

Abstract
Wilms' tumor 1 (WT1) is a transcription factor with a multitude of downstream targets that have wide-ranging effects in non-glioma cell lines. Though its expression in glioblastomas is now well-documented, the role of WT1 in these tumors remains poorly defined. We hypothesized that WT1 functions as an oncogene to enhance glioblastoma viability and chemoresistance. WT1's role was examined by studying the effect of WT1 silencing and overexpression on DNA damage, apoptosis and cell viability. Results indicated that WT1 silencing adversely affected glioblastoma viability, at times, in synergy with 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU) and cisplatin. To investigate other mechanisms through which WT1 could affect viability, we measured cell cycle distribution, senescence, and autophagy. WT1 silencing had no effect on these processes. Lastly, we examined WT1 regulation of IGF-1R expression. Counterintuitively, upregulation of IGF-1R was evident after WT1 silencing. In conclusion, WT1 functions as a survival factor in glioblastomas, possibly through inhibition of IGF-1R expression.
Keywords: Wilms' tumor 1, Glioblastoma, RNA interference, Chemosensitivity, IGF-1R
Introduction
Glioblastomas are one of the most common and deadliest types of brain tumors [1]. Even with the combination of surgery, chemotherapy and radiation therapy, median life expectancy is only 12–14 months [2, 3]. Despite significant gains in the understanding of glioblastoma biology, the prognosis of the disease has not changed significantly over the last 20 years. Wilms' tumor 1 (WT1) was recently identified as a possible oncogene in glioblastomas and numerous other cancers including those arising from the breast and hematopoietic system [4–8]. WT1 is expressed in approximately 80% of glioblastoma specimens and over 50% of malignant astrocytoma cell lines [9]. WT1 is a particularly exciting target for therapy because it can regulate many aspects of tumorigenesis; further, its expression in the normal brain is essentially absent [9–11].
Isolating how WT1 functions as an oncogene has been difficult because WT1 has a bewildering array of over 24 isoforms [12–14]. The four main isoforms are generated by alternative splicing, which causes the inclusion or exclusion of the 17 amino acid sequence coding for exon 5 or the three amino acid KTS region in exon 9 [12]. (From this point on, the different isoforms will be designated by “±17a.a.” in combination with “±KTS.”) The KTS region lies within four Kruppel C2H2 class zinc fingers. The presence of the zinc fingers, reminiscent of those in the DNA binding protein early growth response (EGR), identified WT1 as a transcription factor [15]. As a transcription factor, WT1 regulates over 30 different target genes that mediate diverse cellular activities such as cell cycle control, apoptosis, growth factor signaling and differentiation [16, 17]. Additionally, WT1 has a putative role in splicing [18–20]. Furthermore, WT1 has been co-immunoprecipitated with numerous other proteins including tumor protein p53 (p53), prostate apoptosis response protein 4 (par-4), and tumor protein p73 (p73) [21–23]. Many of these binding partners can be used to modulate WT1's transcriptional activities. In short, WT1 is an extremely complex gene capable of exerting significant influence on cell biology.
The function of WT1 in glioblastomas is currently unknown. Oji et al. [7] reported that application of antisense oligonucleotides targeting WT1 decreased glioblastoma survival. However, one of the cell lines in their study, U87-MG, does not express WT1 according to work performed in our laboratory, therefore, raising the possibility that non-specific antisense effects account for Oji's results [9]. WT1 antisense experiments have been shown to impede proliferation and induce apoptosis in K562 and MM6 myeloid leukemia, pancreatic, gastric AZ-521, lung OS3, ovarian TYK-nu, prostate and multiple breast cancer lines [24–29]. Taken together, these studies strongly suggest that, in glioblastomas, WT1 will behave as an oncogene rather than as a tumor suppressor.
Because WT1 regulates many genes, there are a multitude of mechanisms through which WT1 can diminish cell viability. Several investigators have opined that WT1 influences survival by regulating apoptosis through transcriptional control of B-cell lymphoma 2 (Bcl-2) family members [30–33]. Thus, we initially hypothesized that WT1 regulation of Bcl-2 causes an increase in glioblastoma viability and tolerance to chemotherapeutic agents. Our results indicate that WT1 does increase glioblastoma viability and chemoresistance; however, the underlying mechanism does not involve the Bcl-2 family members that were examined.
In fact, we were not able to demonstrate that WT1 silencing significantly alters cell cycle progression or enhances either type I (apoptosis) or type II (autophagy) programmed cell death. In agreement with studies performed in other cell lines, our results confirm that WT1 is a strong transcriptional repressor of insulin-like growth factor receptor 1 (IGF-1R) in glioblastomas [34–36]. We suggest that WT1 silencing causes IFG-1R overexpression, which is the mechanism of a recently described form of non-apoptotic, non-autophagic programmed cell death termed “paraptosis” [37–41].
Results
WT1 expression results in cell-line dependent increases in chemoresistance
Previous studies performed in our laboratory indicated that only the −17a.a./+KTS and +17a.a./+KTS isoforms of WT1 were expressed in glioblastoma cell lines and patient specimens [9]. We hypothesized that stable expression of −17a.a./+KTS or +17a.a./+KTS WT1 in LN229, LNZ308 and U87MG (all non-WT1 expressing cell lines) would result in increased chemoresistance compared to parental controls. In preliminary screening, expression of −17a.a./+KTS WT1 in the LNZ308 cell line appeared to confer a small survival advantage (Fig. 1a–c). Further analysis of the LNZ308 cell line confirmed those findings (P < 0.05) (Fig. 1d).
Fig. 1.
