

Abstract
Previous studies have identified the 3,6-dialkyl-4-hydroxy-pyran-2-one marine microbial metabolites pseudopyronines A and B to be modest growth inhibitors of Mycobacterium tuberculosis and a range of tropical diseases including Plasmodium falciparum and Leishmania donovani. In an effort to expand the structure-activity relationship of this compound class towards. infectious diseases, a library of natural product and natural product-like 4-methoxy-6-styryl-pyran-2-ones and a subset of catalytically reduced examples were synthesised. In addition, the photochemical reactivity of several of the 4-methoxy-6-styryl-pyran-2-ones were investigated yielding head-to-head and head-to-tail cyclobutane dimers as well as examples of asymmetric aniba-dimer A-type dimers. All compounds were evaluated for cytotoxicity and activity against M. tuberculosis, P. falciparum, L. donovani, Trypanosoma brucei rhodesiense and T. cruzi. Of the styryl-pyranones, natural product 3 and non-natural styrene and naphthalene substituted examples 13, 18, 21, 22 and 23 exhibited antimalarial activity (IC50 < 10 μM) with selectivity indices (SI) > 10. Δ7 Dihydro analogues were typically less active or lacked selectivity. Head-to-head and head-to-tail photodimers 5 and 34 exhibited moderate IC50s of 2.3 to 17 μM towards several of the parasitic organisms, while the aniba-dimer-type asymmetric dimers 31 and 33 were identified as being moderately active towards P. falciparum (IC50 1.5 and 1.7 μM) with good selectivity (SI ∼ 80). The 4-tert-butyl aniba-dimer A analogue 33 also exhibited activity towards L. donovani (IC50 4.5 μM), suggesting further elaboration of this latter scaffold could lead to the identification of new leads for the dual treatment of malaria and leishmaniasis.
Keywords: Malaria, Tuberculosis, Natural product, Pyran-2-one, Photochemical dimerisation
1. Introduction
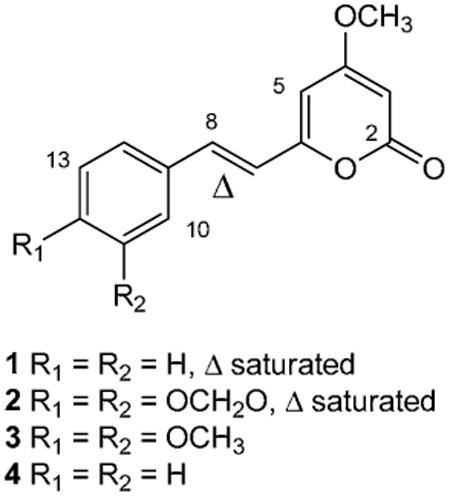
4-Methoxy-6-styryl-2H-pyran-2-ones are polyketide synthase-derived natural products commonly reported from fungi and primitive angiosperms.1,2 The most well known representatives of this class of compounds are the kavapyrones being neuroactive and potentially hepatotoxic metabolites of Kava (Piper methysticum).3 The related dihydrostyryl- and styryl-pyrones 1, 2 and 3, isolated from a number of sources, including Alpinia speciosa4 and Polygala sabulosa,5 exhibit plant growth inhibiting properties4 as well as anxiolytic6 and antinociceptive7 (visceral pain) effects in mice. There are however few reports of the evaluation of this family of natural products for activity towards infectious diseases such as trypanosomiasis, leishmaniasis, malaria or tuberculosis.8-10 Pyrone 3 displays weak activity against American Trypanosoma cruzi blood trypomastigotes,9 while 2 and 4, isolated from Piper sanctum, were growth inhibitors of Mycobacterium tuberculosis H37Rv with MICs of 32 μg/mL and 4 μg/mL respectively.10
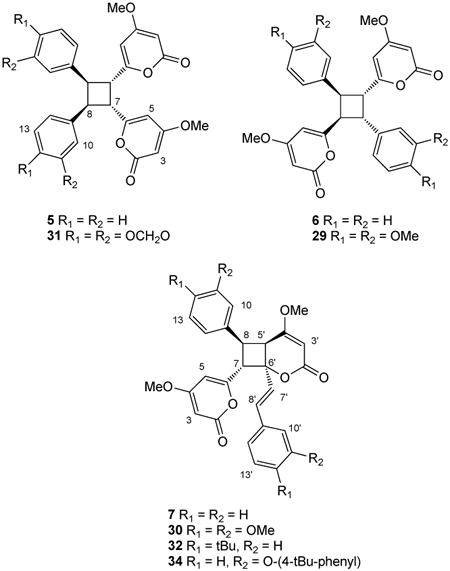
Styrylpyrones such as 4 are also known to undergo photochemically-induced dimerization, to form symmetrical head-to-head (5)11,12 or head-to-tail (6)13,14 cyclobutane dimers or unsymmetrical dimers (7).15,16 Although examples of each dimer class have been reported as natural products, it is still unclear whether they are true natural products or artifacts generated during the process of isolation and purification. Head-to-tail and asymmetric dimers related to 6 and 7, isolated from extracts of the Colombian medicinal plant Achyrocline bogotensis, were recently reported to modulate cytokine production by chemically stimulated human peripheral blood mononuclear cells.17
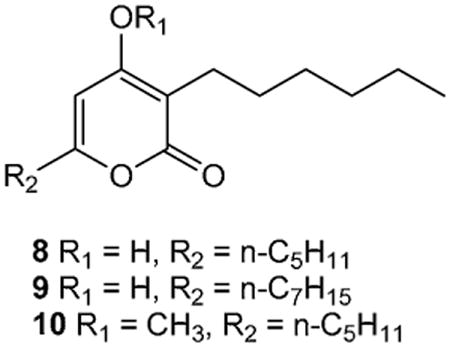
During our recent study of the structures and anti-infective bioactivities of the marine microbial pyrones pseudopyronines A (8) and B (9), the latter was found to be a modest growth inhibitor of Mycobacterium tuberculosis (MIC 2.7 μM), Trypanosoma brucei rhodesiense (IC50 42 μM), Leishmania donovani (IC50 4.8 μM) and Plasmodium falciparum (K1 strain, chloroquine resistant) (IC50 48 μM) while the methylether analogue of pseudopyronine A 10 was similarly active towards L. donovani, less active towards M tuberculosis (MIC 45 μM) but more potent towards both T. brucei rhodesiense (IC50 14 μM) and P. falciparum (IC50 11 μM).18 In continuation of our interest in this class of natural products, we have synthesized a library of natural and unnatural 4-methoxy-6-styryl-2H-pyran-2-ones, and a selection of Δ7 dihydro and photochemically dimerized analogues, and evaluated their abilities to act as growth inhibitors of the neglected disease target organisms M. tuberculosis, L. donovani, T. brucei rhodesiense, T. cruzi and P. falciparum.
2. Chemistry
Target styrylpyrones were chosen to explore the effects of steric bulk, lipophilicity and extended conjugation on biological activity. To this end, the pyrones were prepared by aldol condensation of 4-methoxy-6-methyl-2H-pyran-2-one 1119 and a variety of aryl aldehydes (Scheme 1, Table 1).20 Thus a solution of pyrone 11 and the appropriate aldehyde in anhydrous MeOH was added to a suspension of magnesium methoxide in anhydrous methanol and the resulting mixture heated at reflux for 6 hours. Upon cooling, the solvent was removed in vacuo and the crude product purified by either recrystallization or silica gel column chromatography.
Scheme 1.
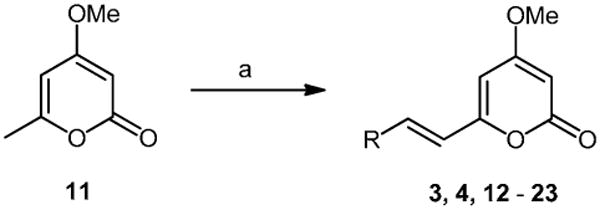
Reagents and conditions: (a) RCHO, Mg(OMe)2, MeOH
Table 1. 6-Styryl-pyran-2H-ones and their biological activities.
| entry | No. | structure | M. tb.a | T.b.rhod.b | T. cruzic | L.don.d | P. falc.e | L6f | SI Pf.g |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 3 |
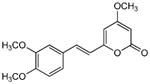
|
>350 | 31 | 170 | 270 | 3.0 | 160 | 54 |
| 2 | 4 |
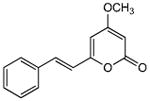
|
>440 | 120 | 64 | 190 | 13 | 160 | 12 |
| 3 | 12 |
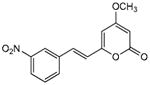
|
>370 | 290 | >330 | 270 | 12 | 180 | 15 |
| 4 | 13 |
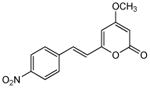
|
>370 | 38 | 240 | 49 | 6.0 | 190 | 32 |
| 5 | 14 |
|
>350 | 44 | 26 | 20 | 5.3 | 24 | 4 |
| 6 | 15 |
|
>370 | 47 | 180 | 230 | 5.6 | 16 | 3 |
| 7 | 16 |
|
>310 | 8.8 | 43 | 30 | 4.9 | 13 | 3 |
| 8 | 17 |
|
>270 | 34 | 40 | 25 | 6.3 | 64 | 10 |
| 9 | 18 |
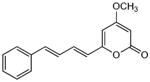
|
>390 | 150 | >350 | 220 | 6.7 | 190 | 29 |
| 10 | 19 |
|
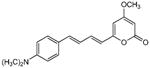
170 | 290 | >300 | >300 | 7.2 | 26 | 4 |
| 11 | 20 |
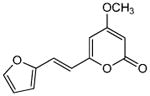
|
>460 | 76 | >410 | 240 | 17 | 140 | 8 |
| 12 | 21 |
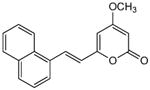
|
>360 | 18 | 34 | 47 | 4.6 | 110 | 24 |
| 13 | 22 |
|
>360 | 2.7 | >320 | >320 | 1.3 | 28 | 22 |
| 14 | 23 |
|
>260 | 119 | >240 | >240 | 8.6 | >240 | >28 |
Mycobacterium tuberculosis H37Rv grown in GAST medium, MIC (μM). Highest test concentration was 100 μg/mL;
Trypanosoma brucei rhodesiense. IC50 (μM);
Trypanosoma cruzi. IC50 (μM);
Leishmania donovani. IC50 (μM);
Plasmodium falciparum. IC50 (μM);
L6 rat skeletal myoblast cell line. IC50 (μM);
Selectivity index for Plasmodium falciparum against L6. All data is the mean value from duplicate assays.
