

Abstract
Transformations between epithelial and mesenchymal cells are widespread during normal development and adult disease, and transforming growth factor-β1 (TGF-β1) has been implicated in some of these phenotypic switches. Dysplastic kidneys are a common cause of chronic kidney failure in young children and result from perturbed epithelial-mesenchymal interactions. In this study, we found that components of the TGF-β1 axis were expressed in these malformations: TGF-β1 mRNA and protein were up-regulated in dysplastic epithelia and surrounding mesenchymal cells, whereas TGF-β receptors I and II were expressed in aberrant epithelia. We generated a dysplastic kidney epithelial-like cell line that expressed cytokeratin, ZO1, and MET, and found that exogenous TGF-β1 inhibited proliferation and decreased expression of PAX2 and BCL2, molecules characterizing dysplastic tubules in vivo. Furthermore, addition of TGF-β1 specifically induced morphological changes compatible with a shift to a mesenchymal phenotype, accompanied by loss of ZO1 at cell borders and up-regulation of the mesenchymal markers α-smooth muscle actin and fibronectin. The descriptive and functional data presented in this report potentially implicate TGF-β1 in the pathobiology of dysplastic kidneys and our results provide preliminary evidence that an epithelial-to-mesenchymal phenotypic switch may be implicated in a clinically important developmental aberration.
Phenotypic transformation between epithelial and mesenchymal cells is widespread in normal development.1 Blastocyst epithelia from the inner cell mass, for example, generate primitive streak mesenchyme, allowing cells to migrate to new organogenetic sites. Epithelial to mesenchymal transformations also occur when neural crest cells delaminate from the neuroepithelium and when cardiac endothelia differentiate into endocardial cushions. Examples of the converse transformation, from mesenchyme to epithelium, occur in somite and eye development.1
Mesenchymal to epithelial transformation is essential for kidney development. The precursor of the adult human organ, the metanephros, arises during the fifth week of human gestation and is comprised of mesenchyme and ureteric bud epithelia.2 Based on organ culture experiments, Grobstein3 demonstrated that mutual inductions caused mesenchymal cells to transform into proximal nephron epithelia and interstitial stromal cells, whereas the bud branched to form collecting ducts. Studies using Lac-Z tagged mouse metanephric precursor cells by one group, however, suggested that renal cell lineages may be more complex, because ureteric bud contributed to proximal nephron epithelia and surrounding stroma, whereas some mesenchymal cells were incorporated into collecting ducts.4
Congenital renal malformations are frequent causes of childhood chronic kidney failure.5 One form is renal dysplasia which represents a prime example of perturbed epithelial-mesenchymal interactions.6 These organs do not contain functional nephrons but comprise dysplastic tubules and cysts surrounded by mesenchymal-like cells and metaplastic cartilage.7 Dysplastic cytokeratin-positive epithelia maintain a high rate of proliferation postnatally, as assessed by proliferating cell nuclear antigen (PCNA) expression, accompanied by expression of the epithelial marker MET,8 the PAX2 transcription factor,9 and BCL2 survival factor.9 The latter molecules are functionally implicated in proliferation and survival of nephrogenic precursor cells.10,11 In contrast, cells around dysplastic tubules have a low rate of proliferation with low or absent PAX2 and BCL2 expression and increased apoptosis,9,12 and a subpopulation that forms collarettes around tubules express α-smooth muscle actin (α-SMA).6,7 Some dysplastic kidneys are associated with obstructed urinary tracts and experimental urinary flow impairment in fetal sheep reproduces some anatomical and molecular features of the human condition.13,14 Edith Potter2 suggested that dysplastic tubules represent incompletely arborized ureteric bud derivatives, whereas surrounding cells were renal mesenchyme which had failed to differentiate into functional nephrons. However, in view of the lineage-tracing experiments reported in normal metanephric development by one group,4 it is conceivable that dysplastic epithelia may also contribute to some of the mesenchymal tissue by phenotypic transformation.
Transforming growth factor-β1 (TGF-β1) is a prototypic molecule implicated in epithelial-to-mesenchymal transformation. For example, in culture, this cytokine causes both mammary epithelia and endothelia to acquire mesenchymal characteristics.15,16 This factor also has antiproliferative effects on epithelia.17 TGF-β1 signaling is transduced via cell surface type I and type II receptors (TGF-βR1 and TGF-βR2).18 TGF-βR2 binds the ligand and forms a complex with TGF-βR1 which activates intracellular signaling cascades.18 These molecules are expressed during normal rodent nephrogenesis19-21 and TGF-β1 is up-regulated in an ovine model of fetal kidney obstruction.13 Interestingly, urinary tract obstruction is frequently associated with human renal dysplasia but neither the mechanism by which obstruction causes maldevelopment, nor the expression of the TGF-β1 signaling system has yet been established in dysplastic kidneys.
In this study we provide preliminary data to support the hypothesis that TGF-β1 is implicated in the pathogenesis of human renal dysplasia. We sought the expression of the ligand and its receptors in human malformations and investigated the potential of TGF-β1 to modulate proliferation and differentiation of cultured human dysplastic epithelia. Our results suggest that up-regulated TGF-β1 signaling may induce epithelial to mesenchymal transformation in human kidney malformations.
Materials and Methods
Reagents were obtained from Sigma (Poole, UK) unless specified.
Tissues and Cells
Tissue Collection and Derivation of Cell Lines
This project was approved by the Hospitals’ Research Ethics Committee. Kidneys were collected as previously described9,12 and comprised: three normal and three dysplastic fetal samples at 17 to 23 weeks of gestation, six normal areas adjacent to, but unaffected by, Wilms’ tumor at 5 to 19 months of age, and 10 dysplastic kidneys at 3 to 36 months of age. One of the prenatal and three of the postnatal samples were attached to obstructed urinary systems. Two postnatal dysplastic kidneys were also used for cell culture: specimens were diced; placed into Dulbecco’s modified Eagle’s medium F-12 (Gibco BRL, Paisley, UK) with 10% fetal calf serum, 1,000 U/L penicillin G, 1 mg/L streptomycin, and 25 mg/L amphotericin; and grown at 37°C in 5% CO2/air. Cultures from one kidney predominantly contained mesenchymal-like cells whereas cultures from the other contained epithelial-like cells. In preliminary experiments, cells stopped proliferating and appeared senescent after two passages. To expand the cell populations, we transduced first passages with a temperature sensitive (tsA58-U19) Simian virus 40 large T antigen (SV40TAg) construct.22 This extends the proliferative lifespan of healthy and diseased human cells including mammary epithelia, fibroblasts, and myoblasts.23,24 Cells were transduced as described,24 selected in 0.5 g/L G418 at 37°C, and transferred to the permissive temperature of 33°C; expression of the SV40TAg was determined using an anti-SV40TAg antibody, kindly supplied by Parmjit Jat, Ludwig Institute, London, UK. G418 was then withdrawn and the cells cultured at the permissive temperature in baseline medium consisting of RPMI 1640 medium (Gibco BRL), 10% fetal calf serum, 2 mmol/L glutamine, 1 mg/L insulin, and antibiotics. Subsequent experiments were performed between passages 8 to 20 on tissue culture plastic or glass chamber slides (BDH, Poole, UK).
[3H]Thymidine Incorporation
Cells (105 cells/ml) were plated into 48-well plates in baseline medium. After 12 hours, the medium was changed to serum- and insulin-free medium, and cells were left for 48 hours to achieve quiescence. Medium was then changed to control medium, consisting of RPMI 1640, 1% fetal calf serum, 2 mmol/L glutamine, and antibiotics, with or without 0.5 to 10.0 ng/ml added TGF-β1 (240-B; R&D Systems, Abingdon, UK) for 48 hours. Subconfluent cells were pulsed with 1 μCi/ml [3H]thymidine (Amersham Life Science Ltd., Little Chalfont, UK) for the last 6 hours of this culture period. Cells were washed three times with phosphate-buffered saline (PBS), pH 7.4, then with ice-cold 5% trichloroacetic acid for 30 minutes, and solubilized in 0.2 ml 0.25 mol/L NaOH and 0.1% sodium dodecyl sulfate (SDS) for 30 minutes at 37°C. Samples were neutralized with acetic acid, suspended in scintillation fluid, and [3H]thymidine incorporation was measured on a MicroBeta Trilux counter (EG&G Wallac, Helsinki, Finland) and analyzed statistically using the Student’s t-test.
