in Europeans33 and then subsequently detected in a GWAS on asthma in African Americans.4 The remaining genes were previously selected as candidate genes on the basis of their known functions and were subsequently reported to be associated with asthma in at least one published study.17 All of these genes had an estimated probability of purifying selection at nonsynonymous sites >80% in both European Americans and African Americans and fell within the 92nd percentile of >10,000 genes included in a genome-wide scan for natural selection.22
DNA Sequencing
Sanger sequencing (on both the forward and reverse strands), variant detection, and annotation to coding and noncoding regions of each gene were performed at the NHLBI-supported Resequencing and Genotyping (RS&G) Service at the J. Craig Venter Institute (JCVI). PCR primers were designed to cover all coding exons with amplicon sizes ranging from 350–800 bp and with a 100 bp overlap between adjacent amplicons. We compared all primer sequences to the whole-genome assembly to verify their uniqueness against pseudogenes and gene families. The coordinates of all amplicons are available in Document S2. Chromatograms were base and quality checked with Applied Biosystems KB Basecaller v1.2 (on a 3730xl sequencer) and TraceTuner with custom calibration for 3730xl (see Web Resources), and they were mixed-base-called with in-house custom software. We annotated variants to coding and non-coding regions by using the Ensemble database v.50 (July 2008). Noncoding regions were intronic, 5′ upstream of the transcription start site, and 3′ downstream of the transcription stop site.
Data Analysis
All variants and subjects passing quality control (QC) at the JCVI were included in the analysis. Additional QC on each variant was performed with PLINK,34 including an assessment of call rates and deviation from the Hardy-Weinberg equilibrium. MAFs of previously identified variants were compared to the HapMap (phase 2, release 24) CEU (Utah residents with ancestry from northern and western Europe from the CEPH collection) and YRI (Yoruba in Ibadan, Nigeria) samples 35 and to pilot data from the 1,000 Genomes Project.36 We inferred ancestral states of each variant on the basis of sequence identity to the chimpanzee by using syntenic net alignments of the human (hg18) and chimpanzee (PanTro2) genomes downloaded from the UCSC genome browser.37,38 We created plots of the site-frequency spectrum by resampling the cases and controls for N = 100 chromosomes across each variant to account for missing data and uneven sampling. For variants with >1 derived state, the derived states were pooled and compared to the single ancestral state.
Statistical analyses were performed with PLINK34 and the R statistical package. We performed tests for allelic association at individual variants by using Fisher's exact test to compare counts of ancestral versus derived alleles in the cases versus controls. For variants with >1 derived state, derived states were pooled and compared to the single ancestral state. Odds ratios (ORs) were estimated with a shrinkage estimator that we obtained by applying the standard OR estimator to allele counts modified by adding 1/239. For the African American sample, we estimated the local European admixture at each gene by using genotypes from the Illumina 1M and 650K platforms and the LAMP (Local Ancestry in adMixed Populations) program.40 Admixture was modeled under seven generations of admixture with a two-population model of 80% ancestry from Africa and 20% ancestry from Europe. Windows were offset by a factor of 0.2, a cutoff for linkage was set to 0.1, and a constant recombination rate was set to 10−8. We repeated tests for allelic association in the African American sample by using local ancestry as a covariate, and we used stratified tests of association in individuals with and without local European admixture for each gene. Using the C-alpha test, we performed gene-based tests of association on nonsynonymous, synonymous, and noncoding variants to investigate the contributions of rare variants to asthma susceptibility (MAF <5% in cases or controls).41 We used a total of 50,000 permutations to evaluate statistical significance by shuffling case and control labels to maintain haplotypes and patterns of missing data. We used 100 permutations in a similar manner to test for an enrichment of small p values across all tests performed. We combined p values by using Fisher's method across the two populations.
Results
Our study population consisted of 108 cases and 248 controls of European American descent and 342 cases and 267 controls of African American descent. A total of 80,302 base pairs, including 17,931 coding sites and 62,371 noncoding sites (5′ and 3′ UTRs, introns, 5′ upstream regions, and 3′ downstream regions) flanking the coding exons, were sequenced across the nine genes. The average coverage of coding sites across all genes was 93%, ranging from 84% in IL12RB1 (MIM 601604) to 100% in both AGT (MIM 106150) and CHIA (MIM 606080) (Table 2). A total of 1,225 variants (303 coding and 922 noncoding) were detected across all nine genes, and 903 (74%) of these variants were absent in dbSNP version 129 (the last release prior to the addition of SNPs identified in the 1,000 Genomes Project). Of these 903 SNPs, 81 were subsequently identified in pilot data from the 1,000 Genomes Project. A total of 657 variants were unique to the African Americans, 184 variants were unique to the European Americans, and 384 variants were shared in both populations. The number of variants specific to the cases was 275, and the number of variants specific to the controls was 285. The majority of variants were rare (frequency <5%), and there was a higher proportion of rare variants in the African Americans (83%) than in the European Americans (71%) (Figure 1). The total counts of nonsynonymous, synonymous, and noncoding variants in cases and controls in each population are shown in Table S2, and the distributions of the number of rare variants carried at the individual level are shown in Figures S2 and S3.