The synthesis and characterisation of target pyrones 3,5,21,22 4,4,23 12,4 13,4 15,22,24 and 1820 have been previously reported and in all cases the spectroscopic and spectrometric data observed in the current study agreed with the previously reported values. The remaining new compounds were fully characterized by MS and NMR, and the data observed was consistent with the structures (see Experimental). Diffusion of hexane into a solution of 3 in acetone and slow evaporation of a methanolic solution of 14 yielded crystals of a suitable quality for X-ray analysis. The crystal structures of 3 and 14 and details of the crystallographic studies are given in the Supplementary data. Noteworthy in both cases is that these particular pyrones pack in the crystal unit cell in head-to-tail fashion, with alignment of Δ5 and Δ7 at intermolecular distances of 3.6-3.7 Å and 3.5-3.9 Å for 3 and 14, respectively (see Supplementary data). Previous studies have concluded that 3.6-4.1 Å is a typical intermolecular distance for cinnamic acid analogues to achieve [2 + 2] cycloaddition.25 The alignment and intermolecular distances have a direct bearing on the observed solid state photochemical reactivities of these pyrones to yield aniba-dimer-type products (see later).
Catalytic hydrogenation20 of pyrones 3, 4, 14, 15, 17, 21, 22, and 23 with 10% Pd/C under a hydrogen atmosphere yielded exclusively the Δ7 dihydro analogues 255, 126, 24, 25, 26, 27, 28, and 29, respectively, in yields of 49-96% (Table 2). The new compounds prepared were fully characterized by MS and NMR, and the data observed was consistent with the structures (see Experimental). Slow evaporation of a H2O-MeOH solution of 2 gave crystals suitable for X-ray analysis, the structure and details of which are given in Supplementary data. Reduction of Δ7 led to loss of overall planarity of the molecule, compared to styryl pyrones 3 and 14.
Table 2. Alkyl-pyran-2H-ones and their biological activities.
| entry | No. | R/structure | M. tb.a | T.b.rhod.b | T. cruzic | L.don.d | P. falc.e | L6f | SI Pfg |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 |
|
>430 | 280 | 240 | 190 | >22 | 290 | - |
| 2 | 2 |
|
>360 | >330 | >330 | >330 | >18 | 150 | - |
| 3 | 24 |
|
170 | 78 | 55 | 24 | 11 | 95 | 9 |
| 4 | 25 |
|
>340 | 200 | >310 | 270 | >17 | >310 | - |
| 5 | 26 |
|
130 | 65 | 36 | 15 | 7.2 | 50 | 7 |
| 6 | 27 |
|
270 | 85 | 76 | 55 | >18 | 110 | - |
| 7 | 28 |
|
180 | 72 | 65 | 42 | 16 | 91 | 6 |
| 8 | 29 |
|
>260 | 200 | >240 | >240 | 3.6 | 50 | 14 |
Mycobacterium tuberculosis H37Rv grown in GAST medium, MIC (μM). Highest test concentration was 100 μg/mL;
Trypanosoma brucei rhodesiense. IC50 (μM);
Trypanosoma cruzi. IC50 (μM);
Leishmania donovani. IC50 (μM);
Plasmodium falciparum. IC50 (μM);
L6 rat skeletal myoblast cell line. IC50 (μM);
Selectivity index for Plasmodium falciparum against L6. All data is the mean value from duplicate assays.
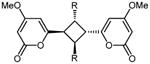
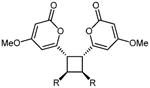
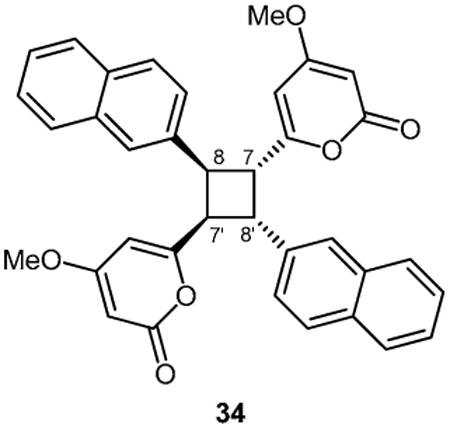
As noted earlier, the solid state photodimerization of 4-methoxy-6-styryl-2H-pyran-2-one 4 has been extensively studied and is known to yield three distinct dimer products, 5 - 7.11-16 The three established photocyclisation products arise from [2+2] cycloadditions of styryl-pyrones in either head-to-head (5) or head-to-tail (6) orientations or cycloaddition between Δ5 of one molecule with Δ7 of a second molecule (7). Preliminary studies of the propensity of styryl pyrones 3, 4, 12 – 23 to undergo photochemical reactions identified 3, 4, 14, 15, 17 and 22 as being reactive and yielding of stable products. Samples of these pyrones were each suspended in water (∼1 mL) with vigorous stirring and exposed to a 300 W sunlamp from a distance of 15 cm for 5 hrs. Under these conditions pyrone 4 afforded dimers 511,12 and 714-16 in isolatable quantities. Trace quantities of dimer 613,14 in the crude product mixture were detected by 1H NMR spectroscopy but could not be purified from the other major products of the reaction. The 1H NMR data observed for 5 agreed with those reported for the cis-trans-cis head-to-head dimer previously obtained as a natural product from the leaves of Aniba gardneri (though the structure was originally assigned as 6-cis-styryl-4-methoxy-2-pyrone)15 or as a synthetic photodimerization product.11,12 The 1H NMR data observed for 7 also agreed with those previously reported for aniba-dimer-A.14 Photodimerization of pyrone 3 gave the previously reported natural products 30 and 31,22 while pyrones 14, 15, 17 and 22 each gave a single product of 33 (asymmetric), 32 (head-to-head), 35 (asymmetric), and 34 (head-to-tail) respectively (Table 3). The structures and relative configuration of asymmetric dimers 33 and 35 were readily established by interpretation of 2D NMR data and comparison with NMR chemical shifts observed for aniba-dimer A (7). Dimer 32 was judged to be of head-to-head orientation by comparison of NMR chemical shifts with that of 5, and by ESI-MS-MS analysis, which identified a major fragment of the pseudomolecular parent ion to be m/z 277.0690 ([M+H]+, calcd for C14H13O6 277.0707), which is only possible for a head-to-head dimer. A similar ion was observed in ESI-MS-MS analysis of 5. The cis-trans-cis relative configuration of 32 was assumed to match that of 5, which has been rigorously established.12 In contrast, 2-naphthalene dimer 34 was determined to be of head-to-tail orientation, based upon ESI-MS-MS analysis which only detected a fragment at m/z 279.1005 ([M+H]+, calcd for C18H15O3 279.1021). Similar retro-[2+2] fragmentation was observed for dimers 6 and 30 (see Experimental).
Table 3. Dimeric pyranones and their biological activities.
| entry | No. | R | M. tb.a | T.b.rhod.b | T. cruzic | L.don.d | P. falc.e | L6f | SI Pfg |
|---|---|---|---|---|---|---|---|---|---|
|
|||||||||
| 1 | 30 | 3,4-dimethoxyphenyl | >170 | 6.2 | 38 | 75 | 3.1 | 22 | 7 |
| 2 | 34 | 2-naphthyl | >180 | 4.9 | 90 | 10 | 2.3 | 95 | 42 |
|
| |||||||||
|
|||||||||
| 3 | 5 | phenyl | >220 | 15 | 32 | 17 | 3.8 | 51 | 13 |
| 4 | 32 | benzo[3,4]dioxomethylene | >180 | ndh | nd | nd | nd | nd | nd |
|
| |||||||||
|
|||||||||
| 5 | 7 | phenyl | >220 | 13 | 28 | 27 | 7.1 | >200 | >28 |
| 6 | 31 | 3,4-dimethoxyphenyl | >170 | 61 | 130 | 79 | 1.5 | 120 | 80 |
| 7 | 33 | 4-tert-butyl-phenyl | >180 | 21 | 71 | 4.5 | 1.7 | 130 | 76 |
| 8 | 35 | 3-(4-tert-butyl-phenoxy)-phenyl | >130 | >120 | >120 | 66 | 4.4 | >120 | >27 |
Mycobacterium tuberculosis H37Rv grown in GAST medium, MIC (μM). Highest test concentration was 100 μg/mL;
Trypanosoma brucei rhodesiense. IC50 (μM);
Trypanosoma cruzi. IC50 (μM);
Leishmania donovani. IC50 (μM);
Plasmodium falciparum. IC50 (μM);
L6 rat skeletal myoblast cell line. IC50 (μM);
Selectivity index for Plasmodium falciparum against L6. All data is the mean value from duplicate assays.
3. Biological results and discussion
3.1. Overview
The resultant library of 6-styryl-pyran-2-ones, dihydro analogues and photodimers were evaluated for anti-tuberculosis, anti-parasitic and cytotoxic activities. Anti-tuberculosis testing was carried out against the Mycobacterium tuberculosis H37Rv strain grown in GAST medium. The reported MIC was determined as the minimal concentration of test compound leading to 100% growth inhibition. To investigate the potential of the compounds to act as drug leads against human African trypanosomiasis (HAT), American trypanosomiasis, leishmaniasis or malaria, their abilities to inhibit the growth of Trypanosoma brucei rhodesiense (strain STIB 900, trypomastigote stage), T. cruzi (strain Tulahuen C2C4, amastigote stage), Leishmania donovani (strain MHOM-ET-67/L82, amastigote/axenic stage) and Plasmodium falciparum (strain K1, IEF stage) were evaluated. In order to establish selectivity indices, the cytotoxicity of compounds towards the rat skeletal myoblast cell line L6 were also determined. Summaries of these assay results are presented in Tables 1, 2 and 3, with compounds grouped into the three different classes of 6-styryl-pyran-2-ones (Table 1), their dihydro analogues (Table 2) and photodimers (Table 3).
3.2. Anti-tuberculosis activity
Overall, none of the library compounds exhibited any appreciable level of anti-tuberculosis activity. This finding was somewhat surprising given the reported activities of the styryl-pyranone 4 and dihydro analogue 2 natural products towards M. tuberculosis H37Rv.10 When compared to the moderately antimycobacterial activities observed for 3,6-dialkyl-pyranones 8 - 10 and other 6-alkyl substituted pyranones18 the current results indicate that the presence of aryl-alkenyl/alkyl substitution at C-6 is detrimental to activity.
3.3 Anti-parasitic activity
On the whole, the library of 6-styryl-pyran-2-ones exhibited moderate activity towards Plasmodium falciparum, with IC50s varying between 1.3 to 17 μM (Table 1). Of particular note was the potency of antimalarial activity coupled with lack of cytotoxicity towards the L6 cell line of dimethoxy 3 (entry 1), para-nitro 13 (entry 4), aryl-diene 18 (entry 9), both naphthalene analogues 21 and 22 (entries 12 and 13) and dipyrone 23 (entry 14). In all cases, these pyrones exhibited antimalarial activity with IC50 < 10 μM, and with a selectivity index greater than 10, identifying them as potentially useful starting points for further structure-activity optimisation studies. Only two examples of styryl-pyran-2-ones, 16 and 22, achieved activity towards Trypanosoma brucei rhodesiense with IC50 < 10 μM. In the case of the former compound, broad ranging anti-parasitic activity was observed suggesting a general mechanism related to toxicity. Collectively, the styryl-pyran-2-ones exhibited only modest to poor activity towards T. cruzi and L. donovani. There was no apparent correlation of specific structural features with biological activity in this series of compounds.