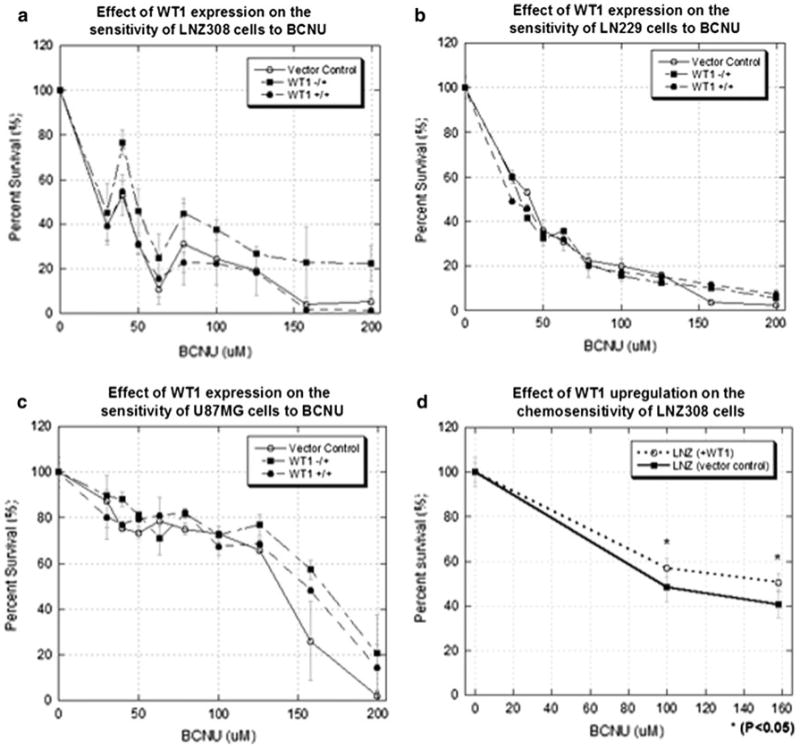
The effect of expression of −17a.a./+KTS and +17a.a./+KTS WT1 isoforms on glioma chemosensitivity to BCNU. ATP assays, performed 5 days after treatment, were used as a surrogate of cell survival. Percent survival was normalized to untreated controls. a WT1 (−17a.a./+KTS) LNZ308 cells were consistently more chemoresistant compared to the other cells, particularly in the 100–200 μM range. b LN229 and c U87MG cells show little or no benefit from WT1 expression. d Expression of −17a.a./+KTS WT1 in LNZ308 cells was investigated further. Additional experiments performed with more replicates confirm a survival benefit at 100 or 150 μM of BCNU
WT1 silencing decreases survival and chemoresistance
The modest survival benefit associated with WT1 expression occurred in only one out of three cell lines. Therefore, RNA interference experiments were performed to test the mirror hypothesis that silencing WT1 would decrease viability. First, we examined the efficacy of our pooled WT1 siRNA in T98G cells. Using scrambled short interfering RNA (siRNA) as a control, WT1 mRNA was decreased by more than 70% from 24 to 168 h after transfection (Fig. 2a). Likewise, WT1 protein levels were significantly decreased after 24 h, and by 96 h WT1 was almost completely absent (Fig. 2b). A lower dose of WT1 siRNA was also examined. Compared to 100 nM, 25 nM of WT1 siRNA had similar efficacy at 24 h, but at 168 h the knockdown was less than 50% (Fig. 2a). Therefore, the 100 nM dose was used for the remainder of this study. The efficacy of WT1 siRNA in the LN18 and VC95G cells lines was similar (data not shown).
Fig. 2.
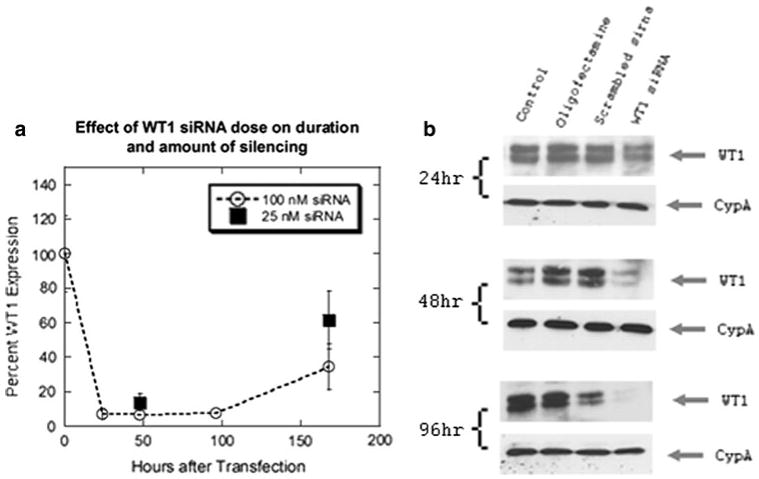
WT1 mRNA and protein silencing induced by siRNA in T98G cells. a This graph depicts the amount of WT1 mRNA expression as a percent of WT1 expression in scrambled controls. The effect of decreasing siRNA dose from 100 to 25 nM is also shown. b Western blot of WT1 at 24, 48 and 96 h reveal that as time progresses, WT1 siRNA caused WT1 levels to decrease significantly compared to untreated, oligofectamine and scrambled siRNA controls. Expression levels of cyclophilin A (CypA) are demonstrated in parallel blots from the same samples as loading controls
Next, we examined the effect on cell survival of WT1 silencing in the T98G, LN18 and VC95G glioblastoma cell lines. In those cell lines, WT1 downregulation alone resulted in decreased viability (P < 0.05) compared to the effect of the scrambled siRNA control (Fig. 3a–c). Tumor cells were then treated with the IC50 dose of 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU) or cisplatin. In all three cell lines, the combination of chemotherapy and WT1 silencing resulted in a further decrease in viability (Fig. 3a–c). Differences were significant (P < 0.05) in all groups, except the VC95G cells that were subjected to cisplatin.