Table 2.
Total Number of Sites Resequenced in Nine Candidate Asthma-Associated Genes, Including Percent Coverage of Coding Sites, and the Total Number of Variants Identified in European Americans and African Americans
| Gene | Coding Sites (% Coverage) | Noncoding Sites | Total Sites | Total Variants (EA/AA) | Novel dbSNPa(EA/AA) | Novel TGPb(EA/AA) | Total Shared | Specific to EA | Specific to AA |
|---|---|---|---|---|---|---|---|---|---|
| ADRB2 | 1,206 (97) | 674 | 1,880 | 37 (19/30) | 19 (8/13) | 19 (8/13) | 12 | 7 | 18 |
| AGT | 1,451 (100) | 1,624 | 3,075 | 69 (20/63) | 52 (11/46) | 47 (11/41) | 14 | 6 | 49 |
| DPP10 | 2,142 (85) | 12,228 | 14,370 | 221 (98/190) | 179 (70/149) | 153 (64/123) | 67 | 31 | 123 |
| CFTR | 3,910 (89) | 13,318 | 17,228 | 178 (87/139) | 127 (45/94) | 119 (41/86) | 48 | 39 | 91 |
| CHIA | 1,417 (100) | 5,326 | 6,743 | 115 (53/105) | 83 (25/73) | 68 (20/58) | 43 | 10 | 62 |
| IKBKAP | 3,760 (95) | 16,945 | 20,705 | 333 (166/284) | 247 (91/201) | 228 (82/183) | 117 | 49 | 167 |
| IL12RB1 | 1,748 (84) | 4,718 | 6,466 | 100 (43/86) | 65 (23/51) | 58 (21/45) | 29 | 14 | 57 |
| PLA2G7 | 1,289 (98) | 5,728 | 7,017 | 117 (51/97) | 86 (30/68) | 86 (30/68) | 31 | 20 | 66 |
| TGFB1 | 1,008 (97) | 1,810 | 2,818 | 55 (31/47) | 45 (25/38) | 44 (24/37) | 23 | 8 | 24 |
The following abbreviations are used: TGP, 1,000 Genomes Project; EA, European Americans; and AA, African Americans.
Variants that are absent in dbSNP build 129 (last version prior to the addition of pilot data from the 1,000 Genomes Project).
Variants that are absent from dbSNP build 129 and the 1,000 Genomes Project pilot data.
Figure 1.
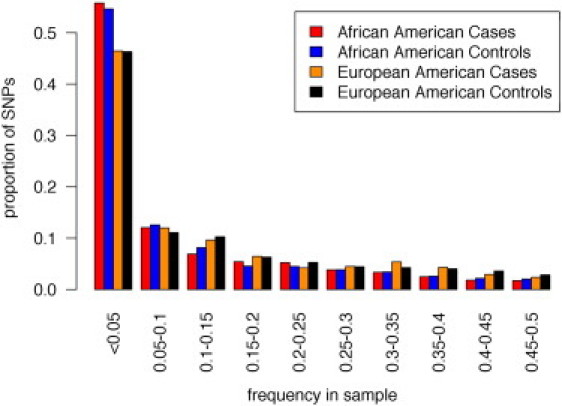
Combined Site-Frequency Spectrum of European American and African American Cases and Controls
Site-frequency spectrum of 1,225 variants identified through the resequencing of coding exons and flanking noncoding regions of nine candidate asthma-associated genes in 450 cases (108 European American and 342 African American) and 515 controls (248 European American and 267 African American). We resampled a total of 100 chromosomes in each of the European American and African American cases and controls to account for missing data and differences in sample size. Derived alleles were pooled for variants with >2 alleles; we used data from the chimpanzee to infer ancestral states. Plots for individual genes are shown in Figure S1.