Cell Morphology
Cells were initially plated at subconfluent density in baseline medium for 12 hours. Medium was then changed to control medium for up to 72 hours with varying concentrations of exogenous factors, including 0.5 to 10.0 ng/ml of TGF-β1 and 20.0 ng/ml of hepatocyte growth factor (HGF) (294-HG-005; R&D Systems). TGF-β1 neutralizing antibodies (AF-101-NA; R&D Systems) were also used in some experiments at a concentration of 500 ng/ml, both with and without additional TGF-β1. This concentration, calculated from the data sheet, was sufficient to block the effects of a minimum of 12.0 ng/ml of exogenous TGF-β1. Cells were examined and photographed under phase-contrast illumination on a Nikon TMS inverted microscope (Nikon, Kingston, UK).
RNA Analysis
In Situ Hybridization
The human TGF-β1 cDNA plasmid (kindly provided by Dr. Y. Sun, National Heart and Lung Institute, London, UK) was linearized with restriction enzymes, and sense and anti-sense uridine triphosphate-digoxigenin-labeled riboprobes were generated with the appropriate RNA polymerase, according to the manufacturer’s instructions (digoxigenin RNA labeling kit; Boehringer Mannheim, Lewes, UK). In situ hybridization was performed as described25 with minor modifications. Paraffin-embedded tissue sections (7 μm) were dewaxed, treated with proteinase K (20 μg/ml) at 37°C for 10 minutes, and postfixed in 4% paraformaldehyde. Sections were covered with 50 μl of prehybridization mix that consisted of 50% v/v formamide, 5× standard saline citrate (SSC), 1× Denhardt’s reagent, heat-denaturated salmon sperm DNA 0.1 mg/ml, and 10% w/v dextran sulfate, for 30 minutes at 50°C, followed by a further 50 μl containing the digoxigenin-labeled riboprobe. Glass coverslips were then applied and slides were left to hybridize at 50°C overnight. Sections were washed at 50°C with 25% formamide in 2× SSC for 1 hour, 1× SSC and 0.1% SDS for 30 minutes, and 0.1× SSC and 0.1% SDS for 30 minutes. Hybridized probe was detected by incubation with anti-digoxigenin antibody conjugated to alkaline phosphatase, followed by the chromogen solution, nitroblue tetrazolium, and 5-bromo-4-chloro-3-indolylphosphate toluidinum. Slides were washed and mounted with Citifluor (Chemical Labs, University of Kent, UK). Controls included hybridization without the riboprobe added or with the sense riboprobe.
Northern Blotting
Total RNA was isolated using Tri-reagent, electrophoresed through a 1% formaldehyde-denatured agarose gel, transferred onto a Hybond-N membrane (Amersham Life Science Ltd.) and fixed with UV-Stratalinker (Stratagene, La Jolla, CA). Probes were prepared from TGF-β1 cDNA26 (1040 bp; accession number X02812), TGF-βR1 cDNA (bp 331 to 1351; accession number L11695), and TGF-βR2 cDNA (full length; accession number NM-003242). Inserts were isolated with restriction enzymes and random primer labeling was performed with Prime-a-Gene labeling system (Promega, Southampton, UK). Unincorporated labeled-dCTP was removed with a push-column (Stratagene). Blots were prehybridized with Quick-Hyb solution (Stratagene) at 65°C for 30 minutes and hybridized with the probes at 65°C for 2 hours. Filters were washed twice with 2× SSC/0.1% SDS at room temperature for 30 minutes and once with 0.1× SSC/0.1% SDS at 65°C for 30 minutes. Blots were then exposed to radiograph films for 12 to 48 hours at −70°C.
Protein Analysis
For protein analysis, antibodies were directed against 1) components of the TGF-β1 axis; 2) molecules characteristically expressed in a wide range of epithelia including pancytokeratin, ZO1, a component of epithelial tight junctions,27 and MET, a receptor tyrosine kinase expressed in normal developing renal epithelia28 ; 3) proteins that we have previously found to characterize dysplastic epithelia in vivo including PAX2, BCL2, and PCNA9 ; and 4) mesenchymal markers including α-SMA, which is expressed in smooth muscle collarettes around dysplastic tubules6 and fibronectin, an extracellular molecule which is particularly prominent in metanephric mesenchyme.29
Western Blotting and Immunoprecipitation
For Western blotting, cells cultured in either control medium alone or with additional 5.0 ng/ml of TGF-β1 for up to 72 hours were rinsed with PBS, scraped into 0.6 ml of ice-cold RIPA buffer (PBS, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS) plus protease inhibitors (30 μg/ml Aprotinin, 100 mmol/L sodium orthovanadate, 100 mmol/L phenylmethyl sulfonyl fluoride), and passed repeatedly through a 21-gauge needle. Cell lysates were incubated on ice for 30 minutes and supernatants were collected after 15 minutes centrifugation at 13,000 rpm. Protein concentration was determined using the bicinchoninic acid protein assay (Pierce, Rockford, IL). Samples were boiled for 5 minutes, 30 to 50 μg of proteins were loaded per well and then electrophoresed using an 8 or 12% SDS-polyacrylamide gel electrophoresis gel. Equality of loading was determined by staining representative gels with Coomassie blue. After electrophoresis, proteins were transferred to nitrocellulose membranes (Amersham Life Science Ltd.) by electroblotting (Bio-Rad, Richmond, CA) and left overnight at 4°C in blocking solution consisting of 5% (w/v) fat-free milk powder, 0.3% (v/v) Tween-20 in PBS. They were then incubated with primary antibodies (1:1,000 to 1:2,000 dilution) including PAX2 (71-6000; Zymed, San Francisco, CA), BCL2 (M887; DAKO, Ely, UK), PCNA (Ab1; Oncogene Science, Cambridge, MA), α-SMA (A2547, Sigma), and fibronectin (F0791, Sigma), for 2 hours at 4°C. After washing in blocking solution, blots were incubated for 30 minutes with appropriate horseradish peroxidase-conjugated second antibodies diluted 1:1,000 or 1:1,500 in blocking solution. Blots were washed three times with blocking solution and once with PBS. The blot was then developed using the enhanced chemiluminescence detection kit (Amersham Life Science Ltd.). Rainbow markers were used to determine protein size. Expected sizes were: PAX2, 46 kd; BCL2, 28 kd; PCNA, 37 kd; α-SMA, 43 kd; and fibronectin, 220 to 250 kd.9,14,15
For MET and MET-phosphotyrosine immunoprecipitation experiments, protein was isolated from cells, as above, after they were serum-starved for 24 hours and then incubated with or without HGF (20.0 ng/ml) for 30 seconds, 5 minutes, and 15 minutes. Subsequent steps were at 4°C, unless stated. One mg of the protein was precleared with 10 μg of normal rabbit IgG and 30 μl of protein A-agarose (Santa Cruz Biotechnology, Santa Cruz, CA) for 30 minutes. The sample was then centrifuged at 3,000 rpm for 5 minutes. The supernatant was incubated for 1 hour with 10 μg of rabbit anti-MET antibody, raised against a peptide mapping at the carboxy terminus of the 140-kd human β subunit protein8 (sc161; Santa Cruz Biotechnology). Thirty μl of protein A-agarose was then added and incubated, with agitation, overnight. Beads were washed four times with RIPA and collected by centrifugation at 3,000 rpm for 5 minutes. Pellets were resuspended in 30 μl of electrophoresis buffer and boiled for 5 minutes, before undergoing electrophoresis in a 6% SDS-polyacrylamide gel electrophoresis gel and detection using anti-MET or anti-phosphotyrosine (sc7020; Santa Cruz Biotechnology) antibodies.
Immunohistochemistry
For immunohistochemistry, kidneys were paraffin-embedded, sectioned, and dewaxed through Histo-Clear (National Diagnostics, Atlanta, GA) twice for 10 minutes, followed by rehydration through graded alcohols (Hayman Ltd., Witham, UK). After washing in PBS for 5 minutes, they were treated with trypsin (1 mg/ml) for 10 minutes at 37°C. Endogenous peroxidase was quenched with 3% H2O2 in methanol for 30 minutes at room temperature. Sections were washed with blocking buffer (PBS, 10% goat serum, 0.1% Tween-20) and then incubated with the primary antibody, at 1:50 to 1:100 dilution, in blocking buffer overnight at 4°C. After thorough washing in PBS/0.1% Tween-20, primary antibodies were detected with appropriate second antibodies (1:100) using a streptavidin-biotin-peroxidase system (ABC kit; DAKO) followed by diaminobenzidine or alkaline phosphatase-fast red systems (DAKO). Sections were counterstained with hematoxylin and mounted in dextropropoxyphene (BDH). Sections wee examined and photographed on a Zeiss Axioplan microscope (Carl Zeiss, Oberkochen, Germany).