Fig. 3.
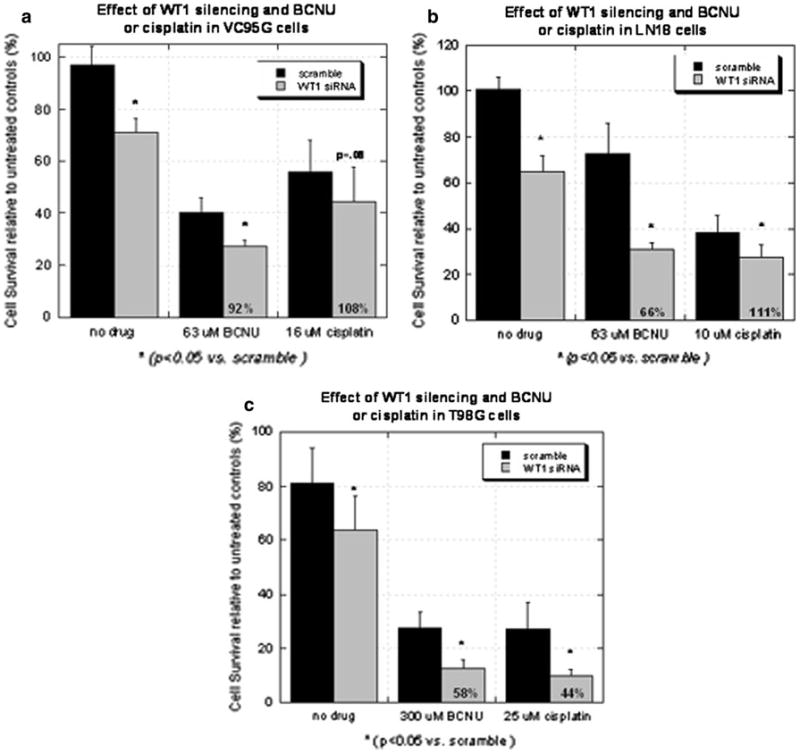
Graphs depicting the effect of WT1 silencing alone or in combination with BCNU or cisplatin in the (a) VC95G, (b) LN18 and (c) T98G cell lines. BCNU and cisplatin data were respectively gathered 3 and 5 days after drug treatment due to differences in drug kinetics. Survival, measured using ATP assays, was normalized to untreated controls. Annotated in each column representing the survival of cells treated with the combination of WT1 siRNA and a chemotherapeutic agent is the actual survival taken as a percentage of the predicted survival. A value less than 70% is suggestive of a synergistic effect
Calculations were then performed to determine if the combined effect of WT1 silencing and the chemotherapeutic agents was additive or synergistic. By definition, synergy occurred when the survival of the combined treatments was less than 70% of survival calculated to occur if toxicity was only additive [8, 42]. Synergy was evident in T98G cells treated with BCNU or cisplatin and in LN18 cells treated with BCNU (Fig. 3).
To validate that WT1 silencing decreased cell viability, and not off-target siRNA effects, the non-WT1 expressing cell line, LNZ308, was treated with WT1 siRNA. There were no significant differences in survival of LNZ308 cells exposed to BCNU with WT1 siRNA or scrambled siRNA (data not shown). Collectively, these experiments indicate that WT1 is a pro-survival factor in glioblastomas and that silencing WT1 has the potential to synergistically enhance the toxicity of chemotherapeutic drugs.
WT1 silencing does not affect chemotherapy-induced DNA damage
We then wanted to determine whether WT1 silencing increases BCNU or cisplatin related DNA damage or alters a subsequent response to the generated death signals. Studies were performed in T98G cells, in which synergy was the most striking. Immunocytochemistry for phospho-53BP1, which binds to regions flanking doublestranded DNA breaks, revealed that silencing of WT1 resulted in no obvious changes in the amount of foci (Fig. 4a–e) [43]. Accordingly, Western blot of phospho-H2AX, a histone involved in DNA repair and damage signaling, showed no change in chemotherapy induced upregulation of phospho-H2AX levels as a result of WT1 silencing (Fig. 4f) [44]. Together, these results indicate that WT1 prevents cell death in response to DNA damage, rather than preventing the DNA damage from occurring in the first place.
Fig. 4.
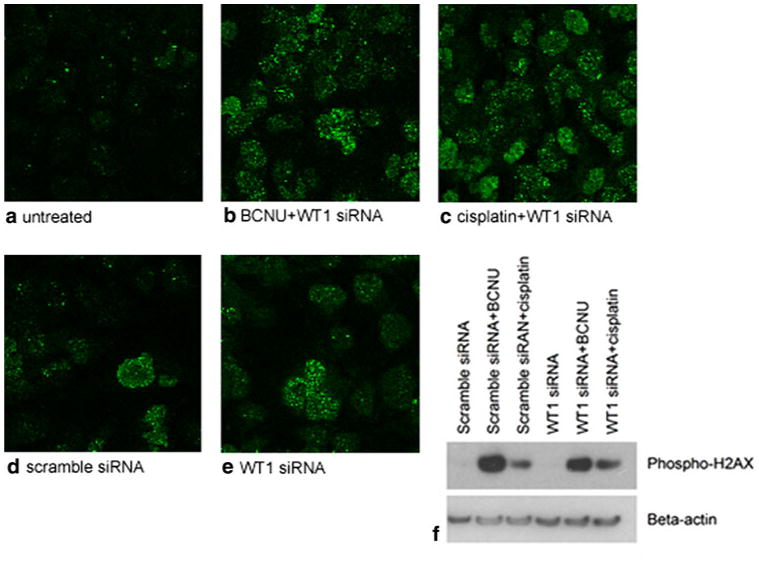
The effect of WT1 silencing in combination with 600 μM BCNU or 40 μM cisplatin on phospho-53BP1 immunostaining and phospho-H2AX protein levels in T98G cells. a Representative confocal images (×63) show minimal phospho-53BP1 staining in untreated cells. Prominent increases in phospho-53BP1 foci are associated with b BCNU and c cisplatin treatment in combination with WT1 silencing. However, differences between d scrambled and e WT1 siRNA groups were not apparent. f Western blot for phospho-H2AX reveals upregulation in response to both chemotherapeutic agents. Cells treated with scrambled or WT1 siRNA possessed similar levels of phospho-H2AX. Experiments were performed in triplicate. Expression levels of beta-actin are demonstrated in parallel blots from the same samples as loading controls
WT1 silencing has no effect on apoptosis
WT1 has been hypothesized to alter viability and chemoresistance through control of apoptosis via transcriptional regulation of Bcl-2 family members [30–32]. However, this mechanism does not appear to be active in our cell lines. WT1 silencing did not detectably change mRNA levels of Bcl-2, Bcl-Xl and Bad in the three WT1 expressing glioblastoma cell lines (Fig. 5a).