No individual variant was significantly associated with asthma in either European Americans or African Americans after a Bonferroni correction for 1,225 tests (p < 4 × 10−5) (Figures S4 and S5, and see Document S3). However, in African Americans, there was a trend toward more rare variants in the asthmatic cases than in the controls (Figure S6). Population structure in African Americans might be a confounding factor in genetic tests of association; however, patterns of local European admixture were not significantly different between cases and controls for any of the genes (Table S3). Consistent with this finding, results in the African Americans were unchanged when we included local ancestry as a covariate or stratified cases and controls by the presence or absence of local European admixture (not shown).
We investigated gene-based tests for the potential involvement of rare variants and asthma susceptibility by using the C-alpha test,41 which allows for mixed effects of variants at a single locus (i.e., protective or risk alleles). Overall, we performed a total of 54 gene-based tests (9 genes × 3 site classes × 2 populations) and expected to see three or fewer p values <0.05 by chance. However, seven tests had a p value < 0.05 (Table 3), suggesting that there were at least four true positive associations in our data. Furthermore, in 100 random permutations of our data, only two resulted in seven or more p values <0.05 (permuted p = 0.02), indicating a significant enrichment of small p values in our data. Assuming that there are 2–3 false-positive results in our data, the pattern of signals seen in Table 2 implies that we expect rare variants in at least two genes to be true associations with asthma. Note that there are two genes (DPP10 and IL12RB1) with multiple signals, making them more likely to be truly associated.
Table 3.
C-alpha Test41 Results of Gene-Based Tests of Association between Rare Variants and Asthma Susceptibility
| Gene |
Nonsynonymous |
Synonymous |
Non-Coding |
|||
|---|---|---|---|---|---|---|
| N | p value | N | p value | N | p value | |
| African Americans | ||||||
| ADRB2 | 6 | 0.63 | 5 | 0.46 | 11 | 0.58 |
| AGT | 21 | 0.060 | 6 | 0.46 | 31 | 0.038 |
| DPP10 | 14 | 0.023 | 8 | 0.80 | 151 | 0.040 |
| CFTR | 23 | 0.82 | 5 | 0.60 | 91 | 0.11 |
| CHIA | 18 | 0.88 | 12 | 0.73 | 46 | 0.74 |
| IKBKAP | 23 | 0.44 | 20 | 0.67 | 186 | 0.047 |
| IL12RB1 | 13 | 0.65 | 15 | 0.84 | 45 | 0.0015 |
| PLA2G7 | 7 | 0.60 | 4 | 0.90 | 72 | 0.67 |
| TGFB1 | 4 | 0.12 | 7 | 0.39 | 34 | 0.16 |
| All | 129 | 0.41 | 82 | 0.95 | 667 | 0.0029 |
| European Americans | ||||||
| ADRB2 | 4 | 0.32 | 3 | 0.32 | 4 | 0.86 |
| AGT | 6 | 0.22 | 2 | 1.0 | 7 | 0.47 |
| DPP10 | 6 | 0.47 | 3 | 0.074 | 70 | 0.56 |
| CFTR | 24 | 0.32 | 4 | 0.59 | 44 | 0.38 |
| CHIA | 5 | 0.64 | 3 | 0.38 | 15 | 0.67 |
| IKBKAP | 14 | 0.79 | 9 | 0.28 | 96 | 0.47 |
| IL12RB1 | 6 | 0.034 | 2 | 1.0 | 18 | 0.022 |
| PLA2G7 | 3 | 0.65 | 1 | 1.0 | 37 | 0.57 |
| TGFB1 | 2 | 0.46 | 2 | 0.51 | 24 | 0.083 |
| All | 70 | 0.73 | 29 | 0.24 | 315 | 0.35 |
p values are based on 50,000 permutations and are in bold font for values <0.05. The seven gene-set combinations showing p < 0.05 represent an enrichment over the null expectation of no association (p = 0.02 based on 100 permutations). The following abbreviation is used: N, number of rare variants included in each comparison.
In African Americans, we observed an association between rare variants and asthma in four of the nine genes, including nonsynonymous rare variants in DPP10 (Table 4) (p = 0.023) and noncoding rare variants in AGT (p = 0.038), DPP10 (p = 0.040), IKBKAP (p = 0.047 [MIM 603722]), and IL12RB1 (p = 0.0015). In European Americans, we observed an association between rare variants and asthma in both nonsynonymous (Table 4) and noncoding variants in IL12RB1 (p = 0.034 and 0.022, respectively). The combined evidence that noncoding rare variants in IL12RB1 contribute to the risk of developing asthma in both African Americans and European Americans yielded a meta-analysis p value of 3.7 × 10−4, which surpasses a more stringent Bonferroni correction for 27 tests (9 genes × 3 site classes).