Antibodies against TGF-β1, TGF-βR1, and TGF-βR2 were all obtained from Santa Cruz Biotechnology. Anti-TGF-β1 antibody (sc146) recognizes an epitope in the human carboxy terminus. This antibody does not cross-react with TGF-β2 or 3,31 and a recent study has confirmed specific staining patterns versus TGF-β2 and TGF-β3 in human tissues.32 We used specific anti-TGF-βR1 (sc402) and TGF-βR2 (sc220) antibodies raised against the carboxy terminus of the human proteins.33 Other antibodies were directed against pancytokeratin (C2562; Sigma) and α-SMA (A5691; Sigma). Controls were omission of primary antibody or preincubation with a 10-fold excess of the appropriate peptide for 4 hours at room temperature.
Immunocytochemistry
Cells were initially cultured on glass chamber slides, as described in Cell Morphology above and then fixed for 2 minutes in 4% paraformaldehyde or methanol. After washing with blocking buffer, primary antibodies, including anti-pancytokeratin, anti-α-SMA, and anti-ZO1 (61-7300; Zymed), were applied for 1 hour at room temperature at dilutions between 1:100 and 1:500. These were detected using fluorescein isothiocyanate-conjugated second antibodies (ACC10F; Serotec Ltd., Kidlington, UK; F 250, F0205; DAKO) (1:500 to 1:1,000). Controls were omission of the primary antibody. For double staining of cytokeratin and α-SMA, cells were incubated with anti-α-SMA (1:500) for 1 hour followed by tetramethylrhodamine isothiocyanate-labeled second antibody (T2659; Sigma) for 1 hour (1:1,000). Slides were then incubated with anti-pancytokeratin-fluorescein isothiocyanate (F3418; Sigma) for 1 hour (1:1,000). Coverslips were mounted on slides in Citifluor (Chemical Labs). The slides were examined and photographed under fluorescence on a Zeiss Axioplan microscope (Carl Zeiss) and on a Leica confocal laser scanning microscope (CLSM Aristoplan-Leica, Heidelberg, Germany).
Results
Gene Expression in Normal Kidneys
Positive TGF-β1 immunostaining was not detected in control sections in which the primary antibody was omitted or preabsorbed with TGF-β1 peptide (Figure 1A)▶ . During normal nephrogenesis TGF-β1 immunoreactivity was detected in the muscular walls of arteries (Figure 1B)▶ . Maturing medullary and cortical collecting ducts, nephron precursors, and stromal cells were negative using this methodology (Figure 1, B and C)▶ . Postnatally, positive immunostaining was only detected in vessels (not shown). In the developing kidney, weak TGF-βR1 and -βR2 immunoreactivity was observed in vessel walls and collecting ducts (Figure 1, D–G)▶ : the latter were considered by Edith Potter to represent the lineage in common with dysplastic tubules.2 Some prenatal glomeruli were also weakly positive (Figure 1, D and F)▶ . Receptor immunoreactivity was restricted to vessels postnatally (not shown).
Figure 1.

Localization of TGF-β1 and its receptors in normal developing kidneys. Antibodies were directed against TGF-β1 (A–C), TGF-βR1 (D and E), and TGF-βR2 (F and G) on sections of normal midgestation human kidneys. A: No immunoreactivity was detected when anti-TGF-β1 antibody was preincubated with excess peptide; vessels (arrowheads) and a glomerulus (g) are indicated. B and C: In sections in which the TGF-β1 antibody was not pre-absorbed, vessels were strongly immunoreactive, but fetal collecting ducts (d) did not show significant staining. D–G: In sections reacted with TGF-β1 receptor I and II antibodies, immunostaining was detected in fetal kidney vessels and collecting ducts. Scale bar, 15 μm.
Gene Expression in Dysplastic Kidneys
A similar pattern of TGF-β1 and receptor immunostaining was detected in the prenatal and postnatal dysplastic kidneys. TGF-β1 protein was most prominent in compact cells proximate to dysplastic tubules and cysts (Figure 2, A–C)▶ . Immunoreactivity for the factor was also observed in the epithelia of larger cysts (Figure 2C)▶ , especially in prenatal samples (not shown). Vascular reactivity was noted, as for the normal samples (not shown). Dysplastic epithelia uniformly stained for both TGF-βR1 and TGF-βR2 (Figure 2, D–G)▶ . Using a sense probe for TGF-β1 mRNA, no signal was observed in dysplastic epithelia or surrounding tissue (Figure 3A)▶ , whereas an antisense probe revealed positive signals in dysplastic tubule and cyst epithelia (Figure 3, B–D)▶ and in a subset of surrounding mesenchyme-like cells (arrowheads in Figure 3, B–D▶ ). The same sense and antisense probes demonstrated a specific signal for TGF-β1 transcripts in renal vessel walls from these organs (Figure 3, E and F)▶ . In contrast with dysplastic tissues, using the same methodology, normal prenatal and postnatal kidneys had no significant signal apart from vessels (not shown).
Figure 2.
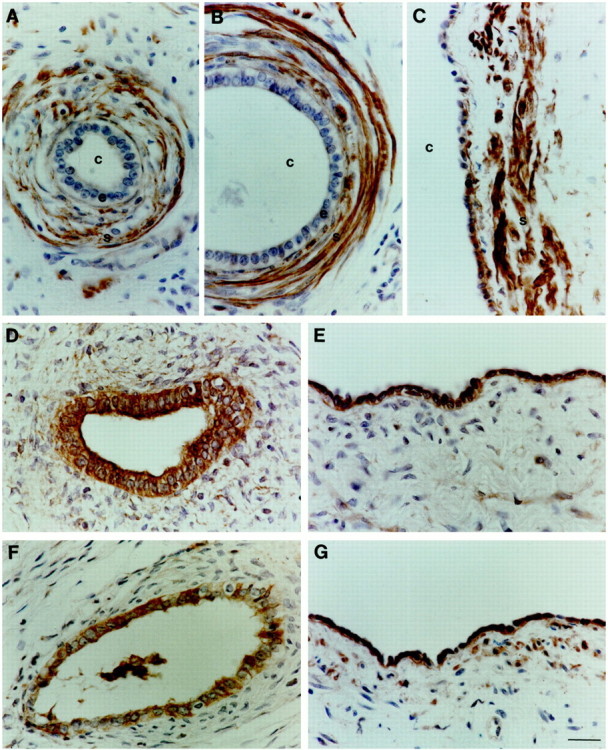
Localization of components of the TGF-β1 axis in dysplastic kidneys. Antibodies were directed against TGF-β1 (A–C), TGF-βR1 (D and E), and TGF-βR2 (F and G) on sections of postnatal dysplastic kidneys. Note that these kidneys lack normal renal structures such as glomeruli. No signal was detected on omission of primary antibody or pre-absorption with the appropriate peptide (not shown). A–C: TGF-β1 immunoreactivity was detected in compact stromal cells (s) proximate to dysplastic tubules and cysts (c), with some epithelia (e) staining in larger cysts, as shown in C. D–G: TGF-βR1 and TGF-βR2 proteins were detected in dysplastic epithelia of all sizes, ranging from small undilated tubules to large cysts. Scale bar, 15 μm.
Figure 3.
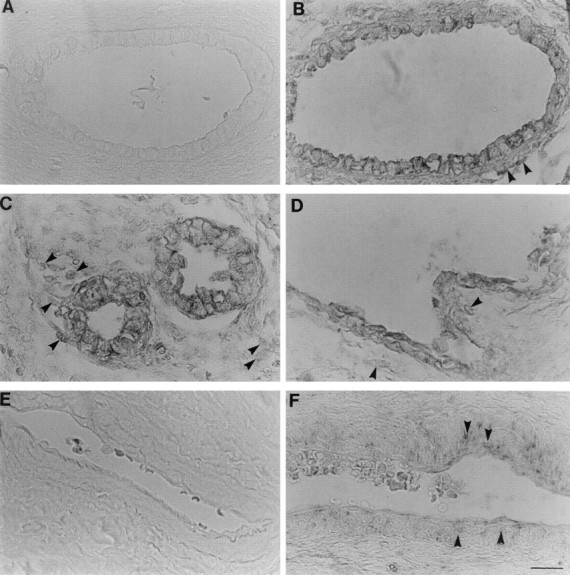
In situ hybridization for TGF-β1 in dysplastic kidneys. A and E: TGF-β1 sense (control) probes, the remaining panels used antisense probes. C and D: Prenatal dysplastic kidneys and remaining panels show postnatal dysplastic samples. A and B: Serial sections of a small cyst. No signal was detected using the sense probe but transcripts were detected in the epithelia and in a subset of surrounding mesenchyme-like cells (arrowheads). C and D: Dysplastic tubules and a large cyst, respectively; note TGF-β1 mRNA in epithelial and adjacent cells (arrowheads). Together E and F confirm a specific signal for TGF-β1 mRNA in vessel walls (arrowheads). Scale bar, 15 μm.