The Δ7 dihydro analogues 1, 2, and 24-29 were typically less potent and lacked selectivity against the parasitic targets (Table 2) compared to their Δ7 counterparts. Three examples were identified as being more active growth inhibitors of P. falciparum (24, 26, 29; entries 3, 5 and 8) with IC50s of 11, 7.2 and 3.6 μM respectively, however they lacked selectivity (SI 9, 7 and 14 respectively) versus the L6 cell line, limiting their therapeutic potential.
Symmetrical head-to-tail dimers 30 and 34 (Table 3, entries 1 and 2) and head-to-head dimer 5 (Table 3, entry 3) were found to be moderate growth inhibitors of two organisms, P. falciparum and T. brucei rhodesiense. In the cases of 5 and 34, limited cytotoxicity towards the L6 cell line identifies both classes of dimers as being useful starting points for further development. In contrast, the asymmetric aniba-dimer A-type compounds 7, 31, 33 and 35 exhibited antimalarial activity (IC50 1.5 to 7.1 μM) with moderate selectivity versus the L6 cell line. Of special note is the identification of tert-butyl-phenyl dimer 33 as a dual organism growth inhibitor exhibiting anti-leishmanial (IC50 4.5 μM) and anti-malarial (IC50 1.7 μM) activities. Although limited in scope, the results presented in Table 3 suggest that enhanced lipophilicity analogues of asymmetric aniba-dimer A-type compounds have the potential to act as novel scaffolds for the development of anti malarial or dual organism anti-malarial and anti-leishmanial drugs.
4. Conclusions
A library of natural product and natural product-like 4-methoxy-6-styryl-pyran-2-ones, selected Δ7 dihydro analogues and photodimers have been synthesized and several candidates identified as potential anti-malarial or dual organism anti-malarial / anti-T. brucei rhodesiense or anti-malarial / anti-leishmanial agents. Overall the styryl-pyranones were moderate growth inhibitors of P. falciparum but in many cases lacked selectivity. Similar conclusions were drawn for the Δ7 dihydro analogues. Cyclobutane head-to-tail and head-to-head photodimers were found to represent a new dual organism P. falciparum / T. brucei rhodesiense inhibiting scaffold while asymmetric dimers related to aniba-dimer A inhibited P. falciparum and in one example, also inhibited L. donovani. Further exploration of structure-activity relationships of these latter photodimers is somewhat constrained by the method of their synthesis – the crystal packing of styryl-pyranone subunits dictates the nature of the resultant dimeric product. We are currently exploring how the judicious choice of functionalization of the benzenoid ring of 4-methoxy-6-styryl-pyran-2-ones can be used to direct crystal packing and to facilitate the preparation of new leads for the treatment of neglected diseases.
5. Experimental
5.1. Chemistry: general methods
Mass spectra were recorded on either a VG-7070 or a a Bruker micrOTOF Q II mass spectrometer. Infrared spectra were run as dry films on sodium chloride or ATR crystal and acquired with a Perkin Elmer Spectrum One Fourier Transform infrared spectrometer with a Universal ATR Sampling Accessory. Melting points were obtained on an Electrothermal melting point apparatus and are uncorrected. 1H NMR (300.13 or 400.13 MHz) and 13C NMR (75.47 or 100.62 MHz) spectra were run on a Bruker Avance 300 MHz or a Bruker DRX 400 MHz spectrometer. Chemical shifts are expressed in parts per million (ppm) relative to TMS in 1H NMR and to deuterated solvent in 13C NMR (CDCl3: 13C 77.0 ppm). Complete assignment of 1H and 13C NMR resonances was based on interpretation of standard 2D NMR data. Pressurized (flash) column chromatography was performed on Kieselgel 60 0.063 - 0.200 mesh (Merck) silica gel. Analytical thin layer chromatography (TLC) was carried out on 0.2 mm thick plates of Kieselgel F254 (Merck). Reactions were heated by immersion in oil or by use of DrySyn™ MULTI reaction block kit while the temperature was taken from a thermometer touching the bottom of the pyrex bath. Microanalyses were carried out by the Campbell Laboratory, University of Otago, Dunedin, New Zealand. 4-Methoxy-6-methyl-2-pyrone (11) was prepared by a literature method.19 The 1H NMR data observed for pyrones 3 5, 4 26, 12 4, 13 4, 15 24, 18 20, dihydro styryl pyrones 1 26 and 2 5, and dimers 5, 11,12 7 14, 30 22, and 31 22 agreed with those previously reported in the literature.
5.2. General procedure for the preparation of 6-substituted 4-methoxy-2H-pyran-2-ones
To a suspension of magnesium methoxide, freshly prepared by gently heating Mg (104 mg) in anhydrous MeOH (10 mL), were added 4-methoxy-6-methyl-2-pyrone (11)19 (200 mg, 1.4 mmol) and aldehyde (1.7 mmol) under nitrogen.20 The reaction mixture was heated at reflux for 6 hrs, allowed to cool and dried in vacuo. The solid was then suspended in acetic acid (3.3 M, 20 mL) and extracted with dichloromethane (4 × 50 mL), and the combined organic layers washed with water (2 × 50 mL) and dried in vacuo. Purification of the resultant pyrone was achieved either by trituration with diethylether followed by recrystallization from MeOH or by silica gel column chromatography eluting with 0-1% MeOH in dichloromethane.
5.2.1. 6-[2-(3,4-Dimethoxyphenyl)ethenyl]-4-methoxy-2H-pyran-2-one (3)
From pyrone (201 mg, 1.4 mmol) and 3,4-dimethoxybenzaldehyde (285 mg, 1.7 mmol). Repeated recrystallization from MeOH gave the product as a yellow crystalline solid (115 mg, 28%). Mp 162-163 °C (lit.22 160-162 °C); Rf = 0.42 (3% MeOH:CH2Cl2); IR (ATR) 1700, 1550, 1408, 1252, 1153 cm−1; 1H NMR (CDCl3, 300 MHz) δ 7.42 (1H, d, J = 15.8 Hz, H-8), 7.05 (1H, dd, J = 8.3, 1.9 Hz, H-14), 7.00 (1H, d, J = 1.9 Hz, H-10), 6.84 (1H, d, J = 8.3 Hz, H-13), 6.43 (1H, d, J = 15.8 Hz, H-7), 5.88 (1H, d, J = 2.2 Hz, H-5), 5.44 (1H, d, J = 2.2 Hz, H-3), 3.90 (3H, s, OMe-11), 3.88 (3H, s, OMe-12), 3.79 (3H, s, OMe-4);13C NMR (CDCl3, 75 MHz) δ 171.2 (C-4), 164.1 (C-2), 158.9 (C-6), 150.4 (C-12), 149.2 (C-11), 135.6 (C-8), 128.2 (C-9), 121.6 (C-14), 116.5 (C-7), 111.2 (C-13), 109.3 (C-10), 100.5 (C-5), 88.4 (C-3), 55.9 (2×OMe), 55.8 (OMe); (+)-ESIMS m/z 289 [M+H]+, (+)-HRESIMS m/z 289.1070 (calcd for C16H17O5 289.1071).
Crystallographic data (excluding structure factors) for the structure of compound 3 have been deposited with the Cambridge Crystallographic Data Centre as Supplementary Publication No. CCDC 831983. Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK, (fax: +44-(0)1223-336033 or e-mail: deposit@ccdc.cam.ac.uk).
Crystal data
C16H16O5, M 288.30, T 93(2) K, Triclinic, P-1, a = 8.6990(7) Å, b = 9.3889(8) Å, c = 10.0485(8) Å, α = 89.624(7)°, β= 68.010(6)°, γ = 67.643(6)°, V = 694.90(10) Å3, Z = 2, Dc = 1.378 Mg/m3, μ = 0.103 mm−1, F(000) = 304, crystal size 0.18 × 0.16 × 0.10 mm3, Θmax = 28.17°, index ranges −11<=h<=11, −12<=k<=12, −13<=l<=13, reflections collected = 8711, independent reflections = 3359 [R(int) = 0.0688], data / restraints / parameters 3359 / 0 / 193. All non hydrogen atoms were identified after isotropic refinement of the initial solution. Full matrix least-squares refinement on F2 was carried out to give R indices [I>2σ(I)], R1 = 0.0730, wR2 = 0.2232 and GOF = 1.084.