Table 4.
Frequencies of Rare Nonsynonymous Variants in DPP10 in African American Cases and Controls and in IL12RB1 in European American Cases and Controls
| Position (hg18) | Allelesa | Frequency in Cases | Frequency in Controls |
|---|---|---|---|
| DPP10 Chromosome 2 in African Americans | |||
| 115,636,295 | G/t | 0.0018 | 0 |
| 116,242,430 | A/g | 0.090 | 0.094 |
| 116,220,141 | G/a | 0.040 | 0.028 |
| 115,783,333 | A/c | 0 | 0.0019 |
| 116,201,862 | A/g | 0.0015 | 0 |
| 116,213,789 | A/c | 0.0089 | 0.0076 |
| 116,251,275 | G/a | 0 | 0.0019 |
| 116,288,939 | G/a | 0.033 | 0.0075 |
| 116,288,964 | T/g | 0.013 | 0.011 |
| 116,289,003 | T/c | 0 | 0.0019 |
| 116,289,012 | A/g | 0.0030 | 0.0019 |
| 116,289,750 | C/t | 0.0017 | 0 |
| 116,310,579 | G/a | 0 | 0.0020 |
| 116,314,867 | C/a | 0.0030 | 0 |
| IL12RB1 Chromosome 19 in European Americans | |||
| 18,041,373 | G/a | 0 | 0.0020 |
| 18,044,095 | C/t | 0.019 | 0.0041 |
| 18,031,808 | C/t | 0.0093 | 0 |
| 18,058,613 | C/t | 0.0046 | 0 |
| 18,031,874 | C/a | 0 | 0.0040 |
| 18,044,068 | G/a | 0 | 0.0020 |
The major allele is capitalized, and the minor allele is lowercase.
Discussion
We resequenced the coding exons and flanking noncoding regions of nine candidate asthma-associated genes that showed signatures of weak purifying selection, and we investigated the contributions of rare variants to asthma susceptibility. The majority (75%) of variants identified were absent from dbSNP build 129; however, some (9%) of them were present in pilot data from the 1,000 Genomes Project. No individual rare variant was significantly associated with asthma after a multiple-testing correction; this finding is not unexpected, given the low statistical power to detect an association for variants with MAFs <5% in samples of this size. However, gene-based tests of association suggested a contribution of rare variants to asthma susceptibility in four (AGT, DPP10, IKBKAP, and IL12RB1) of the nine genes studied. Only IL12RB1 showed a significant contribution of rare variants to asthma susceptibility in European Americans, whereas four genes were identified in African Americans. However, because the European American sample was smaller than the African American sample and European populations are expected to harbor less rare variation overall than African populations,42 we cannot rule out that the differences we observe between these samples are due to differences in statistical power.
Our original expectation was to discover associations between rare nonsynonymous variants and asthma risk. However, we found more evidence of rare noncoding variants in the regions flanking coding exons. For example, five of the seven p values <0.05 were due to associations with rare noncoding variants, and associations with noncoding variants in IL12RB1 were significantly associated with asthma in both African Americans and European Americans. Interestingly, 71% of the rare noncoding variants in IL12RB1 were specific to either one of the populations. Furthermore, nonsynonymous sites on IL12RB1 were associated with asthma in European Americans (p = 0.034) but not in African Americans (p = 0.65). Therefore, even when susceptibility-associated genes are shared between the two populations, different sets of rare variants might contribute to the risk of developing asthma.
Although we targeted coding exons, the majority of sites that were resequenced were flanking noncoding sites. In African Americans, only noncoding rare variants showed a significant contribution to asthma susceptibility when pooled across all genes (p = 0.0029), even though nonsynonymous substitutions are predicted to have larger effects on disease. Indeed, two genes (DPP10 in African Americans and IL12RB1 in European Americans) show evidence of rare nonsynonymous variants that are involved in asthma susceptibility. However, both of these genes also showed significant contributions of noncoding rare variation. Although it is possible that the most highly penetrant nonsynonymous mutations are too rare to be detected in studies of only hundreds of cases, our results suggest that rare noncoding variation in regions flanking exons play a more prominent role in asthma susceptibility.