Fig. 5.
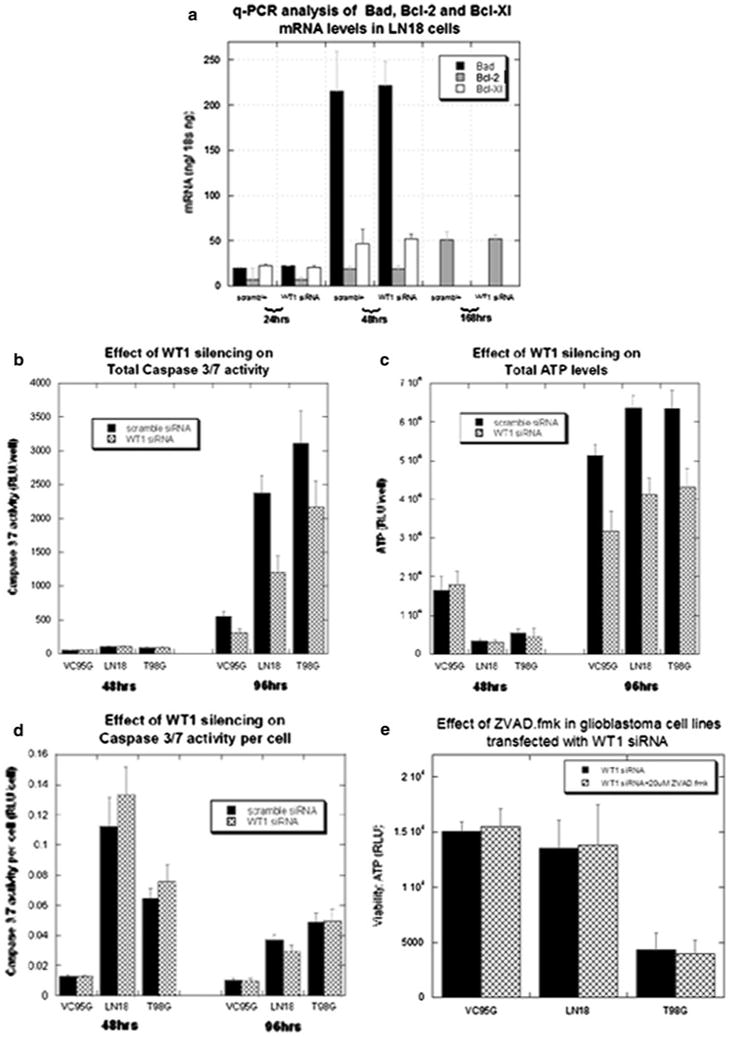
The effect of WT1 silencing on apoptosis. a Quantitative RT–PCR measurement of mRNA levels of Bcl-2, Bcl-Xl and Bad at several time points did not reveal significant differences between the effects of scrambled and WT1 siRNA in the VC95G, LN18 and T98G cell lines. The most comprehensive analysis was performed in the LN18 cell line and shown here. b Caspase 3/7 levels were assayed 2 and 4 days after treatment. To account for toxic effects which influenced cell viability, caspase 3/7 activity was normalized to cell number with parallel ATP assays (c). The ATP assays were correlated to cell counts (data not shown). d Caspase 3/7 activity per cell was unaffected by WT1 downregulation, but was significantly increased by BCNU and cisplatin co-treatment. e Caspase inhibition with ZVAD.fmk did not alter the viability of cells treated with WT1 siRNA 4 days after transfection
We also examined the activity of caspases 3 and 7, which are effectors of the apoptotic pathway. Four days after treatment, total caspase 3/7 activity was significantly (P < 0.05) decreased in the WT1 siRNA group (Fig. 5b). However, this difference reflected the number of viable cells in each well, as reflected by ATP content utilizing a chemiluminescent assay (Fig. 5c). When normalized to cell count, caspase 3/7 activity per cell was not increased after 2 or 4 days of WT1 silencing in T98G, LN18 or VC95G cells (Fig. 5d). Correspondingly, there was no effect on survival when cells were pre-incubated with the pan-caspase inhibitor carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone (ZVAD.fmk) (Fig. 5e). Increasing dosage from 20 μM to either 40 or 60 μM of ZVAD.fmk in the LN18 cell line also had no effect on survival in WT1 silenced cells (data not shown).