5.2.2. 4-Methoxy-6-styryl-2H-pyran-2-one (4)
From pyrone (200 mg, 1.4 mmol) and benzaldehyde (180 μL, 1.8 mmol). Recrystallization from MeOH gave the product as a white microcrystalline solid (153.1 mg, 47%). Mp 137-138 °C (lit.23 134-136 °C); Rf = 0.78 (3% MeOH:CH2Cl2); IR (smear) vmax 3076, 1719, 1635, 1607, 1555, 1446, 1406, 1256, 1152, 1001, 954, 831, 748, 685 cm−1; 13C NMR (CDCl3, 100 MHz) δ 171.1 (C-4), 164.0 (C-2), 158.6 (C-6), 135.8 (C-8), 135.2 (C-9), 129.4 (C-12), 128.9 (C-11), 127.4 (C-10), 118.6 (C-7), 101.3 (C-5), 88.9 (C-3), 55.9 (OMe); EIMS m/z 228 [M]+; HREIMS m/z 228.0788 (calcd for C14H12O3 228.0786); Anal.Calcd for C14H12O3: C, 73.67; H, 5.30. Found: C, 73.63; H, 5.44. 1H NMR data agreed with literature.23
5.2.3. 4-Methoxy-6-(3-nitrostyryl)-2H-pyran-2-one (12)
From pyrone (202 mg, 1.4 mmol) and 3-nitrobenzaldehyde (259 mg, 1.7 mmol). Recrystallization from MeOH gave the product as a yellow amorphous solid (115 mg, 29%). Mp 235-236 °C; Rf = 0.76 (3% MeOH:CH2Cl2); IR (ATR) vmax 3078, 1726, 1526, 1347, 1257 cm−1; 1H NMR (CDCl3, 300 MHz) δ 8.37 (1H, t, J = 1.8 Hz, H-10), 8.17 (1H, ddd, J = 8.1, 2.2, 1.0 Hz, H-12), 7.75 (1H, br d, J = 7.7 Hz, H-14), 7.55 (1H, t, J = 8.0 Hz, H-13), 7.52 (1H, d, J = 16.0 Hz, H-8), 6.70 (1H, d, J = 16.0 Hz, H-7), 6.02 (1H, d, J = 2.2 Hz, H-5), 5.53 (1H, d, J = 2.2 Hz, H-3), 3.84 (3H, s, OMe); 13C NMR (CDCl3, 75 MHz) δ 170.7 (C-4), 163.5 (C-2), 157.4 (C-6), 148.8 (C-11), 137.0 (C-9), 133.6 (C-14), 132.9 (C-8), 130.0 (C-13), 123.6 (C-12), 121.5 (C-7), 121.1 (C-10), 102.9 (C-5), 89.7 (C-3), 56.1 (OMe); EIMS m/z 273 [M]+; HREIMS m/z 273.0637 (calcd for C14H11NO5 273.0637). 1H NMR data agreed with literature.4
5.2.4. 4-Methoxy-6-(4-nitrostyryl)-2H-pyran-2-one (13)
From pyrone (199 mg, 1.4 mmol) and 4-nitrobenzaldehyde (258 mg, 1.7 mmol). Recrystallization from MeOH gave the product as an orange solid (21 mg, 5%). Mp 213-214 °C (lit4 211.5-214 °C); Rf = 0.77 (3% MeOH:CH2Cl2); IR (smear) vmax 3081, 1694, 1610, 1591, 1553, 1513, 1448, 1337, 1254, 1154, 955, 810 cm−1; 1H NMR (CDCl3, 400 MHz) δ 8.23 (2H, d, J = 8.8 Hz, H-11), 7.62 (2H, d, J = 8.8 Hz, H-10), 7.51 (1H, d, J = 16.0 Hz, H-8), 6.70 (1H, d, J = 16.0 Hz, H-7), 6.03 (1H, d, J = 2.1 Hz, H-5), 5.54 (1H, d, J = 2.1 Hz, H-3), 3.84 (3H, s, OMe); 13C NMR (CDCl3, 100 MHz) δ 170.6 (C-4), 163.4 (C-2), 157.3 (C-6), 147.8 (C-12), 141.4 (C-9), 132.9 (C-8), 127.9 (C-10), 124.2 (C-11), 122.6 (C-7), 103.3 (C-5), 89.9 (C-3), 55.1 (OMe); EIMS m/z 273 [M]+; HREIMS m/z 273.06323 (calcd for C14H11NO5 273.06372). 1H NMR data agreed with literature.4
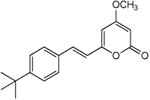
5.2.5. 4-Methoxy-6-(4-tert-butyl-styryl)-2H-pyran-2-one (14)
From pyrone (201 mg, 1.4 mmol) and 4-tert-butylbenzaldehyde (290 μL, 1.7 mmol). Repeated recrystallization from MeOH gave the product as a white microcrystalline solid (104 mg, 25%). Mp 146-147 °C; Rf = 0.77 (3% MeOH:CH2Cl2); IR (ATR) vmax 2960, 1713, 1638, 1552, cm−1; 1H NMR (CDCl3, 400 MHz) δ 7.48 (1H, d, J = 16.0 Hz, H-8), 7.43 (2H, m, H-10), 7.39 (2H, m, H-11), 6.54 (1H, d, J = 16.0 Hz, H-7), 5.92 (1H, d, J = 2.2 Hz, H-5), 5.47 (1H, d, J = 2.2 Hz, H-3), 3.81 (3H, s, OMe), 1.32 (9H, s, H-14); 13C NMR (CDCl3, 100 MHz) δ 171.1 (C-4), 164.1 (C-2), 158.9 (C-6), 152.9 (C-12), 135.7 (C-8), 132.4 (C-9), 127.3 (C-10), 125.9 (C-11), 117.8 (C-7), 100.9 (C-5), 88.6 (C-3), 55.9 (OMe), 34.8 (C-13), 31.2 (C-14); EIMS m/z 284 [M]+; HREIMS m/z 284.1412 (calcd for C18H20O3 284.1412); Anal. Calcd for C18H20O3: C, 76.03; H, 7.09. Found: C, 75.87; H, 7.35.
Crystallographic data (excluding structure factors) for the structure of compound 14 in this paper have been deposited with the Cambridge Crystallographic Data Centre as Supplementary Publication No. CCDC 831984. Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK, (fax: +44-(0)1223-336033 or e-mail: deposit@ccdc.cam.ac.uk).
Crystal data
C18H20O3, M 284.34, T 93(2) K, Triclinic, P21/n, a = 14.6633(5) Å, b = 7.1228(3) Å, c = 15.6152(6) Å, α= 90°, β= 113.468(3)°, γ = 90°, V = 1496.01(10) Å3, Z= 4, Dc = 1.262 Mg/m3, μ = 0.085 mm−1, F(000) = 608, crystal size 0.44 × 0.12 × 0.12 mm3, Θmax = 27.99°, index ranges −19<=h<=19, −9<=k<=9, −20<=l<=20, reflections collected = 18895, independent reflections = 3588 [R(int) = 0.0656], data / restraints / parameters 3588 / 0 / 202. All non hydrogen atoms were identified after isotropic refinement of the initial solution. Full matrix least-squares refinement on F2 was carried out to give R indices [I>2σ(I)], R1 = 0.0465, wR2 = 0.1007 and GOF = 1.013.
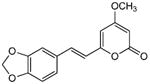
5.2.6. 6-[2-(1,3-Benzodioxo-5-yl)ethenyl]-4-methoxy-2H-pyran-2-one (15)
From pyrone (201 mg, 1.4 mmol) and piperanal (260 mg, 1.7 mmol). Recrystallization from MeOH gave the product as a yellow crystalline solid (155 mg, 40%). Mp 231-232 °C (lit.24 234-235°C); Rf = 0.43 (3% MeOH:CH2Cl2); 13C NMR (CDCl3, 100 MHz) δ 171.1 (C-4), 164.0 (C-2), 158.8 (C-6), 148.9 (C-12), 148.3 (C-11), 135.5 (C-8), 129.7 (C-9), 123.5 (C-14), 116.8 (C-7), 108.6 (C-10), 105.9 (C-13), 101.4 (C-15), 100.7 (C-5), 88.5 (C-3), 55.9 (OMe); EIMS m/z 272 [M]+; HREIMS m/z 272.0686 (calcd for C15H12O5 272.0685); Anal. Calcd for C14H12O3: C, 66.17; H, 4.44. Found: C, 66.12; H, 4.53. IR and 1H NMR data agreed with literature.24
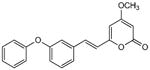
5.2.7. 4-Methoxy-6-(3-phenoxy-styryl)-2H-pyran-2-one (16)
From pyrone (200 mg, 1.4 mmol) and 3-phenoxybenzaldehyde (295 μL, 1.7 mmol). Purification by repeated silica gel column chromatography eluting with 1% MeOH in dichloromethane gave the product as a yellow oil (104 mg, 23%). Rf = 0.58 (2% MeOH:CH2Cl2); IR (ATR)vmax 3078, 1721, 1641, 1607, 1554, 1484, 1233 cm−1; 1H NMR (CDCl3, 400 MHz) δ 7.42 (1H, d, J = 16.0 Hz, H-8), 7.35 (2H, m, H-18), 7.32 (1H, m, H-13), 7.21 (1H, m, H-14), 7.13 (1H, m, H-19), 7.11 (1H, m, H-10), 7.01 (2H, m, H-17), 6.97 (1H, m, H-12), 6.51 (1H, d, J = 16.0 Hz, H-7), 5.92 (1H, d, J = 2.0 Hz, H-5), 5.48 (1H, d, J = 2.0 Hz, H-3), 3.81 (3H, s, OMe); 13C NMR (CDCl3, 100 MHz) δ 171.0 (C-4), 163.8 (C-2), 158.3 (C-6), 157.9 (C-11), 156.7 (C-16), 137.0 (C-9), 135.1 (C-8), 130.2 (C-13), 129.9 (C-18), 123.6 (C-19), 122.5 (C-14), 119.7 (C-12), 119.3 (C-7), 119.1 (C-17), 116.9 (C-10), 101.6 (C-5), 89.0 (C-3), 55.9 (OMe); EIMS m/z 320 [M]+; HREIMS m/z 320.1051 (calcd for C20H16O4 320.1049).
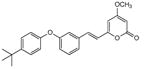
5.2.8. 4-Methoxy-6-(3-(4-tert-butyl-phenoxy)-styryl)-2H-pyran-2-one (17)
From pyrone (202 mg, 1.4 mmol) and 3-(4-tert-butyl-phenoxy)benzaldehyde (434 mg, 1.7 mmol). Repeated recrystallization from MeOH gave the product as a white microcrystalline solid (121 mg, 22%). Mp 138-139 °C; Rf = 0.64 (2% MeOH:CH2Cl2); IR (ATR) vmax 2959, 1717, 1638, 1565, 1505, 1442, 1242 cm−1; 1H NMR (CDCl3, 400 MHz) δ 7.42 (1H, d, J = 16.0 Hz, H-8), 7.36 (2H, m, H-18), 7.31 (1H, m, H-13), 7.20 (1H, m, H-14), 7.10 (1H, br m, H-10), 6.97 (1H, m, H-12), 6.95 (2H, m, H-17), 6.52 (1H, d, J = 16.0 Hz, H-7), 5.92 (1H, d, J = 2.2 Hz, H-5), 5.48 (1H, d, J = 2.2 Hz, H-3), 3.81 (3H, s, OMe), 1.33 (9H, s, H-21); 13C NMR (CDCl3, 100 MHz) δ 171.0 (C-4), 163.9 (C-2), 158.3 (C-6), 158.2 (C-11), 154.1 (C-16), 146.6 (C-19), 136.9 (C-9), 135.2 (C-8), 130.1 (C-13), 126.7 (C-18), 122.0 (C-14), 119.4 (C-12), 119.2 (C-7), 118.7 (C-17), 116.8 (C-10), 101.6 (C-5), 89.0 (C-3), 55.9 (OMe), 34.3 (C-20), 31.5 (C-21); EIMS m/z 376 [M]+; HREIMS m/z 376.1674 (calcd for C24H24O4 376.1675); Anal. Calcd for C24H24O4: C, 76.57; H, 6.43. Found: C, 76.69; H, 6.58.
5.2.9. 4-Methoxy-6-(4-phenyl-1,3-butadienyl)-2H-pyran-2-one (18)
From pyrone (201 mg, 1.4 mmol) and cinnamaldehyde (450 μL, 3.6 mmol). Recrystallization from MeOH gave the product as a yellow solid (23 mg, 6%). Mp 189-190 °C (lit.20 188.5-190 °C); Rf = 0.81 (3% MeOH:CH2Cl2); 13C NMR (CDCl3, 100 MHz) δ 171.0 (C-4), 164.0 (C-2), 158.7 (C-6), 138.3 (C-10), 136.4 (C-11), 136.3 (C-8), 128.8 (C-13), 128.6 (C-14), 127.1 (C-9), 127.0 (C-12), 122.0 (C-7), 100.9 (C-5), 88.7 (C-3), 55.9 (OMe); EIMS m/z 254 [M]+; HREIMS m/z 254.0936 (calcd for C16H14O3 254.0943); Anal. Calcd for C14H12O3: C, 75.57; H, 5.55. Found: C, 75.28; H, 5.68. IR and 1H NMR data agreed with literature.20
5.2.10. 4-Methoxy-6-(4-(4-dimethylaminophenyl)-1,3-butadienyl)-2H-pyran-2-one (19)
From pyrone (200 mg, 1.4 mmol) and 4-dimethylaminocinnamaldehyde (298 mg, 1.7 mmol) and heating with magnesium methoxide in MeOH under nitrogen for 22 hrs. Recrystallization from MeOH gave the product as a red solid (21 mg, 5%). Mp 239-240 °C; Rf = 0.86 (3% MeOH:CH2Cl2); IR (ATR) vmax 1720, 1595, 1541, 1523, 1258 cm−1; 1H NMR (CDCl3, 400 MHz) δ 7.33 (2H, m, H-12), 7.28 (1H, m, H-8), 6.75 (1H, m, H-10), 6.70 (1H, m, H-9), 6.65 (2H, m, H-13), 6.02 (1H, d, J = 15.2 Hz, H-7), 5.79 (1H, d, J = 2.0 Hz, H-5), 5.41 (1H, d, J = 2.0 Hz, H-3), 3.78 (3H, s, OMe), 2.98 (6H, s, H-16); 13C NMR (CDCl3, 100 MHz) δ 171.2 (C-4), 164.3 (C-2), 159.4 (C-6), 150.7 (C-14), 139.1 (C-10), 137.4 (C-8), 128.4 (C-12), 124.6 (C-11), 122.7 (C-9), 119.2 (C-7), 112.1 (C-13), 99.7 (C-5), 87.9 (C-3), 55.8 (OMe), 40.2 (C-16); EIMS m/z 297 [M]+; HREIMS m/z 297.1365 (calcd for C18H19NO3 297.1365).