There is currently great debate as to whether more of the heritability of common diseases, such as asthma, is explained by rare or common variants. Recent GWASs on asthma have identified common variants that explain very little of the heritability of asthma—their estimated effect sizes range from 1.1 to 1.3.1–10 These findings are consistent with either or both of the following scenarios: a large number of common variants that individually confer only a modest increase in the risk of asthma (such as for height43) and/or a large number of rare variants with larger effect sizes but which are not well tagged by commercial genotyping platforms. In the current study, we find associations between rare variants and asthma susceptibility in four of the nine genes studied, but we do not find associations between common variants and asthma susceptibility in any of the nine genes (see Document S3). Ongoing exome-sequencing studies in larger populations will better illuminate the relative contributions of rare and/or common variants to the heritability of asthma, but such studies might neglect the majority of noncoding variants. Our results suggest that noncoding variants in the exon-flanking sequences should not be ignored in future sequencing studies.
On the basis of theoretical predictions11 and because Mendelian-disease-associated genes have a greater proportion of rare variants20 and show strong signatures of purifying selection compared to other genes,21 we hypothesized that complex-disease genes harboring rare variants with larger effect sizes might show similar evolutionary patterns. We therefore selected candidate genes for this study on the basis of their evidence of weak purifying selection, and we identified significant associations between rare variants and asthma in up to four of the nine genes selected (Table S1). Future resequencing studies will illuminate whether selecting candidate genes on the basis of evidence of purifying selection increases the chances of finding genes harboring rare variants contributing to disease risk or whether we could have achieved similar results by selecting nine candidate asthma-associated genes at random from the list of 53. Lastly, regardless of whether there is a direct relationship between natural selection and asthma susceptibility, genes subjected to purifying selection might be more relevant in a broader range of processes and might therefore harbor a greater proportion of rare variation with functional effects.11
Overall, our results suggest that rare variants play an important role in asthma susceptibility in both African Americans and European Americans and that multiple rare variants at a single locus can contribute to a common disease etiology. Additional studies will need to address whether using signatures of purifying selection to prioritize candidate genes for resequencing studies or to assign weights to genes in exome-sequencing studies is an effective way of identifying novel rare variants that contribute to a common disease.
Acknowledgments
The authors gratefully acknowledge Dana Busam and Ewen Kirkness at the J. Craig Venter Institute (JCVI), the National Heart, Lung, and Blood Institute (NHLBI)-funded Resequencing and Genotyping Service at the JCVI, and all of the patients, investigators, coordinators, and research teams for the CAMP (Childhood Asthma Management Program) Genetic Ancillary Study, the Childhood Asthma Research and Education network, the Severe Asthma Research Program network, the Collaborative Studies on the Genetics of Asthma study, and the Genomic Research on Asthma in the African Diaspora study. These studies were supported by U01 HL49596 and R01 HL072414 to C.O.; RC2 HL101651 to C.O. and D.L.N.; R01 HL087665 to D.L.N.; U10 HL064307, U10 HL064288, U10 HL064295, U10 HL064287, U10 HL064305, and U10 HL064313 to F.D.M.; U01 HL075419, U01 HL65899, P01 HL083069, R01 HL086601, and RC2 HL101651 to S.T.W.; R01 HL69167 and HL101487 to D.A.M. and E.R.B.; and R01 HL087699, U01 HL49612, R01 AI50024, and R01 AI44840 to K.C.B. K.C.B. was supported in part by the Mary Beryl Patch Turnbull Scholar Program. Resequencing services were provided by the JCVI under U.S. Federal Government contract number N01-HV-48196 from the NHLBI.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