Similarly, in T98G cells, the amount of DNA fragmentation, assessed using terminal deoxynucleotidyltransferase dUTP nick end labeling (TUNEL) analysis and flow cytometry, was not significantly different in cells treated with scrambled or WT1 siRNA (Fig. 6 b–c). Interestingly, despite the synergistic toxicity, WT1 silencing did not increase the amount of apoptosis caused by BCNU or cisplatin (Fig. 6 d, e). Taken together, these experiments indicate that WT1 silencing in glioblastoma cells does not increase apoptosis.
Fig. 6.
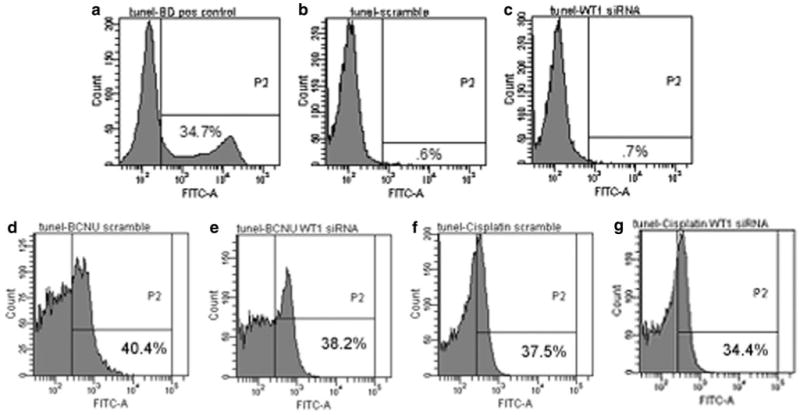
Representative flow cytometry experiments which indicate the amount of TUNEL staining in lymphoma and T98G cells. a The lymphoma cells were used as a positive control and show a classic second peak indicative of DNA fragmentation. Negative staining controls were performed without terminal deoxynucleotidyltransferase and used to set thresholds (<1%). Four days after transfection, T98G cells treated with (b) scrambled or (c) WT1 siRNA showed no significant increase in TUNEL staining. d–g Although treatment with BCNU and cisplatin alone resulted in DNA fragmentation, additional DNA fragmentation was not noted with WT1 downregulation. The percentage of cells with increased FITC-dUTP signaling is annotated in the lower right hand quadrant
WT1 silencing has no effect on autophagy
Autophagy has recently been identified as a type of programmed cell death that commonly occurs in response to a wide variety of stimuli in glioblastomas [45–51]. Because apoptosis does not account for the additional cytotoxicity due to WT1 silencing, we examined the influence of WT1 on autophagy. Protein levels of beclin 1, an autophagy associated protein, were unaffected by WT1 siRNA in the T98G cell line 4 days after transfection (data not shown). Likewise, acridine orange staining in VC95G, LN18 and T98G cells for characteristic acidic vesicular organelles, visualized with fluorescent microscopy, demonstrated no obvious effects from WT1 downregulation (Fig. 7a, b). To quantify acridine orange staining, flow cytometry was performed in the T98G cell line. Again, differences between WT1 siRNA and scrambled siRNA groups were not apparent; although significant staining was evident in groups treated with BCNU or cisplatin (Fig. 7c). These results indicate that WT1 silencing has no effect on autophagy.
Fig. 7.
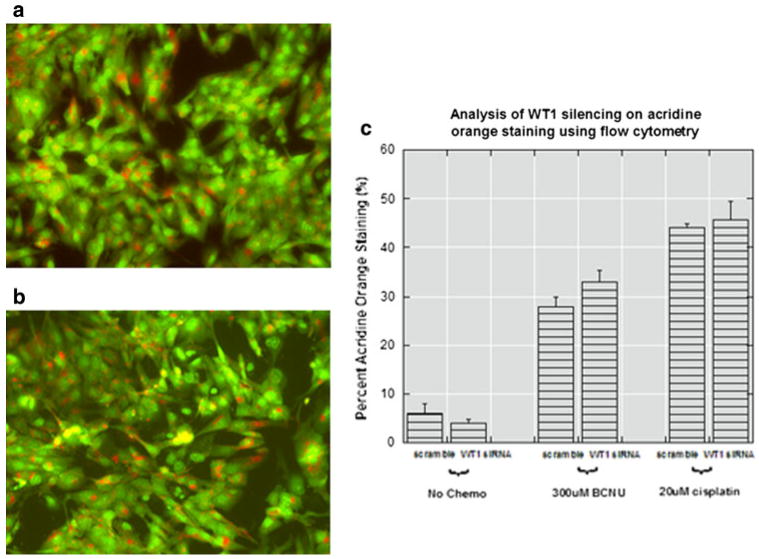
Effect of WT1 silencing on autophagy. Representative fluorescent microscopy images (×20) of acridine orange staining in T98G cells treated with (a) scrambled or (b) WT1 siRNA. The number of orange autophagosomes in each group is approximately the same by visual inspection so flow cytometry was used for quantification. c Flow cytometry confirmed that with or without chemotherapy, silencing WT1 did not significantly alter the amount of red fluorescence per cell. The Q2 quadrant represents cells containing red (acidic vesicular organelles) and green (DNA) fluorescence
WT1 silencing has no effect on the cell cycle and senescence
Deceleration of the cell cycle from WT1 downregulation, which occurs in breast cancer cell lines, could have caused decreased proliferation in our study resulting in diminished cell counts [26, 27]. In LN18 and VC95G cells, flow cytometry analysis of propidium iodide staining revealed that WT1 downregulation caused no significant differences in the percentage of cells in the G0/G1, G2 or S phases 4 days after transfection (data not shown). However, in the T98G cell line, WT1 silencing after 4 days significantly decreased the distribution of cells in the G0G1 phase from 86.7 ± 1.0% to 79.2 ± 0.5%; increased the distribution of cells in the G2 phase from 4.5 ± 0.7% to 6.9 ± 0.3%; and increased the distribution of cells in the S-phase from 8.8 ± 0.3% to 13.8 ± 0.8% (P < 0.05) (Fig. 8a, b). To further test this finding, we examined the effect of WT1 silencing in T98G cells 2 days after transfection and found that: the percent of cells decreased from 76.1 ± 0.6% to 72.9 ± 0.4% in the G0G1 phase (P < 0.05); increased from 8.1 ± 0.3% to 10.2 ± 0.4% in the G2 phase (P < 0.05); and increased from 15.8 ± 0.6% to 16.9 ± 0.3% in the S-phase (P > 0.05).