Immunohistochemistry for cytokeratin revealed positive staining in all dysplastic epithelia (Figure 4, A and B)▶ . A minor population of cytokeratin-positive cells were detected in the interstitium proximate to, but quite distinct from, large cysts (arrowheads in Figure 4B▶ ) and these cells had an elongated appearance, consistent with a motile-type phenotype.34 α-SMA was detected in compact cells around dysplastic tubules and cysts (Figure 4, C and D)▶ , a similar location to mesenchymal TGF-β1 immunoreactivity. Occasional cells (<1%) were also weakly positive for α-SMA in dysplastic epithelia (Figure 4C)▶ . In the normal prenatal kidneys, α-SMA was not detected in renal mesenchyme, or interstitial stromal cells between maturing tubules, although it was detected in vessels in normal and dysplastic samples (not shown).
Figure 4.

Localization of cytokeratin and α-SMA in dysplastic kidneys. Dysplastic tubules are shown in A and C , whereas B and D show large cysts. Antibodies were directed against cytokeratin detected using diaminobenzidine (A and B), or α-SMA detected using fast red (C and D). No signal was detected on omission of primary antibody (not shown). A and B: All cells in dysplastic epithelia were positive for cytokeratin. A minor population of cytokeratin-positive cells were also detected in the interstitium proximate to large cysts (arrowheads in B); note the elongated appearance of these cells. C and D: α-SMA was detected in compact cells around dysplastic tubules and cysts. Rare cells were also weakly positive for α-SMA in dysplastic tubule epithelia in C. Scale bar, 15 μm.
Characterization of Cultured Cells
Using tissues from two postnatal dysplastic kidneys we generated two transduced cell lines. Both expressed SV40TAg by Western blotting (not shown). One had a mesenchymal-like morphology and was not studied further. The other comprised cells with an epithelial-like morphology: sparse cells appeared cuboidal whereas confluent cultures had an irregular cobblestone appearance. This phenotype was maintained >30 passages without evidence of senescence or blast crisis. These cultured cells expressed classical epithelial markers including cytokeratin and ZO1, the receptor tyrosine kinase MET, which we have previously described in normal and abnormal developing renal epithelia,8 and other proteins up-regulated in dysplastic epithelia in vivo, including PAX2, BCL2, and PCNA (Table 1)▶ .
Table 1.
Characterization of Dysplastic Kidneys and Cells in Culture
| Dysplastic kidneys in vivo | Cultured dysplastic cells | |||
|---|---|---|---|---|
| Epithelia | Mesenchyme | Control media | Added TGF-β1 | |
| PAX2 | ++ | +/− | ++ | + |
| BCL2 | ++ | − | ++ | + |
| PCNA | ++ | +/− | ++ | + |
| Cytokeratin | ++ | +/− | ++ | + |
| α-SMA | +/− | ++ | +/− | ++ |
| Fibronectin | na | na | + | ++ |
The PAX2, BCL2, and PCNA data for dysplastic kidneys in vivo are from our previous studies.9 Immunohistochemistry for cytokeratin and α-SMA are depicted in Figure 4▶ of this study. Data for dysplastic cells were generated in the present study and represent a synthesis of Western blotting for PAX2, BCL2, PCNA, α-SMA, and fibronectin plus immunocytochemistry for cytokeratin and α-SMA. Note the similarities in protein expression between the dysplastic cells in this study and dysplastic epithelia in vivo. Key: −, no protein detected; +/−, rare positive cells by immunohistochemistry or immunocytochemistry, or barely detectable signals by Western blotting; + and ++, increasing intensity of either the immunohistochemical or Western blotting signal; na is not assessed. See Figures 1 to 9▶ ▶ ▶ for detailed results.
Effects of TGF-β1 on Dysplastic Cells
TGF-β1, TGF-βR1, and TGF-βR2 transcripts were detected in the cultured dysplastic epithelia by Northern blot (Figure 5A)▶ , consistent with the in vivo expression of these genes by dysplastic tubules and cysts, described above. Proliferation, as assessed by [3H]thymidine incorporation, was significantly (P < 0.01; Student’s t-test) down-regulated at 48 hours by exogenous TGF-β1 concentrations between 0.5 and 10.0 ng/ml versus time-matched controls (Figure 5B)▶ . Maximal effects were observed between 2.0 and 10.0 ng/ml, with no significant difference between these concentrations. For the rest of the experiments in this study, therefore, we used concentrations within this range. We also examined how TGF-β1 affected the molecular phenotype of these cells. Figure 5C▶ shows representative Western blots for three experiments in which cells were grown in either control medium or in medium with 5.0 ng/ml TGF-β1 for 2 days. Exogenous TGF-β1 induced a reproducible decrease in immunoreactive PAX2, BCL2, and PCNA proteins, molecules characteristically expressed in dysplastic epithelia.
Figure 5.

TGF-β1, TGF-βR1, and TGF-βR2 expression, and effects of TGF-β1 on proliferation and protein expression in cultured dysplastic cells. A: Northern blot demonstrated that dysplastic cells express transcripts for TGF-β1 (2.3 kb), TGF-βR1 (5.1 kb), and TGF-βR2 (4.6 kb). B: Exogenous TGF-β1 reproducibly inhibited [3H]thymidine incorporation of the dysplastic cells at concentrations between 0.5 and 10.0 ng/ml versus time-matched cells cultured in control medium, with significant effects (*P < 0.01, Student’s t-test) between 0.5 and 10.0 ng/ml. C: In this Western blot, representative of three separate experiments, PAX2, BCL2, and PCNA bands at the expected sizes were detected in cells cultured in control medium (−), but there was a reproducible decrease in expression of these molecules 48 hours after exposure to 5.0 ng/ml TGF-β1 (+).
Dysplastic cells cultured in control medium had an irregular epithelial-like morphology in monolayer culture (Figure 6A)▶ . After exposure to added 2.0 ng/ml TGF-β1 for only 1 day, we recorded reproducible effects comprising elongation and increase in area of individual cells (see Figure 9, A and C▶ ). In semiconfluent and confluent cultures, TGF-β1 exposure for 48 to 72 hours elicited more pronounced changes, with formation of multilayered aggregates (Figure 6B)▶ . Cell density was relatively sparse between aggregates, but close inspection revealed individual cells with filopodia and lamellipodia characteristic of migratory cells (Figure 6B)▶ .34 These effects of exogenous TGF-β1 were completely abrogated by addition of TGF-β1 blocking antibody as described in the Materials and Methods. Conversely, culture of the cells with only the blocking antibody had no effects on the irregular epithelial-like morphology, perhaps suggesting that, in vitro, endogenous secretion of TGF-β1 protein is not sufficient to alter the baseline phenotype. After exposure to TGF-β1, changes in cell shape and aggregation were reversible on switching to control medium for 48 hours (not shown). Under control conditions, cells formed tight junctions with neighboring cells, as evidenced by immunodetection of ZO1 protein at the lateral cell junctions (Figure 6C)▶ 27 and, after exposure to TGF-β1, there was loss of junctional ZO1 protein (Figure 6D)▶ .
Figure 6.

Effects of TGF-β1 on morphology and ZO1 expression. Gross morphology is shown in A and B, whereas C and D show ZO1 immunocytochemistry (with propidium-iodide nuclear counterstaining) of epithelial-like dysplastic cells cultured in either control medium (A and C) or with exogenous TGF-β1 (B and D). A: Cells cultured in control medium had an epithelial-like morphology in monolayer culture. B: Morphological changes were observed after exposure to 2.0 ng/ml TGF-β1 for 72 hours: multilayered aggregates formed in semiconfluent and confluent cultures and individual cells between aggregates became larger and developed filopodia and lamellipodia characteristic of a motile phenotype.34 C and D: In control medium ZO1 was immunolocalized to lateral cell junctions (arrowheads) but immunostaining at cell borders was lost after culture with TGF-β1. Scale bar: 40 μm (A and B); 15 μm (C and D).
Figure 9.