5.2.11. 4-Methoxy-6-[2-(2-furyl)ethenyl]-2H-pyran-2-one (20)
From pyrone (200 mg, 1.4 mmol) and furfuraldehyde (145 μL, 1.8 mmol). Purification by recrystallization from MeOH gave the product as a yellow solid (66 mg, 21%). Mp 174-175 °C; Rf = 0.46 (3% MeOH:CH2Cl2); IR (ATR) vmax 3091, 2917, 1697, 1634, 1543, 1449, 1145, 943 cm−1; 1H NMR (CDCl3, 400 mHz) δ 7.43 (1H, d, J = 1.5 Hz, H-12), 7.23 (1H, d, J = 15.6 Hz, H-8), 6.49 (1H, m, H-10), 6.47 (1H, m, H-7), 6.44 (1H, m, H-11), 5.89 (1H, d, J = 2.2 Hz, H-5), 5.45 (1H, d, J = 2.2 Hz, H-3), 3.81 (3H, s, OMe); 13C NMR (CDCl3, 100 MHz) δ 171.0 (C-4), 163.9 (C-2), 158.4 (C-6), 151.6 (C-9), 144.0 (C-12), 122.6 (C-8), 116.5 (C-7), 113.3 (C-10), 112.3 (C-11), 101.2 (C-5), 88.7 (C-3), 55.9 (OMe); EIMS m/z 218 [M]+; HREIMS m/z 218.0575 (calcd for C12H10O4 218.0579); Anal. Calcd for C12H10O4: C, 66.05; H, 4.62. Found: C, 66.34; H, 4.85.
5.2.12. 4-Methoxy-6-[2-(1-naphthyl)ethenyl]-2H-pyran-2-one (21)
From pyrone (200 mg, 1.4 mmol) and 1-napthaldehyde (233 μL, 1.7 mmol). Purification by repeated recrystallization from MeOH gave the product as a yellow solid (160 mg, 40%). Mp 120-121 °C; Rf = 0.83 (3% MeOH:CH2Cl2); IR (ATR) vmax 3079, 1724, 1636, 1556, 1411 cm−1; 1H NMR (CDCl3, 400 MHz) δ 8.29 (1H, d, J = 15.7 Hz, H-8), 8.23 (1H, d, J = 8.2 Hz, H-16), 7.85 (2H, m, H-12, 13), 7.72 (1H, d, J = 7.2 Hz, H-10), 7.58 (1H, m, H-15), 7.52 (1H, m, H-14), 7.47 (1H, m, H-11), 6.66 (1H, d, J = 15.7 Hz, H-7), 5.99 (1H, d, J = 2.2 Hz, H-5), 5.53 (1H, d, J = 2.2 Hz, H-3), 3.85 (3H, s, OMe); 13C NMR (CDCl3, 100 MHz) δ 171.0 (C-4), 164.0 (C-2), 158.6 (C-6), 133.7 (C-12a), 132.7 (C-9), 132.6 (C-8), 131.4 (C-16a), 129.8 (C-12), 128.6 (C-13), 126.7 (C-15), 126.2 (C-14), 125.4 (C-11), 124.1 (C-10), 123.6 (C-16), 121.3 (C-7), 101.6 (C-5), 89.0 (C-3), 55.9 (OMe); EIMS m/z 278 [M]+; HREIMS m/z 278.0942 (calcd for C18H14O3 278.0943).
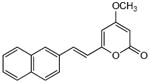
5.2.13. 4-Methoxy-6-[2-(2-naphthyl)ethenyl]-2H-pyran-2-one (22)
From pyrone (201 mg, 1.4 mmol) and 2-napthaldehyde (269 mg, 1.7 mmol). Purification by repeated recrystallization from MeOH gave the product as a yellow crystalline solid (138 mg, 35%). Mp 201-202 °C; Rf = 0.56 (3% MeOH:CH2Cl2); IR (ATR) vmax 3081, 1709, 1638, 1549, 1448, 1408, 1251 cm−1; 1H NMR (CDCl3, 400 MHz) δ 7.87 (1H, d, J = 1.0 Hz, H-16), 7.82 (3H, m, H-11, H-12, H-15), 7.64 (1H, d, J = 15.9 Hz, H-8), 7.63 (1H, m, H-10), 7.48 (2H, m, H-14 and H-13), 6.68 (1H, d, J = 15.9 Hz, H-7), 5.96 (1H, d, J = 2.2 Hz, H-5), 5.49 (1H, d, J = 2.2 Hz, H-3), 3.81 (3H, s, OMe); 13C NMR (CDCl3, 100 MHz) δ 171.0 (C-4), 164.0 (C-2), 158.7 (C-6), 135.8 (C-8), 133.8 (C-11a), 133.4 (C-15a), 132.7 (C-9), 128.9 (C-16), 128.6 (C-11), 128.4 (C-15), 127.7 (C-12), 126.9 (C-13), 126.6 (C-14), 123.2 (C-10), 118.8 (C-7), 101.3 (C-5), 88.8 (C-3), 55.9 (OMe); EIMS m/z 278 [M]+; HREIMS m/z 278.0945 (calcd for C18H14O3 278.0943).
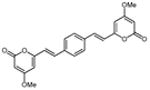
5.2.14. 6,6′-(1E,1′E)-2,2′-(1,4-phenylene)bis(ethene-2,1-diyl)bis(4-methoxy-2H-pyran-2-one) (23)
From pyrone (200 mg, 1.4 mmol) and terephthaldicarboxaldehyde (95 mg, 0.7 mmol). Purification by recrystallization from MeOH gave the product as a yellow solid (72.4 mg, 27%). Mp 330 °C (decomp.); Rf = 0.15 (3% MeOH:CH2Cl2); IR (ATR) vmax 3078, 1726, 1640, 1556, 1409 cm−1; 1H NMR (CDCl3, 400 MHz) δ 7.50 (4H, s, H-10), 7.48 (2H, d, J = 16.0 Hz, H-8), 6.60 (2H, d, J = 16.0 Hz, H-7), 5.96 (2H, d, J = 2.0 Hz, H-5), 5.50 (2H, d, J = 2.0 Hz, H-3), 3.83 (6H, s, OMe); 13C NMR (CDCl3, 100 MHz) δ 171.0 (C-4), 163.9 (C-2), 158.4 (C-6), 136.3 (C-9), 134.8 (C-8), 128.0 (C-10), 119.3 (C-7), 101.8 (C-5), 89.1 (C-3), 56.0 (OMe); FABMS m/z 379 [M+H]+; HRFABMS m/z 379.1188 (calcd for C22H19O6 379.1182).
5.3. General procedure for catalytic hydrogenation of pyrones
Pyrone was dissolved in MeOH/CHCl3 (5 mL, 1:1) and stirred under a hydrogen atmosphere (balloon) for 2 h in the presence of 10% Pd/C. The suspension was filtered and the solvent evaporated in vacuo. Purification by silica gel column chromatography eluting with MeOH (0-1%) in dichloromethane gave the product.
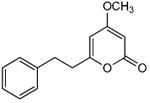
5.3.1. 7,8-Dihydro-4-methoxy-6-styryl-2H-pyran-2-one (1)
From pyrone 4 (24.9 mg, 0.11 mmol) and Pd/C (10%, 6.8 mg) to give product 1 as a white solid (16.9 mg, 67%). Mp 99-100 °C (lit.26 96 - 97 °C); Rf = 0.59 (3% MeOH:CH2Cl2); EIMS m/z 230 [M]+; HREIMS m/z 230.0939 (calcd for C14H14O3 230.0943); Anal. Calcd for C14H14O3: C, 73.03; H, 6.13. Found: C, 73.30; H, 6.11. 1H and 13C NMR data agreed with the literature.26
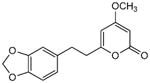
5.3.2. 6-[2-(1,3-Benzodioxo-5-yl)ethyl]-4-methoxy-2H-pyran-2-one (2)
From pyrone 15 (17.9 mg, 0.066 mmol) and Pd/C (10%, 4.8 mg) to give product 2 as white crystals (13.0 mg, 72%). Mp 143-144 °C (lit.5 138-140 °C); Rf = 0.39 (3% MeOH:CH2Cl2); IR (smear) vmax 3108, 2927, 1705, 1645, 1567, 1239 cm−1; 1H NMR (CDCl3, 400 MHz) δ 6.71 (1H, d, J = 7.8 Hz, H-13), 6.64 (1H, d, J = 1.6 Hz, H-10), 6.60 (1H, dd, J = 7.8, 1.6 Hz, H-14), 5.91 (2H, s, H2-15), 5.70 (1H, d, J = 2.3 Hz, H-5), 5.40 (1H, d, J = 2.1 Hz, H-3), 3.77 (3H, s, OMe), 2.88 (2H, t, J = 7.6 Hz, H2-8), 2.68 (2H, t, J = 7.6 Hz, H2-7); 13C NMR (CDCl3, 100 MHz) δ 171.1 (C-4), 164.9 (C-2), 164.2 (C-6), 147.7 (C-11), 146.1 (C-12), 133.6 (C-9), 121.2 (C-14), 108.7 (C-10), 108.3 (C-13), 100.9 (C-15), 100.3 (C-5), 87.7 (C-3), 55.8 (OMe), 35.7 (C-7), 32.6 (C-8); EIMS m/z 274 [M]+; HREIMS m/z 274.0841 (calcd for C15H14O5 274.0841); Anal. Calcd for C15H14O5: C, 65.69; H, 5.15. Found: C, 65.76; H, 5. 23.
Crystallographic data (excluding structure factors) for the structure of compound 2 have been deposited with the Cambridge Crystallographic Data Centre as Supplementary Publication No. CCDC 831985. Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK, (fax: +44-(0)1223-336033 or e-mail: deposit@ccdc.cam.ac.uk).