Fig. 8.
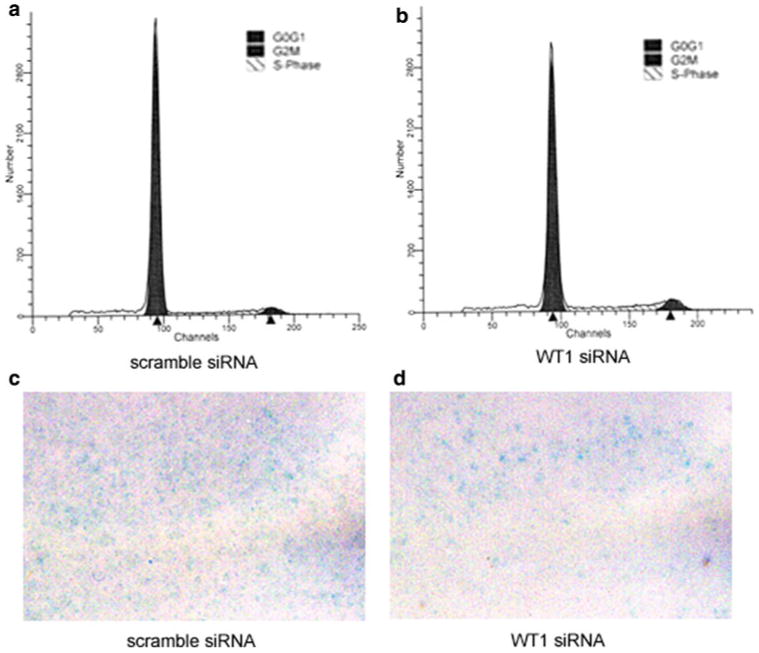
An examination of the effects of WT1 silencing on the cell cycle and senescence. Representative data from flow cytometry experiments for cell cycle analysis show that at posttreatment day 4 scrambled siRNA controls (a) when compared to WT1 silenced T98G cells (b) demonstrated small but statistically significant changes in the percent of cells in the G0G1, G2 and S phases. Representative images of β-gal staining in T98G cells treated with scrambled (c) or WT1 siRNA (d) did not suggest a difference in the number of senescent cells. Note that images were captured from the center of the well where the majority of β-gal staining occurred
The cell cycle effects described above occurred in only one of three cell lines and unexpectedly indicated an acceleration of the cell cycle in association with WT1 silencing. We then questioned whether WT1 could influence chemotoxicity by increasing the proportion of tumor cells undergoing senescence. Our data indicate that the amount of senescence in VC95G, LN18 (data not shown) and T98G cells, determined by β-gal staining, was not noticeably altered by WT1 downregulation (Fig. 8c, d).
WT1 silencing and IGF-1R expression
WT1 is a known repressor of IGF-1R [34–36], the overexpression of which has been associated with non apoptotic cell death [37–41]. We investigated using Western blot the effect of WT1 silencing on IGF-1R levels (Fig. 9a). Our results show that IGF-1R levels were markedly higher in T98G cells treated with WT1 siRNA compared to untreated and scrambled siRNA controls. Light microscopy reveals clear morphological effects of WT1 siRNA treatment (Fig. 9b, c). When WT1 is downregulated, cytoplasmic pleomorphism and vacuolization is prominent.
Fig. 9.
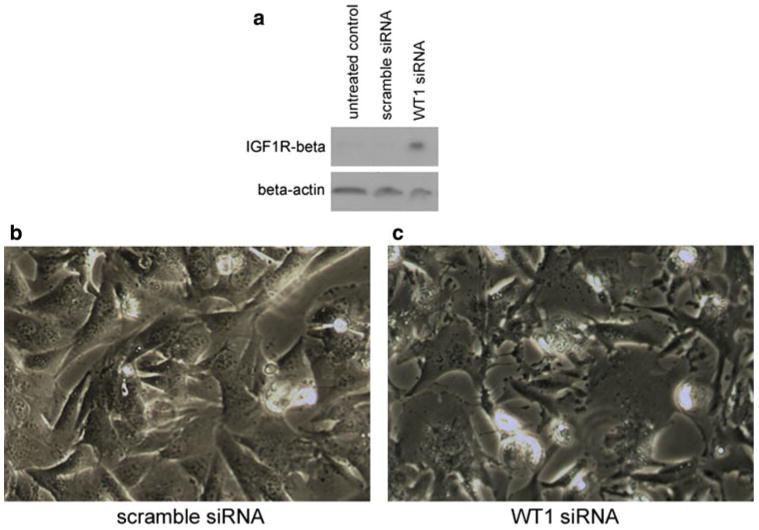
The effect of WT1 silencing on IGF-1R expression and cell morphology. Western blot of protein extracts from T98G cells showed that WT1 silencing markedly upregulated the expression of the IGF-1R beta subunit (a). Blots were performed in triplicate. Expression levels of beta-actin are demonstrated in parallel blots from the same samples as loading controls. Visualization using inverted light microscopy (×20) revealed that the morphology of cells treated with scrambled siRNA (b) is much more normal than T98G cells treated with WT1 siRNA (c)
Discussion
WT1, a gene expressed in a variety of cancers, is interesting in many aspects. However, defining the role of WT1 in glioblastomas is particularly exciting for two reasons. The first is that as a promiscuous transcription factor, WT1 can potentially act as a master switch in tumorigenesis, as it has already been suggested to be in embryogenesis [52]. The second is that WT1 is expressed in a very high frequency in glioblastomas, and is essentially absent in the adult brain [9]. Together, these characteristics make WT1 a potential target for inhibitory or immunologically based therapies. In fact, a phase II WT1-based immunotherapy trial has already been performed in 21 glioblastoma patients in Japan [53]. However, the basic function of WT1 in glioblastoma biology has not been examined in detail up to this point. This study aimed to further examine the role of WT1 in glioblastomas and to obtain an idea of the mechanism(s) though which it acts.