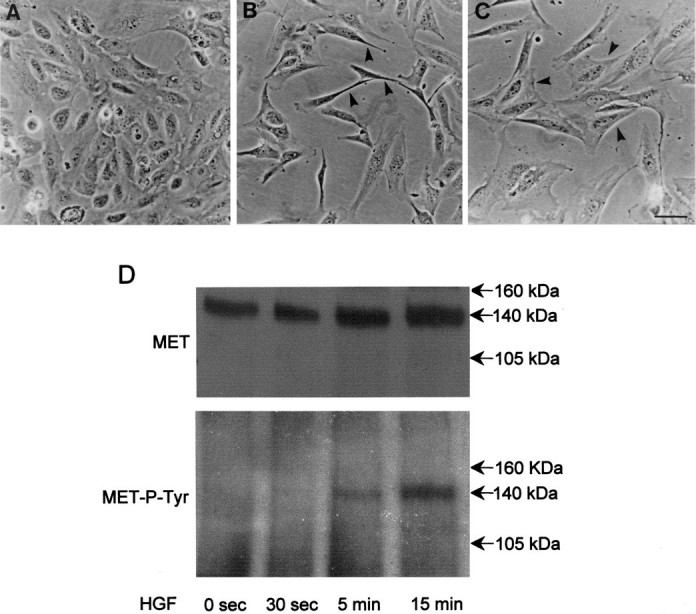
Different effects of HGF and TGF-β1 on dysplastic cells. A–C: Gross morphology of cells cultured in control medium (A), and with additional 20.0 ng/ml of HGF (B) or 2.0 ng/ml of TGF-β1 (C) for 24 hours. Note the epithelial-like appearance of cells in control medium, the separation and elongation of cells on exposure to HGF (arrowheads in B), and the tendency to begin to enlarge and clump with TGF-β1 (arrowheads in C). D: Cell lysates were immunoprecipitated with anti-MET antibody and probed with either anti-MET antibody (top) or antiphosphotyrosine antibody (MET-P-TYR; bottom). Note the expression of MET, which becomes tyrosine phosphorylated within 5 minutes of exposure to HGF. Scale bar, 10 μm (A–C).
Using double immunofluorescence, all cells in control medium expressed cytokeratin (Figure 7A)▶ whereas α-SMA immunoreactivity was present in <1% of cells in representative cultures. In marked contrast, α-SMA protein was detected in >70% of cells in a typical time-matched culture exposed to TGF-β1 for 48 to 72 hours (Figure 7B)▶ . As assessed by confocal laser scanning microscopy (Figure 7, C–H)▶ , the above impressions were confirmed and furthermore, after exposure to exogenous TGF-β1, it was common to detect individual cells that expressed both cytokeratin and α-SMA (Figure 7, F–H)▶ , consistent with an epithelial-mesenchymal transition. As assessed by Western blotting, α-SMA was barely detectable in lysates of cell cultures grown in control medium, whereas there was a reproducible, stepwise increase in expression after addition of 5.0 ng/ml of TGF-β1 for 24, 48, and 72 hours (Figure 8A)▶ . Similar analyses for fibronectin protein revealed that TGF-β1 up-regulated levels of this extracellular matrix molecule which is known to be highly expressed by metanephric mesenchymal cells (Figure 8B)▶ .29
Figure 7.
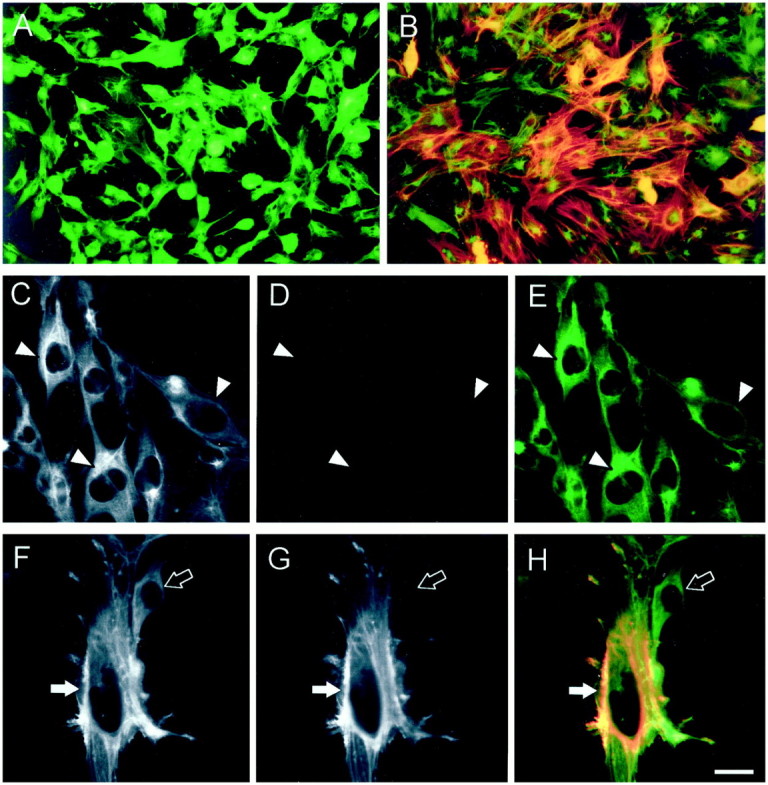
Effects of TGF-β1 on cytokeratin and α-SMA expression. Shown are semiconfluent cells grown in control medium (A and C–E) and cells that have been exposed to exogenous TGF-β1 for 72 hours (B and F–H) . Panels are representative of three experiments. All cultures were double-immunostained with antibodies to cytokeratin and α-SMA, which were, respectively, detected with the fluorochromes fluorescein isothiocyanate (green) and tetramethylrhodamine isothiocyanate (red); double-expressing cells appear orange/yellow. A, B, E, and H: Composites of both signals are shown. C and F: Only the cytokeratin wavelength is depicted. D and G: Only the α-SMA image is shown. All cells expressed cytokeratin in control medium (A, and arrowheads in C and E), but >99% were negative for α-SMA (A, D, and E). Time-matched TGF-β1-treated cells demonstrated marked up-regulation of α-SMA in >70% of cells (B). After culture with exogenous TGF-β1, analysis at the single cell level revealed a major population of larger cells which expressed cytokeratin and α-SMA (solid arrow in F–H), demonstrating evidence of a transitional phenotype, whereas a subset of cells were cytokeratin-positive only (open arrow in F–H). Scale bar: 30 μm (A and B); 10 μm (C–H).
Figure 8.
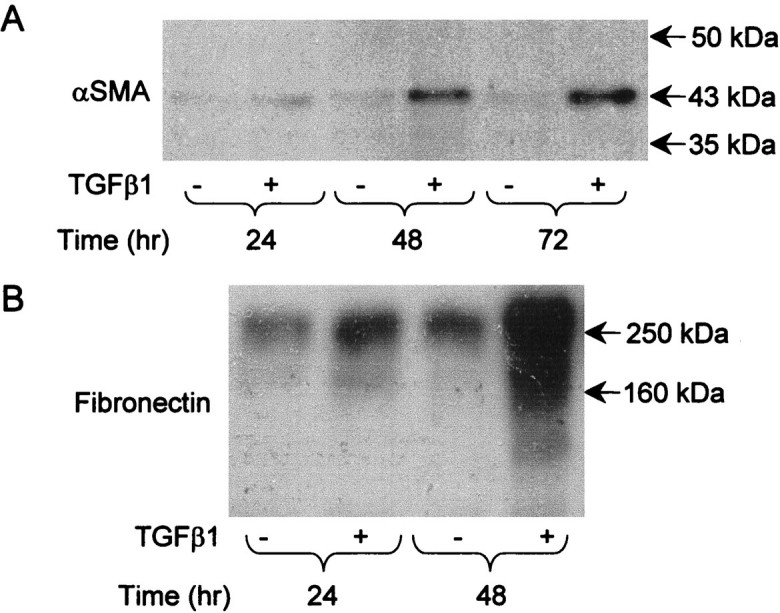
Effects of TGF-β1 on α-SMA and fibronectin protein expression. Western blots, representative of three experiments, for α-SMA (A) and fibronectin (B) in dysplastic cells grown in either control (−) or medium supplemented with 2.0 ng/ml TGF-β1 (+) for up to 72 hours. A: A single band at the expected size of 43 kd for α-SMA was barely detectable at all time points in control cultures, whereas there was a progressive, stepwise increase in immunoreactive protein when cultured with exogenous TGF-β1. B: Low levels of fibronectin were detected at around 250 kd in control cultures but immunoreactive protein increased after 48 hours of culture with exogenous TGF-β1. Similar effects were elicited by levels of TGF-β1 as low as 0.5 ng/ml (not shown).