Crystal data
C15H14O5, M 274.26, T 98(2) K, Triclinic, P-1, a = 8.0036(2) Å, b = 8.3760(3) Å, c = 10.5599(3) Å, α= 90.8060(10)°, β= 105.8980(10)°, γ = 110.4210(10)°. V = 633.31(3) Å3, Z = 2, Dc = 1.438 Mg/m3, μ = 0.109 mm−1, F(000) = 288, crystal size 0.43 × 0.29 × 0.20 mm3, Θmax = 27.98°, index ranges −10<=h<=10, −11<=k<=10, −13<=l<=13, reflections collected = 15272, independent reflections = 3024 [R(int) = 0.0243], data / restraints / parameters 3024 / 0 / 182. All non hydrogen atoms were identified after isotropic refinement of the initial solution. Full matrix least-squares refinement on F2 was carried out to give R indices [I>2σ(I)], R1 = 0.0342, wR2 = 0.0944 and GOF = 1.033.
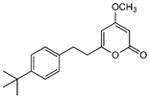
5.3.3. 4-Methoxy-6-[2-(4-tert-butylphenyl)ethyl]-2H-pyran-2-one (24)
From pyrone 14 (19.3 mg, 0.068 mmol) and Pd/C (10%, 5.2 mg) to give product 24 as a white solid (17.4 mg, 90%). Mp 77-79 °C; Rf = 0.42 (3% MeOH:CH2Cl2); IR (ATR) vmax 2959, 1713, 1567, 1451, 1411, 1248 cm−1; 1H NMR (CDCl3, 400 MHz) δ 7.31 (2H, d, J = 8.3 Hz, H-11/13), 7.11 (2H, d, J = 8.3 Hz, H-10/14), 5.75 (1H, d, J = 2.1 Hz, H-5), 5.41 (1H, d, J = 2.1 Hz, H-3), 3.77 (3H, s, OMe), 2.92 (2H, m, H2-8), 2.73 (2H, m, H2-7), 1.30 (9H, s, H-16); 13C NMR (CDCl3, 100 MHz) δ 171.2 (C-4), 165.0 (C-2), 164.6 (C-6), 149.3 (C-12), 136.8 (C-9), 127.9 (C-10/14), 125.5 (C-11/13), 100.1 (C-5), 87.7 (C-3), 55.8 (OMe), 35.4 (C-7), 34.4 (C-15), 32.3 (C-8), 31.3 (C-16); (+)-ESIMS m/z 287 [M+H]+, (+)-HRESIMS m/z 287.1639 (calcd for C18H23O3 287.1642).
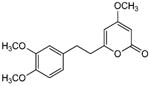
5.3.4. 6-[2-(3,4-Dimethoxyphenyl)ethyl]-4-methoxy-2H-pyran-2-one (25)
From pyrone 3 (20.8 mg, 0.072 mmol) and Pd/C (10%, 3.9 mg) to give 25 as a white solid (20.1 mg, 96%). Mp 71-73 °C; (lit.27 74 – 75 °C); Rf = 0.18 (3% MeOH:CH2Cl2); IR (ATR) vmax 1709, 1651, 1569, 1518, 1453, 1241, 1136 cm−1; 1H NMR (CDCl3, 400 MHz) δ 6.77 (1H, d, J = 8.1 Hz, H-13), 6.69 (1H, dd, J = 8.1, 1.9 Hz, H-14), 6.66 (1H, d, J = 1.9 Hz, H-10), 5.69 (1H, d, J = 2.2 Hz, H-5), 5.40 (1H, d, J = 2.2 Hz, H-3), 3.84 (3H, s, OMe-11*), 3.83 (3H, s, OMe-12*), 3.76 (3H, s, OMe-4), 2.91 (2H, m, H2-8), 2.71 (2H, m, H2-7); 13C NMR (CDCl3, 100 MHz) δ 171.2 (C-4), 165.1 (C-2), 164.3 (C-6), 148.8 (C-11), 147.5 (C-12), 132.4 (C-9), 120.1 (C-14), 111.6 (C-10), 111.3 (C-13), 100.4 (C-5), 87.6 (C-3), 55.8 (OMe), 55.7 (OMe), 35.7 (C-7), 32.4 (C-8); (+)-ESIMS m/z 313 [M+Na]+, (+)-HRESIMS m/z 313.1056 (calcd for C16H18NaO5 313.1046).
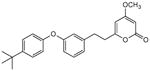
5.3.5. 4-Methoxy-6-[2-(3-(4-tert-butylphenoxy)phenyl)ethyl]-2H-pyran-2-one (26)
From pyrone 17 (21.2 mg, 0.056 mmol) and Pd/C (10% 3.8 mg) to give 26 as a colorless oil (19.6 mg, 92%). Rf = 0.49 (3% MeOHCH2Cl2); IR (smear) vmax 2960, 1721, 1568, 1247 cm−1; 1H NMR (CDCl3, 400 MHz) δ 7.33 (2H, m, H-18/20), 7.22 (1H, t, J = 7.8 Hz, H-13), 6.90 (2H, m, H-17/21), 6.87 (1H, m, H-14), 6.83 (1H, m, H-12), 6.81 (1H, m, H-10), 5.71 (1H, d, J = 2.1 Hz, H-5), 5.40 (1H, d, J = 2.1 Hz, H-3), 3.77 (3H, s, OMe), 2.93 (2H, m, H2-8), 2.72 (2H, m, H2-7), 1.32 (9H, s, H-23);13C NMR (CDCl3, 75 MHz) δ 171.1 (C-4), 164.8 (C-2), 164.2 (C-6), 157.8 (C-11), 154.5 (C-16), 146.2 (C-19), 141.7 (C-9), 129.8 (C-13), 126.5 (C-18,20), 122.9 (C-14), 118.4 (C-17/21), 118.3 (C-10), 116.6 (C-12), 100.3 (C-5), 87.7 (C-3), 55.8 (OMe), 35.2 (C-7), 34.3 (C-22), 32.7 (C-8), 31.5 (C-23); (+)-ESIMS m/z 401 [M+Na]+, (+)-HRESIMS m/z 401.1725 (calcd for C24H26NaO4 401.1723).
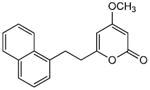
5.3.6. 4-Methoxy-6-[2-(1-naphthyl)ethyl]-2H-pyran-2-one (27)
From pyrone 21 (21.1 mg, 0.076 mmol) to give 27 as a white solid (16.0 mg, 75%). Mp 75-77 °C; Rf = 0.40 (3% MeOH:CH2Cl2); IR (ATR) vmax 3078, 1714, 1647, 1563, 1465, 1257, 1138 cm−1; 1H NMR (CDCl3, 400 MHz) δ 8.02 (1H, d, J = 8.4 Hz, H-16), 7.86 (1H, d, J = 7.8 Hz, H-13), 7.73 (1H, d, J = 8.2 Hz, H-12), 7.50 (2H, m, H-15/H-14), 7.38 (1H, t, J = 7.6 Hz, H-11), 7.30 (1H, d, J = 7.6 Hz, H-10), 5.71 (1H, d, J = 2.2 Hz, H-5), 5.42 (1H, d, J = 2.2 Hz, H-3), 3.76 (3H, s, OMe), 3.42 (2H, m, H2-8), 2.86 (2H, m, H2-7); 13C NMR (CDCl3, 100 MHz) δ 171.1 (C-4), 164.9 (C-2), 164.4 (C-6), 135.9 (C-9), 133.9 (C-12a), 131.5 (C-16a), 128.9 (C-13), 127.3 (C-12), 126.2 (C-10/15), 125.6 (C-11*), 125.5 (C-14*), 123.2 (C-16), 100.3 (C-5), 87.8 (C-3), 55.8 (OMe), 34.8 (C-7), 30.2 (C-8); (+)-ESIMS m/z 281 [M+H]+, (+)-HRESIMS m/z 281.1174 (calcd for C18H17O3 281.1172).
5.3.7. 4-Methoxy-6-[2-(2-naphthyl)ethyl]-2H-pyran-2-one (28)
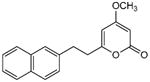
From pyrone 22 (19.4 mg, 0.070 mmol) to give 28 as a white solid (16.3 mg, 84%). Mp 72-74 °C; Rf = 0.42 (3% MeOH:CH2Cl2); IR (ATR) vmax 3082, 1722, 1649, 1564, 1412, 1250, 1142 cm−1; 1H NMR (CDCl3, 400 MHz) δ 7.77 (3H, m, H-11,12,15), 7.61 (1H, br s, H-16), 7.43 (2H, m, H-13,14), 7.29 (1H, dd, J = 8.4, 1.6 Hz, H-10), 5.71 (1H, d, J = 2.1 Hz, H-5), 5.40 (1H, d, J = 2.1 Hz, H-3), 3.74 (3H, s, OMe), 3.13 (2H, m, H2-8), 2.83 (2H, m, H2-7); 13C NMR (CDCl3, 100 MHz) δ 171.2 (C-4), 165.1 (C-2), 164.2 (C-6), 137.2 (C-9), 133.5 (C-15a), 132.1 (C-11a), 128.2 (C-11*), 127.6 (C-12*), 127.5 (C-15*), 126.7 (C-10), 126.6 (C-16), 126.1 (C-13*), 125.5 (C-14*), 100.4 (C-5), 87.7 (C-3), 55.8 (OMe), 35.3 (C-7), 32.9 (C-8); (+)-ESIMS m/z 281 [M+H]+, (+)-HRESIMS m/z 281.1172 (calcd for C18H17O3 281.1172).
5.3.8. 6,6′-(2,2′-(1,4-Phenylene)bis(ethane-2,1-diyl))bis(4-methoxy-2H-pyran-2-one) (29)
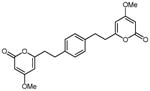
From pyrone 23 (19.7 mg, 0.052 mmol) and Pd/C (10%, 4.9 mg) to give 29 as a white solid (9.8 mg, 49%). Mp 196-198 °C; Rf = 0.06 (3% MeOH:CH2Cl2); IR (ATR) vmax 1710, 1651, 1566, 1450, 1413, 1250, 1143 cm−1; 1H NMR (CDCl3, 400 MHz) δ 7.08 (4H, s, H-10), 5.70 (2H, d, J = 2.2 Hz, H-5), 5.39 (2H, d, J = 2.2 Hz, H-3), 3.77 (6H, s, OMe), 2.92 (4H, m, H2-8), 2.70 (4H, m, H2-7); 13C NMR (CDCl3, 100 MHz) δ 171.1 (C-4), 164.8 (C-2), 164.3 (C-6), 138.0 (C-9), 128.5 (C-10), 100.2 (C-5), 87.7 (C-3), 55.8 (OMe), 35.4 (C-7), 32.4 (C-8); (+)-ESIMS m/z 405 [M + Na]+, (+)-HRESIMS m/z 405.1316 (calcd for C22H22NaO6 405.1309).