CARE Network Clinical Trials, http://www.asthma-carenet.org/trials.html
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
SourceForge Trace Tuner, http://sourceforge.net/projects/tracetuner/
The R Project for Statistical Computing, www.r-project.org
References
- 1.Hancock D.B., Romieu I., Shi M., Sienra-Monge J.J., Wu H., Chiu G.Y., Li H., del Rio-Navarro B.E., Willis-Owen S.A., Weiss S.T. Genome-wide association study implicates chromosome 9q21.31 as a susceptibility locus for asthma in mexican children. PLoS Genet. 2009;5:e1000623. doi: 10.1371/journal.pgen.1000623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Himes B.E., Hunninghake G.M., Baurley J.W., Rafaels N.M., Sleiman P., Strachan D.P., Wilk J.B., Willis-Owen S.A., Klanderman B., Lasky-Su J. Genome-wide association analysis identifies PDE4D as an asthma-susceptibility gene. Am. J. Hum. Genet. 2009;84:581–593. doi: 10.1016/j.ajhg.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li X., Howard T.D., Zheng S.L., Haselkorn T., Peters S.P., Meyers D.A., Bleecker E.R. Genome-wide association study of asthma identifies RAD50-IL13 and HLA-DR/DQ regions. J. Allergy Clin. Immunol. 2010;125 doi: 10.1016/j.jaci.2009.11.018. 328–335.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mathias R.A., Grant A.V., Rafaels N., Hand T., Gao L., Vergara C., Tsai Y.J., Yang M., Campbell M., Foster C. A genome-wide association study on African-ancestry populations for asthma. J. Allergy Clin. Immunol. 2010;125 doi: 10.1016/j.jaci.2009.08.031. 336–346.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moffatt M.F., Gut I.G., Demenais F., Strachan D.P., Bouzigon E., Heath S., von Mutius E., Farrall M., Lathrop M., Cookson W.O., GABRIEL Consortium A large-scale, consortium-based genomewide association study of asthma. N. Engl. J. Med. 2010;363:1211–1221. doi: 10.1056/NEJMoa0906312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moffatt M.F., Kabesch M., Liang L., Dixon A.L., Strachan D., Heath S., Depner M., von Berg A., Bufe A., Rietschel E. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature. 2007;448:470–473. doi: 10.1038/nature06014. [DOI] [PubMed] [Google Scholar]
- 7.Sleiman P.M., Flory J., Imielinski M., Bradfield J.P., Annaiah K., Willis-Owen S.A., Wang K., Rafaels N.M., Michel S., Bonnelykke K. Variants of DENND1B associated with asthma in children. N. Engl. J. Med. 2010;362:36–44. doi: 10.1056/NEJMoa0901867. [DOI] [PubMed] [Google Scholar]
- 8.Torgerson D.G., Ampleford E.J., Chiu G.Y., Gauderman W.J., Gignoux C.R., Graves P.E., Himes B.E., Levin A.M., Mathias R.A., Hancock D.B., Mexico City Childhood Asthma Study (MCAAS) Children's Health Study (CHS) and HARBORS study. Genetics of Asthma in Latino Americans (GALA) Study, Study of Genes-Environment and Admixture in Latino Americans (GALA2) and Study of African Americans, Asthma, Genes & Environments (SAGE) Childhood Asthma Research and Education (CARE) Network. Childhood Asthma Management Program (CAMP) Study of Asthma Phenotypes and Pharmacogenomic Interactions by Race-Ethnicity (SAPPHIRE) Genetic Research on Asthma in African Diaspora (GRAAD) Study Meta-analysis of genome-wide association studies of asthma in ethnically diverse North American populations. Nat. Genet. 2011;43:887–892. doi: 10.1038/ng.888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferreira M.A., Matheson M.C., Duffy D.L., Marks G.B., Hui J., Le Souëf P., Danoy P., Baltic S., Nyholt D.R., Jenkins M., Australian Asthma Genetics Consortium Identification of IL6R and chromosome 11q13.5 as risk loci for asthma. Lancet. 2011;378:1006–1014. doi: 10.1016/S0140-6736(11)60874-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hirota T., Takahashi A., Kubo M., Tsunoda T., Tomita K., Doi S., Fujita K., Miyatake A., Enomoto T., Miyagawa T. Genome-wide association study identifies three new susceptibility loci for adult asthma in the Japanese population. Nat. Genet. 2011;43:893–896. doi: 10.1038/ng.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pritchard J.K. Are rare variants responsible for susceptibility to complex diseases? Am. J. Hum. Genet. 2001;69:124–137. doi: 10.1086/321272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohen J.C., Kiss R.S., Pertsemlidis A., Marcel Y.L., McPherson R., Hobbs H.H. Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science. 2004;305:869–872. doi: 10.1126/science.1099870. [DOI] [PubMed] [Google Scholar]