Dysplastic cells were also cultured with 20.0 ng/ml HGF, the ligand for MET. After 1 day of exposure to this ligand, epithelial-like dysplastic cells (Figure 9A)▶ exhibited the classical scattering response (Figure 9B)▶ , first reported in the Madin Darby canine kidney cell line, prototypic renal epithelial cells.35 During the response the dysplastic cells uniformly separated from each other and became very thin with bipolar elongated processes (Figure 9B)▶ . This is different from the early changes elicited by 1 day of exposure to exogenous TGF-β1, when cells increased in area and tended to form clumps (Figure 9C)▶ . An additional difference was that HGF had no effect on cell proliferation, as assessed by [3H]thymidine incorporation (not shown). Western blot confirmed expression of MET by the dysplastic cells, as well as tyrosine phosphorylation of this receptor after exposure to HGF (Figure 9D)▶ . Hence these cells can respond to more than one factor but the effects of TGF-β1 seems characteristic.
Discussion
Our descriptive and functional studies provide preliminary data to implicate TGF-β1 in the pathobiology of dysplastic kidneys. Moreover, ours is the first study to isolate cells from human renal malformations to assess their behavior in strictly defined milieus. Finally, our results introduce the possibility that an epithelial-to-mesenchymal phenotypic switch is implicated in a clinically important developmental aberration.
TGF-β1 is Up-Regulated in Human Dysplastic Kidneys
We found that in normal human kidney development, TGF-β1 mRNA and protein were expressed in vessels, whereas type I and II TGF-β1 receptors could be detected in these structures and also in maturing collecting ducts. These observations are broadly similar to previous nonhuman studies.19,21,33 In dysplastic kidneys, TGF-β1 mRNA and protein were up-regulated. TGF-β1 transcripts were prominent in dysplastic epithelia and also detected in a subset of the surrounding cells. TGF-β1 protein was strikingly up-regulated and was mainly immunolocalized in swirls of mesenchyme-like cells around dysplastic epithelia although it was also detected in the epithelia of some large cysts. We postulate that the relatively high epithelial TGF-β1 mRNA levels compared to immunoreactive protein may reflect secretion of the factor, either apically or basally. Indeed, immunoreactive TGF-β1 can be detected in the urine after experimental obstruction of the fetal lower urinary tract in sheep.13 We detected TGF-β1 receptor I and II immunoreactivity in all dysplastic epithelia, ranging from small tubules to large cysts. The observed distribution patterns of TGF-β1 and its receptor proteins is consistent with both autocrine signaling systems within epithelia and paracrine effects, with mesenchymal TGF-β1 acting on epithelial receptors.
Our observation of TGF-β1 up-regulation in dysplastic kidney tissues is similar to reports of increased TGF-β1 mRNA in ureteral obstruction in adults,36 and several studies during animal kidney development:13,37 Chevalier and colleagues,37 for example, described increased TGF-β1 in obstructed neonatal rat kidneys that are still undergoing active nephrogenesis, whereas Medjebeur et al13 reported up-regulation of TGF-β1 mRNA in ovine kidney malformations generated by fetal urinary obstruction.
TGF-β1 Affects the Biology of Dysplastic Renal Epithelia in Vitro
We next examined the effects of TGF-β1 on cultured cells which displayed characteristics of dysplastic epithelia in vivo, including TGF-β1, TGF-βR1, and TGF-βR2 expression. At concentrations as low as 0.5 ng/ml, exogenous cytokine inhibited proliferation of these epithelial-like cells, as reported in mature renal epithelia.38 This effect also parallels the inhibition of TGF-β1 on ureteric bud and collecting duct branching in murine metanephric culture39 and in Madin Darby canine kidney cells.40 This data, taken together with reports that kidney development is normal in TGF-β1 null mutants,41 suggests that excess TGF-β1, rather than lack of this factor, has significant biological effects on metanephric growth.
During the same period that TGF-β1 decreased proliferation of cultured dysplastic cells, we recorded down-regulation of PCNA and PAX2 proteins, molecules associated with expansion of metanephric precursor cells in normal development10 and pathological renal cystogenesis.9,42 Of note, Liu et al38 demonstrated that TGF-β1 down-regulated PAX2 mRNA in mature rabbit proximal tubule cells by decreasing transcript stability. In our current study we also found that exogenous TGF-β1 decreased BCL2, a molecule ectopically expressed in hyperproliferative dysplastic kidney epithelia,9 but reduction of this survival factor11 was not associated with fulminant apoptosis, as assessed by serial microscopy and a search for DNA laddering (data not shown).
Exogenous TGF-β1 induced morphological and cytoskeletal changes consistent with a switch from epithelial toward a mesenchymal phenotype. The epithelial characteristics of the dysplastic cells in control media comprised a cuboidal cell shape, expression of cytokeratin and MET, and the localization of ZO1 at lateral cell junctions. Exogenous TGF-β1 initially induced an increase in cell area (see Figure 9C▶ ) and then the formation of multilayered aggregates; between these structures we observed cells with an elongated, motile phenotype (see Figure 6B▶ ).34 These changes were accompanied by the loss of ZO1 at cell borders, and up-regulation of α-SMA and fibronectin.
One could argue that these TGF-β1-induced changes result from selection of a small subpopulation (<1%) of α-SMA-positive cells present before exogenous cytokine was added. It seems unlikely, however, that such a small fraction could expand rapidly enough to comprise >70% of the TGF-β1-treated population within 2 to 3 days, even if selective proliferation was combined with massive apoptosis of the initial epithelial-like cells, especially because we did not observe excess cell death (see above). The reversibility of the morphological changes induced by TGF-β1 after withdrawal of the cytokine for 24 hours also argues against selection, although it would be interesting to determine whether prolonged exposure to the cytokine would elicit a more complete phenotypic transformation in our human cells.
The most compelling argument in favor of TGF-β1-induced phenotypic transformation, however, is our finding of cells that clearly co-expressed α-SMA and cytokeratin after treatment with this factor (see Figure 7H▶ ); we propose that these are epithelial-like cells which have entered a transitional stage during the switch toward a mesenchymal phenotype. It is intriguing, therefore, that dysplastic kidneys contained rare α-SMA-positive cells in dysplastic epithelia and cytokeratin-positive interstitial cells near to cysts. These cells potentially represent the equivalent transitional stage between epithelium and mesenchyme in vivo, but one would have to trace the cell lineage changes over time to prove that they were undergoing phenotypic transformation. It is difficult to conceive of an experimental strategy to do this in humans but it may be possible using an animal model, as follows: first developing renal epithelia would have to be labeled in vivo, for example with a vital dye such as Dil or with a genetic marker such as the Lac-Z reporter gene. Second, these cells would need to be exposed to an increased milieu of TGF-β1, such as that induced by obstruction of the urinary tract. Finally, one would need to demonstrate in tissue sections that labeled epithelial cells had changed shape and position to become mesenchymal cells.
TGF-β1 has previously been implicated in progressive fibrosis which accompanies loss of renal function in chronic adult kidney diseases, where it mediates expansion of the interstitium by increasing α-SMA-positive myofibroblastic cells and extracellular matrix.43 Evidence is accumulating that some of these cells may be derived from mature epithelia, rather than just stromal fibroblasts, by a process termed “transdifferentiation.” For example, in the rat remnant kidney, proximal tubule cells begin to express α-SMA and lose basement membrane integrity, consistent with the acquisition of a mesenchymal phenotype.44 In addition, TGF-β1 promotes transdifferentiation of adult rat kidney tubular epithelial cells into myofibroblasts in vitro.30 The master molecules that drive this TGF-β1-induced phenotypic switch are poorly defined, although Okada and colleagues45 have functionally implicated FSP1, an intracellular calcium-binding protein, in TGF-β1-induced transdifferentiation of mature murine proximal tubule cells; expression of FSP1 has not yet been reported in human dysplastic kidneys. Because TGF-β1 induces epithelial-to-mesenchymal conversion of normal mature as well as dysplastic renal epithelia, one might also expect normal developing renal epithelial cells to react in a similar manner. Proof of this hypothesis requires further experiments.