First, we expressed WT1 in glioblastoma cell lines that do not normally express WT1. One out of the three cell lines showed a modest, but statistically significant increase in chemoresistance. There are several possible interpretations of these findings. One interpretation is that the −17a.a./+KTS and +17a.a./+KTS WT1 isoforms, which were overexpressed, do not play a significant role in glioblastoma biology. Caricasole et al. [19] supplied evidence that supports this line of reasoning. They showed that +KTS isoforms have a greater affinity for RNA than DNA and may be preferentially involved in post-transcriptional events unlike −KTS isoforms, which reportedly have more of a role in transcriptional regulation [19]. The natural assumption here is that WT1's splicing activities do not have as much of an impact on chemoresistance as its transcriptional activities.
Yet, there is a basis to believe that the modest effect of WT1 overexpression is misleading and that WT1 activity could significantly affect glioblastoma biology. One speculation is that the conditions of cell culture actually provide redundancy of activation of some of parallel or alternative pathways for cells which normally do not rely on WT1 for survival. Therefore, adding another pro-survival factor may not create much of an impact. Another explanation is that the two cell lines, which did not respond, may not have possessed the proper milieu for WT1 to function. Studies have consistently documented that WT1's behavior varies in different cell lines [30, 54–59]. Why WT1 acts in this manner is unknown, but a guess is that complex interplay with other genes and its numerous protein binding partners may play a role in modulating WT1's activity.
To further investigate the significance of WT1 expression in glioblastomas, we performed WT1 silencing experiments. Downregulation of WT1 decreased cell survival and chemoresistance to BCNU and cisplatin. That a WT1 antisense strategy caused these effects agrees with Oji's aforementioned findings in glioblastomas and the results of investigators in other cancer cell lines [7, 24–29]. Importantly, the present study confirmed that, in glioblastomas, WT1 acts as an oncogene rather than a tumor suppressor.
It is also interesting to note that the p53 status of the cells (T98G and LN18 cell lines have mutant p53, whereas, the VC95G cell line possesses wild-type p53) did not seem to have an effect, even though WT1 and p53 have been shown to modulate each other's activities [21, 35, 60, 61]. However, this observation does correlate with our previously published results [9]. Apparently, in glioblastomas, p53 does not influence WT1 activity to a significant extent.
Our data also indicates that silencing WT1 was potentially synergistic with the chemotherapeutic agents BCNU and cisplatin. Synergism varied by cell line and agent. The highest amount of synergy was seen in the T98G cell line, while the least was witnessed in the VC95G cell line. Perhaps, the difference was caused by the amount of WT1 in those respective cell lines. In T98G cells, WT1 is expressed in moderate levels; whereas in VC95G cells, WT1 expression is an order of magnitude lower [9]. Nevertheless, synergy was possible and its presence suggests the pathways influenced by WT1 and those responsible for cell death caused by BCNU and cisplatin likely intersect.
The results of this study also suggested that this point of intersection between the WT1 pathway and the effect of the chemotherapeutic drugs is after the occurrence of the DNA damage. The amount of DNA damage caused by BCNU or cisplatin, reflected by levels of phospho-53BP1 and phospho-H2AX, was equal despite WT1 silencing. Thus, it can be inferred that WT1 modulation alters glioblastoma response to these signals. This notion is further supported by Shahrabani-Gargir's observation that ATM (ataxia-telangiectasia mutated gene) utilizes WT1 to mediate cellular response to DNA damage [62].
The mechanism(s) through which WT1 influences glioblastoma cell death have to our knowledge never been investigated. Therefore, a survey of the diverse cellular activities associated with WT1's many transcriptional targets was performed. As alluded to before, WT1 has been shown to influence apoptosis in other cancer types through a variety of mechanisms which include the regulation of Bcl-2 family members and several growth/proliferation factors (EGFR, IGF2, PDGF-A, amphiregulin, and c-myc) [21, 30, 55, 56, 59, 63–67]. Contrary to those results, our data revealed that WT1 silencing in glioblastoma cell lines did not affect typical indicators of apoptosis. Alteration in Bad, Bcl-Xl and particularly Bcl-2 levels was not seen. Caspase 3/7 activity did not increase. There was no response to the pan-caspase inhibitor ZVAD.fmk. TUNEL staining was unchanged. Taken together, these findings suggest that WT1 downregulation decreases cell viability through a mechanism other than apoptosis.
Prior studies have suggested that in glioblastomas autophagy prevents apoptosis by, almost paradoxically, either inhibiting apoptosis as a means of survival or supplanting it as an alternative programmed cell death mechanism [48]. In glioblastomas autophagy occurs in response to a wide variety of stimuli including: ceramide, radiation, adenovirus, mutated Ras, arsenic trioxide, etoposide, temozolomide, BCNU, cisplatin and rapamycin [45–50, 68, 69]. However, in our experiments, silencing WT1 did not increase autophagy.