- 13.Romeo S., Pennacchio L.A., Fu Y., Boerwinkle E., Tybjaerg-Hansen A., Hobbs H.H., Cohen J.C. Population-based resequencing of ANGPTL4 uncovers variations that reduce triglycerides and increase HDL. Nat. Genet. 2007;39:513–516. doi: 10.1038/ng1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cohen J.C., Pertsemlidis A., Fahmi S., Esmail S., Vega G.L., Grundy S.M., Hobbs H.H. Multiple rare variants in NPC1L1 associated with reduced sterol absorption and plasma low-density lipoprotein levels. Proc. Natl. Acad. Sci. USA. 2006;103:1810–1815. doi: 10.1073/pnas.0508483103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kotowski I.K., Pertsemlidis A., Luke A., Cooper R.S., Vega G.L., Cohen J.C., Hobbs H.H. A spectrum of PCSK9 alleles contributes to plasma levels of low-density lipoprotein cholesterol. Am. J. Hum. Genet. 2006;78:410–422. doi: 10.1086/500615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johansen C.T., Wang J., Lanktree M.B., Cao H., McIntyre A.D., Ban M.R., Martins R.A., Kennedy B.A., Hassell R.G., Visser M.E. Excess of rare variants in genes identified by genome-wide association study of hypertriglyceridemia. Nat. Genet. 2010;42:684–687. doi: 10.1038/ng.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ober C., Hoffjan S. Asthma genetics 2006: The long and winding road to gene discovery. Genes Immun. 2006;7:95–100. doi: 10.1038/sj.gene.6364284. [DOI] [PubMed] [Google Scholar]
- 18.Vercelli D. Advances in asthma and allergy genetics in 2007. J. Allergy Clin. Immunol. 2008;122:267–271. doi: 10.1016/j.jaci.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 19.Ober C., Yao T.C. The genetics of asthma and allergic disease: A 21st century perspective. Immunol. Rev. 2011;242:10–30. doi: 10.1111/j.1600-065X.2011.01029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blekhman R., Man O., Herrmann L., Boyko A.R., Indap A., Kosiol C., Bustamante C.D., Teshima K.M., Przeworski M. Natural selection on genes that underlie human disease susceptibility. Curr. Biol. 2008;18:883–889. doi: 10.1016/j.cub.2008.04.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bustamante C.D., Fledel-Alon A., Williamson S., Nielsen R., Hubisz M.T., Glanowski S., Tanenbaum D.M., White T.J., Sninsky J.J., Hernandez R.D. Natural selection on protein-coding genes in the human genome. Nature. 2005;437:1153–1157. doi: 10.1038/nature04240. [DOI] [PubMed] [Google Scholar]
- 22.Torgerson D.G., Boyko A.R., Hernandez R.D., Indap A., Hu X., White T.J., Sninsky J.J., Cargill M., Adams M.D., Bustamante C.D., Clark A.G. Evolutionary processes acting on candidate cis-regulatory regions in humans inferred from patterns of polymorphism and divergence. PLoS Genet. 2009;5:e1000592. doi: 10.1371/journal.pgen.1000592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guilbert T.W., Morgan W.J., Krawiec M., Lemanske R.F.J., Jr., Sorkness C., Szefler S.J., Larsen G., Spahn J.D., Zeiger R.S., Heldt G., Prevention of Early Asthma in Kids Study,Childhood Asthma Research and Education Network The Prevention of Early Asthma in Kids study: Design, rationale and methods for the Childhood Asthma Research and Education network. Control. Clin. Trials. 2004;25:286–310. doi: 10.1016/j.cct.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 24.Lester L.A., Rich S.S., Blumenthal M.N., Togias A., Murphy S., Malveaux F., Miller M.E., Dunston G.M., Solway J., Wolf R.L., Collaborative Study on the Genetics of Asthma Ethnic differences in asthma and associated phenotypes: Collaborative study on the genetics of asthma. J. Allergy Clin. Immunol. 2001;108:357–362. doi: 10.1067/mai.2001.117796. [DOI] [PubMed] [Google Scholar]
- 25.Moore W.C., Bleecker E.R., Curran-Everett D., Erzurum S.C., Ameredes B.T., Bacharier L., Calhoun W.J., Castro M., Chung K.F., Clark M.P., National Heart, Lung, Blood Institute's Severe Asthma Research Program Characterization of the severe asthma phenotype by the National Heart, Lung, and Blood Institute's Severe Asthma Research Program. J. Allergy Clin. Immunol. 2007;119:405–413. doi: 10.1016/j.jaci.2006.11.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.American Thoracic Society Proceedings of the ATS workshop on refractory asthma: Current understanding, recommendations, and unanswered questions. Am. J. Respir. Crit. Care Med. 2000;162:2341–2351. doi: 10.1164/ajrccm.162.6.ats9-00. [DOI] [PubMed] [Google Scholar]