Potential Roles of Other Growth Factors in Human Renal Dysplasia
Aberrant expression of several other soluble signaling factors has also been reported in dysplastic kidneys, including HGF,8 insulin-like growth factors,46 platelet-derived growth factor,47 and tumor necrosis factor-α.48 One could argue that perturbation of these factors might have the same effects as up-regulation of TGF-β1. This seems unlikely, however, because we found that exogenous HGF caused distinct, different effects; after exposure to HGF for only 24 hours, cells exhibited the classical scattering response35 and there was no effect on cell proliferation. This demonstrates that the dysplastic cells have a range of potential responses to signaling molecules in vitro. Ultimately, however, it will be important to ascertain which signaling systems, if any, have critical roles in the biology of dysplasia in the whole animal. In future studies, the potential in vivo roles of individual factors could be tested by altering their tissue levels in animal models of renal dysplasia. For example, to investigate the role of overexpression of TGF-β1 in renal dysplasia one would need to block this factor (eg, using decorin49 ) in an animal model of fetal lower urinary tract obstruction13,14 and assess the resulting histological and biological effects.
Complex Biology of Human Dysplastic Kidneys–A Working Model
How, then, might TGF-β1 fit into the cascade of gene expression and aberrant cell biology observed in human dysplastic kidneys? We propose that it is possible to link up-regulated TGF-β1 expression to fetal urinary tract obstruction, which often accompanies human dysplasia (Figure 10)▶ . In this model, increased hydrostatic pressure from impaired fetal urinary flow has two separate effects. On the one hand, obstruction would trigger cystic epithelial hyperproliferation,9 as reported when MDCK cysts are subjected to increased tension in vitro.50 PAX2 may be implicated in this process because experimental ovine fetal ureteric obstruction causes increased PAX2 expression in renal cysts,14 increased PAX2 expression causes cyst proliferation in transgenic mice42 and this molecule has been implicated in oncogenesis.51 On the other hand, stretch might also up-regulate TGF-β1 in the metanephros, as reported in other renal cells in culture,52 and this would have a number of secondary effects. First, increased TGF-β1 acts as a biological brake on epithelial hyperproliferation as demonstrated in the current study; this effect is potentially beneficial because it limits cyst growth and may be mediated by down-regulation of PAX2.38 Second, based on several in vitro experiments,39,40 excess TGF-β1 inhibits normal branching morphogenesis, a classic feature of dysplastic kidneys reported by Edith Potter2 in microdissection studies. Third, epithelial cells are diverted or lost into a mesenchymal/smooth muscle lineage contributing to the characteristic collarettes around dysplastic tubules.6,7 Most likely, the final biological consequences of fetal obstruction would be determined by a balance between these molecular changes which may be heterogeneous within a single developing organ. For example, in areas where cysts form, the effects of PAX2 may outweigh the inhibitory influence of TGF-β1, but the cytokine may still limit the rate of cyst growth.
Figure 10.

Working model linking obstruction, TGF-β1 and PAX2 in human kidneys. Increased hydrostatic pressure from impairment of fetal urinary flow is postulated to trigger cystic epithelial hyperproliferation, mediated via up-regulation of PAX2, and to directly up-regulate TGF-β1. Increased levels of this cytokine act as a biological brake on epithelial hyperproliferation, inhibit normal branching morphogenesis, and promote phenotypic transformation of epithelial cells and their loss into a mesenchyme-like phenotype. The final biological effects therefore represent a balance between these positive and negative factors. Other triggers such as mutations or teratogens may also initiate this sequence of events. → indicates stimulates or causes; indicates inhibits or prevents.
Finally, it should be noted that overt obstruction was only diagnosed in a subset of our samples, whereas up-regulation of TGF-β1 was uniformly observed. Variants on this model might therefore include other triggers such as mutations or teratogens that initiate the same sequence of events.
Footnotes
Address reprint requests to Dr Paul Winyard,, Nephro-Urology Unit, Institute of Child Health, 30 Guilford St., London, WC1N 1EH, UK. E-mail: pwinyard@ich.ucl.ac.uk.
Supported by project grants from Action Research (S/P/3178), the Wellcome Trust (058005), the National Kidney Research Fund (R18/1/2000), and the Kidney Research Aid Fund.
References
- 1.Hay ED, Zuk A: Transformations between epithelium and mesenchyme: normal, pathological, and experimentally induced. Am J Kidney Dis 1995, 26:678-690 [DOI] [PubMed] [Google Scholar]
- 2.Potter EL: Normal and abnormal development of the kidney. 1972. Year Book Medical Publishers Inc. Chicago
- 3.Grobstein C: Inductive interaction in the development of the mouse metanephros. J Exp Zool 1955, 130:319-340 [Google Scholar]
- 4.Qiao J, Cohen D, Herzlinger D: The metanephric blastema differentiates into collecting system and nephron epithelia in vitro. Development 1995, 121:3207-3214 [DOI] [PubMed] [Google Scholar]
- 5.Warady BA, Hebert D, Sullivan EK, Alexander SR, Tejani A: Renal transplantation, chronic dialysis, and chronic renal insufficiency in children and adolescents. The 1995 Annual Report of the North American Pediatric Renal Transplant Cooperative Study. Pediatr Nephrol 1997, 11:49-64 [DOI] [PubMed] [Google Scholar]
- 6.Woolf AS, Winyard PJD: Gene expression and cell turnover in human renal dysplasia. Histol Histopathol 2000, 15:159-166 [DOI] [PubMed] [Google Scholar]
- 7.Daikha-Dahmane F, Dommergues M, Muller F, Narcy F, Lacoste M, Beziau A, Dumez Y, Gubler MC: Development of human fetal kidney in obstructive uropathy: correlations with ultrasonography and urine biochemistry. Kidney Int 1997, 52:21-32 [DOI] [PubMed] [Google Scholar]
- 8.Kolatsi-Joannou M, Moore R, Winyard PJ, Woolf AS: Expression of hepatocyte growth factor/scatter factor and its receptor, MET, suggests roles in human embryonic organogenesis. Pediatr Res 1997, 41:657-665 [DOI] [PubMed] [Google Scholar]
- 9.Winyard PJD, Risdon RA, Sams VR, Dressler G, Woolf AS: The PAX2 transcription factor is expressed in cystic and hyperproliferative dysplastic epithelia in human kidney malformations. J Clin Invest 1996, 98:451-459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Torres M, Gomex-Pardo E, Dressier GR, Gruss P: Pax-2 controls multiple steps of urogenital development. Development 1995, 121:4057-4065 [DOI] [PubMed] [Google Scholar]
- 11.Veis DJ, Sorenson CM, Shutter JR, Korsmeyer SJ: Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys and hypopigmented hair. Cell 1993, 75:229-240 [DOI] [PubMed] [Google Scholar]
- 12.Winyard PJD, Nauta J, Lirenman DS, Hardman P, Sams VR, Risdon RA, Woolf AS: Deregulation of cell survival in cystic and dysplastic renal development. Kidney Int 1996, 49:135-146 [DOI] [PubMed] [Google Scholar]
- 13.Medjebeur AA, Bussieres L, Gasser B, Gimonet V, Laborde K: Experimental bilateral urinary obstruction in fetal sheep: transforming growth factor-β 1 expression. Am J Physiol 1997, 273:F372-F379 [DOI] [PubMed] [Google Scholar]
- 14.Attar R, Quinn F, Winyard PJ, Mouriquand PD, Foxall P, Hanson MA, Woolf AS: Short-term urinary flow impairment deregulates PAX2 and PCNA expression and cell survival in fetal sheep kidneys. Am J Pathol 1998, 152:1225-1235 [PMC free article] [PubMed] [Google Scholar]
- 15.Miettinen PJ, Ebner R, Lopez AR, Derynck R: TGF-β induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J Cell Biol 1994, 127:2021-2036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Potts JD, Runyan RB: Epithelial-mesenchymal cell transformation in the embryonic heart can be mediated, in part, by transforming growth factor β. Dev Biol 1989, 134:392-401 [DOI] [PubMed] [Google Scholar]