5.4. General procedure for photodimerization of 6-styryl-pyran-2-ones
6-Styryl-pyran-2-one was suspended, with stirring, in water and exposed to a sun lamp (300W, 15 cm from vial) for three 5 hr periods at which time the resulting crude product was purified by repeated column chromatography eluting with 0-5% MeOH in dichloromethane.
5.4.1. 6,6′-(3,4-Diphenylcyclobutane-1,2-diyl)bis(4-methoxy-2H-pyran-2-one) (5) and Aniba-dimer-A (7)
From pyrone 4 (43.0 mg, 0.19 mmol) to give the head-to-tail dimer 5 11,12 (8.0 mg, 19%) and aniba-dimer A14-16 7 (4.2 mg, 10%), both as amorphous colorless solids.
Data for 5
Mp 82-84 °C (lit.11 103-105 °C; lit.12 170-172 °C); Rf = 0.24 (3% MeOH:CH2Cl2); IR (ATR) vmax 2918, 1714, 1646, 1563, 1454, 1409, 1239 cm−1; 1H NMR (CDCl3, 400 MHz) δ 7.12 (4H, m, H-11,13), 7.06 (2H, m, H-12), 6.95 (4H, m, H-10,14), 5.99 (2H, d, J = 2.3 Hz, H-5), 5.36 (2H, d, J = 2.3 Hz, H-3), 4.50 (2H, m, H-8), 4.11 (2H, m, H-7), 3.75 (6H, s, OMe); 13C NMR (CDCl3, 100 MHz) δ 171.0 (C-4), 164.3 (C-2), 162.4 (C-6), 137.9 (C-9), 128.2 (C-11,13), 127.7 (C-10,14), 126.7 (C-12), 101.5 (C-5), 88.0 (C-3), 55.9 (OMe), 44.9 (C-8), 44.0 (C-7); (+)-ESIMS m/z 457 [M+H]+, (+)-HRESIMS m/z 457.1650 (calcd for C28H25O6 457.1646); (+)-HRESIMSMS (parent m/z 457) fragment m/z 277.0701 (calcd for C14H13O6 277.0707), 229.0853 (calcd for C14H13O3 229.0859).
Data for 7
Mp 178-180 °C (lit.15 178-179 °C; lit.14 185-188 oC); Rf = 0.20 (3% MeOH:CH2Cl2); IR (ATR) vmax 3078, 1704, 1647, 1623, 1567, 1244 cm−1; 1H NMR (CDCl3, 400 MHz) δ 7.41 (2H, m, H-10′,14′), 7.29 (8H, m, H-10,11,12,13,14,11′,12′,13′), 6.94 (1H, d, J = 15.9 Hz, H-8′), 6.59 (1H, d, J = 15.9 Hz, H-7), 5.91 (1H, d, J = 2.2 Hz, H-5), 5.34 (1H, d, J = 2.2 Hz, H-3), 5.28 (1H, s, H-3′), 4.35 (1H, t, J = 10.3 Hz, H-8), 4.16 (1H, d, J = 11.1 Hz, H-7), 3.71 (3H, s, OMe-4), 3.59 (1H, d, J = 10.3 Hz, H-5′), 3.27 (3H, s, OMe-4′); 13C NMR (CDCl3, 100 MHz) δ 170.5 (C-4), 169.9 (C-4′), 164.6 (C-2*), 163.9 (C-2′*), 158.6 (C-6), 135.9 (C-9′), 135.6 (C-9), 131.5 (C-8′), 128.7, 128.5, 128.3, 127.8 (C-11,12,13,11′,12′,13′), 127.5 (C-10/14), 126.9 (C-10′/14′), 124.4 (C-7), 102.7 (C-5), 91.8 (C-3′), 88.7 (C-3), 79.4 (C-6′), 55.9 (OMe-4), 55.4 (OMe-4′), 54.5 (C-7), 45.7 (C-5′), 39.2 (C-8); (+)-ESIMS m/z 479 [M+Na]+, (+)-HRESIMS m/z 479.1453 (calcd for C28H24NaO6 479.1465).
5.4.2. 6,6′-(2,4-Bis(3,4-dimethoxyphenyl)cyclobutane-1,3-diyl)bis(4-methoxy-2H-pyran-2-one) (30) and 11,12,11′,12′-Tetramethoxy-aniba-dimer-A (31)
From pyrone 3 (19.9 mg, 0.069 mmol) to give the head-to-tail dimer 30 as an amorphous colorless solid (4.1 mg, 14%) and asymmetric dimer 31 as an amorphous yellow solid (6.9 mg, 35%).
Data for 30
Mp 86-88 °C (lit.22 112-114 °C); Rf = 0.09 (3% MeOH:CH2Cl2); IR (ATR) vmax 2918, 2849, 1706, 1641, 1563, 1515, 1454, 1409, 1249, 1142, 1023 cm−1; 13C NMR (CDCl3, 100 MHz)δ 170.6 (C-4), 164.0 (C-2), 162.9 (C-6), 148.9 (C-11), 148.2 (C-12), 129.8 (C-9), 119.4 (C-14), 111.04 (C-10*), 110.96 (C-13*), 101.3 (C-5), 87.8 (C-3), 56.0 (OMe-11), 55.82 (OMe-12), 55.76 (OMe-4), 45.5 (C-7), 43.5 (C-8); (+)-ESIMS m/z 577 [M+H]+, (+)-HRESIMS m/z 577.2073 (calcd for C32H33O10 577.2068); (+)-HRESIMSMS (parent m/z 577) fragment m/z 289.1055 (calcd for C16H17O5 289.1071). 1H and 13C NMR chemical shifts agreed with the literature, however the previously reported assignments of C-2 and C-4 should be reversed.22
Data for 31
Mp 99-100 °C (lit.22 195-197 °C); Rf = 0.05 (3% MeOH:CH2Cl2); (+)-ESIMS m/z 599 [M+Na]+, (+)-HRESIMS m/z 599.1881 (calcd for C32H32NaO10 599.1888). 1H and 13C NMR data were in agreement with literature values, however the previously reported assignments of C-2 and C-4 should be reversed.22
5.4.3. 6,6′-(3,4-Di(benzo[d][1,3]dioxo-5-yl)cyclobutane-1,2-diyl)bis(4-methoxy-2H-pyran-2-one) (32)
From pyrone 15 (19.3 mg, 0.071 mmol) to give 32 as an amorphous colorless solid (2.0 mg, 10%) and unreacted starting material 15 (5.6 mg, 29%). Mp 101-103 °C; Rf = 0.14 (3% MeOH:CH2Cl2); IR (ATR) vmax 2905, 1706, 1646, 1561, 1490, 1445, 1408, 1237, 1034 cm−1; 1H NMR (CDCl3, 400 MHz) δ 6.62 (2H, d, J = 8.0 Hz, H-13), 6.46 (2H, dd, J = 8.0, 1.7 Hz, H-14), 6.43 (2H, d, J = 1.7 Hz, H-10), 5.96 (2H, d, J = 2.1 Hz, H-5), 5.86 (4H, br s, H2-15), 5.35 (2H, d, J = 2.1 Hz, H-3), 4.35 (2H, m, H-8), 3.96 (2H, m, H-7), 3.75 (6H, s, OMe-4); 13C NMR (CDCl3, 100 MHz) δ 171.0 (C-4), 164.2 (C-2), 162.2 (C-6), 147.7 (C-11), 146.3 (C-12), 131.8 (C-9), 120.8 (C-14), 108.1 (C-10), 101.4 (C-5), 100.9 (C-15), 88.1 (C-3), 55.9 (OMe-4), 44.8 (C-8), 44.4 (C-7); (+)-ESIMS m/z 545 [M+H]+, (+)-HRESIMS m/z 545.1435 (calcd for C30H25O10 557.1959), (+)-HRESIMSMS (parent m/z 545) fragment m/z 277.0690 (calcd for C14H13O6 277.0707).
5.4.4. 12,12′-Di-tert-butyl-aniba-dimer-A (33)
From pyrone 14 (20.6 mg, 0.036 mmol) to give 33 as an amorphous colorless solid (9.5 mg, 46%). Mp 130-131 °C; Rf = 0.26 (3% MeOH:CH2Cl2); IR (ATR) vmax 2960, 1713, 1626, 1567, 1455, 1390, 1244 cm−1; 1H NMR (CDCl3, 400 MHz) δ 7.35 (4H, s, H-10′,11′,13′,14′), 7.34 (2H, d, J = 9.9 Hz, H-11,13), 7.16 (2H, d, J = 8.3 Hz, H-10,14), 6.91 (1H, d, J = 15.9 Hz, H-8′), 6.55 (1H, d, J = 15.9 Hz, H-7), 5.89 (1H, d, J = 2.1 Hz, H-5), 5.33 (1H, d, J = 2.1 Hz, H-3), 5.27 (1H, s, H-3′), 4.33 (1H, t, J = 10.4 Hz, H-8), 4.14 (1H, d, J = 10.4 Hz, H-7), 3.70 (3H, s, OMe-4), 3.53 (1H, d, J = 10.4 Hz, H-5′), 3.24 (3H, s, OMe-4′), 1.30 (9H, s), 1.29 (9H, s, H-16,16′); 13C NMR (CDCl3, 100 MHz) δ 170.5 (C-4), 170.1 (C-4′), 164.8, 164.0 (C-2,2′), 158.8 (C-6), 151.4 (C-12′), 150.9 (C-12), 133.1 (C-9′), 132.6 (C-9), 131.1 (C-8′), 127.2 (C-10,14), 126.6 (C-10′,14′), 125.7 (C-11′,13′), 125.3 (C-11,13), 123.6 (C-7′), 102.6 (C-5), 91.7 (C-3′), 88.7 (C-3), 79.5 (C-6′), 55.8 (OMe-4), 55.2 (OMe-4′), 54.5 (C-7), 45.8 (C-5′), 38.8 (C-8), 34.6, 34.5 (C-15,15′), 31.3, 31.2 (C-16,16′); (+)-ESIMS m/z 591 [M + Na]+, (+)-HRESIMS m/z 591.2711 (calcd for C36H40NaO6 591.2717).
5.4.5. 6,6′-(2,4-Di(naphthalen-2-yl)cyclobutane-1,3-diyl)bis(4-methoxy-2H-pyran-2-one) (34)
From pyrone 22 (21.3 mg, 0.077 mmol) to give 34 as an amorphous colorless solid (4.1 mg, 19%). Mp 130-131 °C; Rf = 0.20 (3% MeOH:CH2Cl2); IR (ATR) vmax 3055, 2946, 1714, 1644, 1564, 1454, 1410, 1246, 1146 cm−1; 1H NMR (CDCl3, 400 MHz) δ 7.80 (8H, m, ArH), 7.45 (6H, m, ArH), 5.77 (2H, d, J = 2.2 Hz, H-5), 5.13 (2H, d, J = 2.2 Hz, H-3), 4.67 (2H, m, H-8), 4.48 (2H, m, H-7), 3.60 (6H, s, OMe-4); 13C NMR (CDCl3, 100 MHz) δ 170.4 (C-4), 163.8 (C-2), 162.6 (C-6), 134.9 (C-9), 133.3, 132.5 (C-11a,15a), 128.3, 127.9, 127.6, 126.2, 126.1, 125.9, 125.8 (C-10,11,12,13,14,15,16), 101.5 (C-5), 87.9 (C-3), 55.6 (OMe-4), 45.3 (C-7), 44.0 (C-8); (+)-ESIMS m/z 557 [M+H]+, (+)-HRESIMS m/z 557.1963 (calcd for C36H29O6 557.1959); (+)-HRESIMSMS (parent m/z 557) fragment m/z 279.1005 (calcd for C18H15O3 279.1021).