- 27.Moore W.C., Meyers D.A., Wenzel S.E., Teague W.G., Li H., Li X., D'Agostino R.J., Jr., Castro M., Curran-Everett D., Fitzpatrick A.M., National Heart, Lung, and Blood Institute's Severe Asthma Research Program Identification of asthma phenotypes using cluster analysis in the Severe Asthma Research Program. Am. J. Respir. Crit. Care Med. 2010;181:315–323. doi: 10.1164/rccm.200906-0896OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Standards for the diagnosis and care of patients with chronic obstructive pulmonary disease (COPD) and asthma. This official statement of the American Thoracic Society was adopted by the ATS Board of Directors, November 1986. Am. Rev. Respir. Dis. 1987;136:225–244. doi: 10.1164/ajrccm/136.1.225. [DOI] [PubMed] [Google Scholar]
- 29.Worldwide variations in the prevalence of asthma symptoms: The International Study of Asthma and Allergies in Childhood (ISAAC) Eur. Respir. J. 1998;12:315–335. doi: 10.1183/09031936.98.12020315. [DOI] [PubMed] [Google Scholar]
- 30.Ober C., Tan Z., Sun Y., Possick J.D., Pan L., Nicolae R., Radford S., Parry R.R., Heinzmann A., Deichmann K.A. Effect of variation in CHI3L1 on serum YKL-40 level, risk of asthma, and lung function. N. Engl. J. Med. 2008;358:1682–1691. doi: 10.1056/NEJMoa0708801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bustamante C.D., Nielsen R., Sawyer S.A., Olsen K.M., Purugganan M.D., Hartl D.L. The cost of inbreeding in Arabidopsis. Nature. 2002;416:531–534. doi: 10.1038/416531a. [DOI] [PubMed] [Google Scholar]
- 32.Tzetis M., Efthymiadou A., Strofalis S., Psychou P., Dimakou A., Pouliou E., Doudounakis S., Kanavakis E. CFTR gene mutations—including three novel nucleotide substitutions—and haplotype background in patients with asthma, disseminated bronchiectasis and chronic obstructive pulmonary disease. Hum. Genet. 2001;108:216–221. doi: 10.1007/s004390100467. [DOI] [PubMed] [Google Scholar]
- 33.Allen M., Heinzmann A., Noguchi E., Abecasis G., Broxholme J., Ponting C.P., Bhattacharyya S., Tinsley J., Zhang Y., Holt R. Positional cloning of a novel gene influencing asthma from chromosome 2q14. Nat. Genet. 2003;35:258–263. doi: 10.1038/ng1256. [DOI] [PubMed] [Google Scholar]
- 34.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frazer K.A., Ballinger D.G., Cox D.R., Hinds D.A., Stuve L.L., Gibbs R.A., Belmont J.W., Boudreau A., Hardenbol P., Leal S.M., International HapMap Consortium A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Durbin R.M., Abecasis G.R., Altshuler D.L., Auton A., Brooks L.D., Durbin R.M., Gibbs R.A., Hurles M.E., McVean G.A., 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karolchik D., Baertsch R., Diekhans M., Furey T.S., Hinrichs A., Lu Y.T., Roskin K.M., Schwartz M., Sugnet C.W., Thomas D.J., University of California Santa Cruz The UCSC Genome Browser Database. Nucleic Acids Res. 2003;31:51–54. doi: 10.1093/nar/gkg129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Agresti A. Second Edition. John Wiley & Sons; Hoboken, NJ: 2007. An Introduction to Categorical Data Analysis (Wiley Series in Probability and Statistics) [Google Scholar]
- 40.Sankararaman S., Sridhar S., Kimmel G., Halperin E. Estimating local ancestry in admixed populations. Am. J. Hum. Genet. 2008;82:290–303. doi: 10.1016/j.ajhg.2007.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Neale B.M., Rivas M.A., Voight B.F., Altshuler D., Devlin B., Orho-Melander M., Kathiresan S., Purcell S.M., Roeder K., Daly M.J. Testing for an unusual distribution of rare variants. PLoS Genet. 2011;7:e1001322. doi: 10.1371/journal.pgen.1001322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boyko A.R., Williamson S.H., Indap A.R., Degenhardt J.D., Hernandez R.D., Lohmueller K.E., Adams M.D., Schmidt S., Sninsky J.J., Sunyaev S.R. Assessing the evolutionary impact of amino acid mutations in the human genome. PLoS Genet. 2008;4:e1000083. doi: 10.1371/journal.pgen.1000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang J., Benyamin B., McEvoy B.P., Gordon S., Henders A.K., Nyholt D.R., Madden P.A., Heath A.C., Martin N.G., Montgomery G.W. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 2010;42:565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.