- 17.Moses HL: TGF-β regulation of epithelial cell proliferation. Mol Reprod Dev 1992, 32:179-184 [DOI] [PubMed] [Google Scholar]
- 18.Wrana JL: TGF-β receptors and signalling mechanisms. Miner Electrolyte Metab 1998, 24:120-130 [DOI] [PubMed] [Google Scholar]
- 19.Schmid P, Cox D, Bilbe G, Maier R, McMaster GK: Differential expression of TGF β 1, β 2 and β 3 genes during mouse embryogenesis. Development 1991, 111:117-130 [DOI] [PubMed] [Google Scholar]
- 20.Pelton RW, Saxena B, Jones M, Moses HL, Gold LI: Immunohistochemical localization of TGFβ1, TGFβ2, and TGFβ3 in the mouse embryo: expression patterns suggest multiple roles during embryonic development. J Cell Biol 1991, 115:1091-1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mariano JM, Montuenga LM, Prentice MA, Cuttitta F, Jakowlew SB: Concurrent and distinct transcription and translation of transforming growth factor-β type I and type II receptors in rodent embryogenesis. Int J Dev Biol 1998, 42:1125-1136 [PubMed] [Google Scholar]
- 22.Jat PS, Sharp PA: Cell lines established by a temperature-sensitive simian virus 40 large T-antigen gene are growth restricted at the nonpermissive temperature. Mol Cell Biol 1989, 9:1672-1681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stamps AC, Davies SC, Burman J, O’Hare MJ: Analysis of proviral integration in human mammary epithelial cell lines immortalized by retroviral infection with a temperature-sensitive SV40 T-antigen construct. Int J Cancer 1994, 57:865-874 [DOI] [PubMed] [Google Scholar]
- 24.Simon LV, Beauchamp JR, O’Hare M, Olsen I: Establishment of long-term myogenic cultures from patients with Duchenne muscular dystrophy by retroviral transduction of a temperature-sensitive SV40 large T antigen. Exp Cell Res 1996, 224:264-271 [DOI] [PubMed] [Google Scholar]
- 25.Yuan HT, Gowan S, Kelly FJ, Bingle CD: Cloning of guinea pig surfactant protein A defines a distinct cellular distribution pattern within the lung. Am J Physiol 1997, 273:L900-L906 [DOI] [PubMed] [Google Scholar]
- 26.Derynck R, Jarrett JA, Chen EY, Eaton DH, Bell JR, Assoian RK, Roberts AB, Sporn MB, Goeddel DV: Human transforming growth factor-β complementary DNA sequence and expression in normal and transformed cells. Nature 1985, 316:701-705 [DOI] [PubMed] [Google Scholar]
- 27.Wang AZ, Ojakian GK, Nelson WJ: Steps in the morphogenesis of a polarized epithelium. I. Uncoupling the roles of cell-cell and cell-substratum contact in establishing plasma membrane polarity in multicellular epithelial (MDCK) cysts. J Cell Sci 1990, 95:137-151 [DOI] [PubMed] [Google Scholar]
- 28.Woolf AS, Kolatsi-Joannou M, Hardman P, Andermacher E, Moorby C, Fine L, Jat PS, Gherardi E: Roles of hepatocyte growth factor/scatter factor and the met receptor in the early development of the metanephros. J Cell Biol 1995, 128:171-184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sariola H, Kuusela P, Ekblom P: Cellular origin of fibronectin in interspecies hybrid kidneys. J Cell Biol 1984, 99:2099-2107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fan JM, Ng YY, Hill PA, Nikolic-Paterson DJ, Mu W, Atkins RC, Lan HY: Transforming growth factor-β regulates tubular epithelial-myofibroblast transdifferentiation in vitro. Kidney Int 1999, 56:1455-1467 [DOI] [PubMed] [Google Scholar]
- 31.Frank S, Madlener M, Werner S: Transforming growth factors β1, β2, and β3 and their receptors are differentially regulated during normal and impaired wound healing. J Biol Chem 1996, 271:10188-10193 [DOI] [PubMed] [Google Scholar]
- 32.Pena JD, Taylor AW, Ricard CS, Vidal I, Hernandez MR: Transforming growth factor β isoforms in human optic nerve heads. Br J Ophthalmol 1999, 83:209-218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu A, Ballermann BJ: TGF-β type II receptor in rat renal vascular development: localization to juxtaglomerular cells. Kidney Int 1998, 53:716-725 [DOI] [PubMed] [Google Scholar]
- 34.Stossel TP: On the crawling of animal cells. Science 1993, 260:1086-1093 [DOI] [PubMed] [Google Scholar]
- 35.Stoker M: Effect of scatter factor on motility of epithelial cells and fibroblasts. J Cell Physiol 1989, 139:565-569 [DOI] [PubMed] [Google Scholar]
- 36.Kaneto H, Ohtani H, Fukuzaki A, Ishidoya S, Takeda A, Ogata Y, Nagura H, Orikasa S: Increased expression of TGF-β1 but not of its receptors contributes to human obstructive nephropathy. Kidney Int 1999, 56:2137-2146 [DOI] [PubMed] [Google Scholar]
- 37.Chevalier RL, Goyal S, Wolstenholme JT, Thornhill BA: Obstructive nephropathy in the neonatal rat is attenuated by epidermal growth factor. Kidney Int 1998, 54:38-47 [DOI] [PubMed] [Google Scholar]
- 38.Liu S, Cieslinski DA, Funke AJ, Humes HD: Transforming growth factor-β 1 regulates the expression of Pax-2, a developmental control gene, in renal tubule cells. Exp Nephrol 1997, 5:295-300 [PubMed] [Google Scholar]
- 39.Rogers SA, Ryan G, Purchio AF, Hammerman MR: Metanephric transforming growth factor-β1 regulates nephrogenesis in vitro. Am J Physiol 1993, 264:F996-F1002 [DOI] [PubMed] [Google Scholar]
- 40.Sakurai H, Barros EJ, Tsukamoto T, Barasch J, Nigam SK: An in vitro tubulogenesis system using cell lines derived from the embryonic kidney shows dependence on multiple soluble growth factors. Proc Natl Acad Sci USA 1997, 94:6279-6284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D: Targeted disruption of the mouse transforming growth factor-β 1 gene results in multifocal inflammatory disease. Nature 1992, 359:693-699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dressler GR, Wilkinson JE, Rothenpieler UW, Patterson LT, Williams-Simons L, Westphal H: Deregulation of Pax-2 expression in transgenic mice generates severe kidney abnormalities. Nature 1993, 362:65-67 [DOI] [PubMed] [Google Scholar]
- 43.Border WA, Noble NA: Transforming growth factor β in tissue fibrosis. N Engl J Med 1994, 331:1286-1292 [DOI] [PubMed] [Google Scholar]
- 44.Ng YY, Huang TP, Yang WC, Chen ZP, Yang AH, Mu W, Nikolic-Paterson DJ, Atkins RC, Lan HY: Tubular epithelial-myofibroblast transdifferentiation in progressive tubulointerstitial fibrosis in 5/6 nephrectomized rats. Kidney Int 1998, 54:864-876 [DOI] [PubMed] [Google Scholar]
- 45.Okada H, Danoff TM, Kalluri R, Neilson EG: Early role of Fsp1 in epithelial-mesenchymal transformation. Am J Physiol 1997, 273:F563-F574 [DOI] [PubMed] [Google Scholar]
- 46.Matsell DG, Bennett T, Armstrong RA, Goodyer P, Goodyer C, Han VK: Insulin-like growth factor (IGF) and IGF binding protein gene expression in multicystic renal dysplasia. J Am Soc Nephrol 1997, 8:85-94 [DOI] [PubMed] [Google Scholar]
- 47.Liapis H, Nag M, Steinhardt G: Effects of experimental ureteral obstruction on platelet-derived growth factor-A and type I procollagen expression in fetal metanephric kidneys. Pediatr Nephrol 1994, 8:548-554 [DOI] [PubMed] [Google Scholar]
- 48.Cale CM, Klein NJ, Winyard PJD, Woolf AS: Inflammatory mediators in human renal dysplasia. Nephrol Dial Transplant 2000, 15:173-183 [DOI] [PubMed] [Google Scholar]
- 49.Border WA, Noble NA, Yamamoto T, Harper JR, Yamaguchi Y, Pierschbacher MD, Ruoslahti E: Natural inhibitor of transforming growth factor-β protects against scarring in experimental kidney disease. Nature 1992, 360:361-364 [DOI] [PubMed] [Google Scholar]
- 50.Tanner GA, McQuillan PF, Maxwell MR, Keck JK, McAteer JA: An in vitro test of the cell stretch-proliferation hypothesis of renal cyst enlargement. J Am Soc Nephrol 1995, 6:1230-1241 [DOI] [PubMed] [Google Scholar]
- 51.Maulbecker CC, Gruss P: The oncogenic potential of Pax genes. EMBO J 1993, 12:2361-2367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Riser BL, Cortes P, Heilig C, Grondin J, Ladson-Wofford S, Patterson D, Narins RG: Cyclic stretching force selectively up-regulates transforming growth factor-β isoforms in cultured rat mesangial cells. Am J Pathol 1996, 148:1915-1923 [PMC free article] [PubMed] [Google Scholar]