5.4.6. 11,11′ Di (4-tert-butyl-phenoxy) aniba-dimer-A (35)
From pyrone 17 (19.9 mg, 0.026 mmol) to give 35 as an amorphous colorless solid (9.7 mg, 49%). Mp 103-104 °C; Rf = 0.36 (3% MeOH:CH2Cl2); IR (ATR) vmax 2960, 1713, 1626, 1568, 1507, 1444, 1240 cm−1; 1H NMR (CDCl3, 400 MHz) δ 7.34 (4H, m, H-18,20,18′,20′), 7.26 (2H, m, H-13,13′), 7.17 (1H, d, J = 7.9 Hz, H-14′), 6.98 (1H, br s, H-10′), 6.90 (8H, m, H-10,12,14,17,21, 12′,17′,21′), 6.85 (1H, m, H-8′), 6.53 (1H, d, J = 15.9 Hz, H-7), 5.89 (1H, d, J = 2.1 Hz, H-5), 5.35 (1H, d, J = 2.1 Hz, H-3), 5.26 (1H, s, H-3′), 4.28 (1H, t, J = 10.2 Hz, H-8), 4.09 (1H, d, J = 10.2 Hz, H-7), 3.72 (3H, s, OMe-4), 3.55 (1H, d, J = 10.2 Hz, H-5′), 3.34 (3H, s, OMe-4′), 1.33 (9H, s, H-23,23′), 1.32 (9H, s, H-23,23′); 13C NMR (CDCl3, 100 MHz) δ 170.4 (C-4), 169.7 (C-4′), 164.3, 163.7 (C-2,2′), 158.4 (C-6), 158.0, 157.8 (C-11,11′), 154.3, 154.3 (C-16,16′), 146.5, 146.3 (C-19,19′), 137.6 (C-9,9′), 131.2 (C-8′), 130.0, 129.6 (C-13,13′), 126.65, 126.60 (C-18,20,18′,20′), 124.8 (C-7′), 121.8 (C-14), 120.8 (C-14′), 118.6, 118.4, 118.2, 117.9, 117.8 (C-10,12,17,21,12′,17′,21′), 117.3 (C-10′), 102.6 (C-5), 91.8 (C-3′), 88.8 (C-3), 79.2 (C-6′), 55.9 (OMe-4), 55.5 (OMe-4′), 54.4 (C-7), 45.6 (C-5′), 38.9 (C-8), 34.3 (C-22,22′), 31.5 (C-23,23′); (+)-ESIMS m/z 753 [M + H]+, (+)-HRESIMS m/z 753.3402 (calcd for C48H49O8 753.3422).
5.5. Biological assays
5.2.1. Anti-tuberculosis Activity
Procedures for the determination of MIC against M. tuberculosis H37Rv in 7H9 broth have been reported elsewhere.28 In the present study, the protocol was modified slightly by utilizing 1:1000 diluted cultures rather than 1:100 and by growth of M tuberculosis H37Rv in GAST medium29 which does not contain bovine serum albumin. Positive control was isoniazid which gave MIC 0.22 μM (0.03μg/mL).
5.2.2. Anti-protozoal Activity
The in vitro activities against the protozoan parasites T. b. rhodesiense, T. cruzi, L. donovani, and P. falciparum and cytotoxicity assessment against L6 cells were determined as reported elsewhere.30 The following strains, parasite forms and positive controls were used: T.b. rhodesiense, STIB900, trypomastigote forms, melarsoprol, IC50 of 0.01 μM (4 ng/mL); T. cruzi, Tulahuen C2C4, amastigote forms in L6 rat myoblasts, benznidazole, IC50 of 1.4 μM (0.352 μg/mL); L.donovani, MHOM/ET/67/L82, axenic amastigote forms, miltefosine, IC50 of 0.5 μM (0.213 μg/mL); P. falciparum, K1 (chloroquine and pyrimethamine resistant), erythrocytic stages, chloroquine, IC50 of 0.20 μM (0.065 μg/mL) and L6 cells, rat skeletal myoblasts, podophyllotoxin, IC50 of 0.01 μM (0.004 μg/mL).
Supplementary Material
Acknowledgments
We thank the University of Auckland and Auckland Medical Research Foundation for funding. This research was supported in part by the Intramural Research Program of the NIH, NIAID. We also thank M. Cal, S. Sax, and C. Stalder (Swiss TPH) for assistance with parasitic assays, Dr M. Schmitz for assistance with NMR data acquisition, and Ms. R Imatdieva for MS data. STM wishes to acknowledge the Tertiary Education Commission of New Zealand for a scholarship.
Footnotes
Supplementary data: Supplementary data associated with this article can be found, in the online version, at doi:
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.McGlacken GP, Fairlamb IJS. Nat Prod Rep. 2005;22:369–385. doi: 10.1039/b416651p. [DOI] [PubMed] [Google Scholar]
- 2.Beckert C, Horn C, Schnitzler JP, Lehning A, Heller W, Veit M. Phytochemistry. 1997;44:275–283. [Google Scholar]
- 3.Olsen LR, Grillo MP, Skonberg C. Chem Res Toxicol. 2011;24:992–1002. doi: 10.1021/tx100412m. [DOI] [PubMed] [Google Scholar]
- 4.Fujita T, Nishimura H, Kaburagi K, Mizutani J. Phytochemistry. 1994;36:23–27. [Google Scholar]
- 5.Pizzolatti MG, Luciano C, Monache FD. Phytochemistry. 2000;55:819–822. doi: 10.1016/s0031-9422(00)00301-0. [DOI] [PubMed] [Google Scholar]
- 6.Duarte FS, Duzzioni M, Mendes BG, Pizzolatti MG, Monteiro De Lima TC. Pharm Biochem Behav. 2007;86:150–161. doi: 10.1016/j.pbb.2006.12.019. [DOI] [PubMed] [Google Scholar]
- 7.Meotti FC, Ardenghi JV, Pretto JB, Souza MM, Moura JA, Junior AC, Soldi C, Pizzolatti MG, Santos ARS. J Pharm Pharmacol. 2006;58:107–112. doi: 10.1211/jpp.58.1.0013. [DOI] [PubMed] [Google Scholar]
- 8.Flores N, Cabrera G, Jimenez IA, Pinero J, Gimennez A, Bourdy G, Cortes-Selva F, Bazzocchi IL. Planta Med. 2007;73:206–211. doi: 10.1055/s-2007-967123. [DOI] [PubMed] [Google Scholar]
- 9.Pizzolatti MG, Mendes BG, Cunha A, Soldi C, Koga AH, Eger I, Grisard EC, Steindel M. Braz J Pharmacognosy. 2008;18:177–182. [Google Scholar]
- 10.Mata R, Morales I, Pérez O, Rivero-Cruz I, Acevedo L, Enriquez-Mendoza I, Bye R, Franzblau S, Timmermann B. J Nat Prod. 2004;67:1961–1968. doi: 10.1021/np0401260. [DOI] [PubMed] [Google Scholar]
- 11.Gottlieb OR, Veloso DP, da Silva Pereira MO. Rev Latinamer Quin. 1975;6:188–190. [Google Scholar]
- 12.Kaloga M, Christiansen I. Z Naturforsch. 1981;36b:505–507. [Google Scholar]
- 13.Mandarino DG, Yoshida M, Gottlieb OR. J Braz Chem Soc. 1990;1:53–54. [Google Scholar]
- 14.Kuroyanagi M, Yamamoto Y, Fukushima S, Ueno A, Noro T, Miyase T. Chem Pharm Bull. 1982;30:1602–1608. [Google Scholar]
- 15.Adrade da Mata Rezende CM, von Bulow MV, Gottlieb OR, Lamego Vieira Pinho S, Da Rocha AI. Phytochemistry. 1971;10:3167–3172. [Google Scholar]
- 16.Mascarenhas YP, Gottlieb OR. Phytochemistry. 1977;16:301–302. [Google Scholar]
- 17.Sagawa T, Takaishi Y, Fujimoto Y, Duque C, Osorio C, Ramos F, Garzon C, Sato M, Okamoto M, Oshikawa T, Ahmed SU. J Nat Prod. 2005;68:502–505. doi: 10.1021/np040187y. [DOI] [PubMed] [Google Scholar]
- 18.Giddens AC, Nielsen L, Boshoff HI, Tasdemir D, Perozzo R, Kaiser M, Wang F, Sacchettini JC, Copp BR. Tetrahedron. 2008;64:1242–1249. [Google Scholar]
- 19.Hansen CA, Frost JW. J Am Chem Soc. 2002;124:5926–5927. doi: 10.1021/ja0176346. [DOI] [PubMed] [Google Scholar]
- 20.Israili ZH, Smissman EE. J Org Chem. 1976;41:4070–4074. doi: 10.1021/jo00888a004. [DOI] [PubMed] [Google Scholar]
- 21.Singh SB, Jayasuriya H, Dewey R, Polishook JD, Dombrowski AW, Zink DL, Guan Z, Collado J, Platas G, Pelaez F, Felock PJ, Hazuda DJ. J Ind Microbiol Biotechnol. 2003;30:721–731. doi: 10.1007/s10295-003-0101-x. [DOI] [PubMed] [Google Scholar]
- 22.Rossi MH, Yoshida M, Maia JGS. Phytochemistry. 1997;45:1263–1269. [Google Scholar]
- 23.Amaral PA, Gouault N, Le Roch M, Eifler-Lima VL, David M. Tetrahedron Lett. 2008;49:6607–6609. [Google Scholar]
- 24.Adam W, Saha-Moller CR, Veit M, Welke B. Synthesis. 1994:1133–1134. [Google Scholar]
- 25.Schmidt GMJ. J Chem Soc. 1964:2014–2021. [Google Scholar]
- 26.Itokawa H, Morita M, Mihashi S. Phytochemistry. 1981;20:2503–2506. [Google Scholar]
- 27.Bu'Lock JD, Leeming PR, Smith HG. J Chem Soc. 1962:2085–2089. [Google Scholar]
- 28.Domenech P, Reed MB, Barry CE. Infect Immun. 2005;73:3492–3501. doi: 10.1128/IAI.73.6.3492-3501.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Voss JJ, Rutter K, Schroeder BG, Su H, Zhu Y, Barry CE. Proc Natl Acad Sci USA. 2000;97:1252–1257. doi: 10.1073/pnas.97.3.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Orhan I, Şener B, Kaiser M, Brun R, Tasdemir D. Mar Drugs. 2010;8:47–58. doi: 10.3390/md8010047. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.