The decrease in cell number caused by WT1 silencing may have also occurred as a result of deceleration of the cell cycle. In two separate experiments performed in breast cancer cell lines, investigators noted that the use of WT1 antisense oligodeoxynucleotides resulted in decreased levels of cyclin D1 and a G1 block [26, 27]. However, based on our results, WT1 silencing affected only one of three cell lines and had no influence on senescence. In the T98G cell line, silencing WT1 caused a modest increase in cell cycle progression, which would lead to increased and not decreased cell number as observed. WT1 is certainly capable of slowing down the cell cycle. Loeb et al. [70] demonstrated that WT1 represses cyclin E. Englert et al. [71] and Wang et al. [65] documented that WT1 upregulated p21Cip1, a cyclin-dependent kinase inhibitor. One plausible explanation to resolve this seeming inconsistency is that silencing WT1 caused cell death in excess of its proliferative cell cycle effects in the T98G cell line.
Our results also indicated that WT1 silencing causes IGF-1R overexpression. This finding corresponds with those of other investigators who have noted that WT1 repressed IGF-1R expression in P69 prostate, G401 renal and Saos-2 osteosarcoma cell lines [34–36, 72]. However, IGF-1R is usually regarded as an anti-apoptotic and proliferative factor; therefore, it is difficult to conceive of IGF-1R overexpression as a cause of the decreased viability seen in our experiments.
Yet, there is evidence to suggest that this scenario is not so far-fetched. Hondo et al. [73] showed that the COOH terminus of IGF-1R induced cell death in the T98G cell line. Furthermore, Plymate et al. [74, 75] demonstrated that IGF-1R overexpression in malignant prostate cell lines caused the same effects. Additionally, Sperandio et al.'s [39, 40] experiments revealed that IGF-1R overexpression in 293T cells and Apaf-1 null mouse embryonic fibroblasts caused a previously undescribed form of cell death that they termed “paraptosis.” Interestingly, cell death secondary to WT1 silencing shared similarities with paraptosis—caspase 3 activity was not increased, ZVAD.fmk did not inhibit cell death, TUNEL staining was negative, and autophagic vacuoles were not present [39]. Additionally, paraptosis has been shown to occur in gliomas in response to macrophage colony-stimulating factor [37, 38].
In conclusion, we showed that WT1 overexpression can increase cell viability and chemosensitivity, and that WT1 silencing has the opposite effect. This confirms that WT1 acts as an oncogene in glioblastomas. Furthermore, our data revealed that the decreased viability resulting from WT1 downregulation was not caused by apoptosis, autophagy, senescence or cell cycle effects. Lastly, we demonstrated an association between WT1 silencing and IGF-1R overexpression that could partially account for WT1's oncogenic behavior. Future studies will need to be performed to fully assess the consequences of WT1 expression in gliomas and to further examine the involvement of IGF-1R overexpression in cell death.
Methods
Cell culture
The T98G, LN18, LNZ308, LN229 and U87MG human glioblastoma cell lines were purchased from the American Type Culture Collection (Manassas, VA). The VC95G cell line, previously used by Van Meter et al. [76] was established from a patient with a type IV astrocytoma. Immunohistochemistry was performed to confirm positive staining for glial fibrillary acidic protein(GFAP). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM), which was supplemented with 10% fetal bovine serum, 4 mM glutamine, 1% non-essential amino acids, and 1% penicillin-streptomycin (Invitrogen, Carlsbad, CA). Cells transfected with the WT1 pcDNA3.1 plasmid were additionally selected with geneticin (Invitrogen). Cells were grown in 6 or 96 well plates in a humidified atmosphere of 5% CO2 at 37.8°C and, for certain experiments, number 1.5 × 10 mm coverslips (Fisher, Pittsburgh, PA) were placed in the wells of 6 well plates. Operating room specimens were obtained with approval of the Virginia Commonwealth University Hospitals internal review board.
Small interfering RNA preparation and transfection
A pool of four small interfering RNAs, purchased from Dharmacon Research, Inc. (Si-Genome, Lafayette, CO), was used to silence WT1. As a control, we used a pool of two, non-targeting, scrambled siRNAs (Dharmacon). All siRNAs were pre-designed by the manufacturer. Cells were plated overnight as described in 6 or 96 well plates. SiRNA was transfected per the manufacturer's protocol. Briefly, 24 h after plating, culture media was removed and replaced with optimem (Invitrogen). A mixture of siRNA, optimem and oligofectamine (Invitrogen) was then added to achieve a final siRNA concentration of 100 ηM. After 4 h of incubation, the cells were supplemented with 30% heat-inactivated fetal bovine serum diluted in optimem. SiRNA concentrations were chosen based on dose–response studies.
Plasmid construction and generation of stable transfectants
WT1 +17a.a./+KTS or WT1 −17a.a./+KTS expression plasmids (pcDNA3.1, Invitrogen) were provided by Dr. Charles T. Roberts, Jr. and have been described previously [77, 78]. LNZ308, LN229 and U87MG cells were plated at a density of 2 × 105 in six well plates and allowed to attach overnight in DMEM without penicillin/streptomycin. Cells were transfected with a mixture of 3 μl Lipofectamine 2000 (Invitrogen) and 1.0 μg of WT1 pcDNA3.1 plasmid diluted in 500 μl DMEM without fetal bovine serum or penicillin-streptomycin and incubated for 4 h at 37°C. After 4 h, cells were supplemented with 1.5 ml DMEM with 10% fetal bovine serum without penicillin-streptomycin. Twenty-four hours after transfection, the conditioned media was replaced with DMEM with 10% fetal bovine serum without antibiotics. Forty-eight hours after transfection, cells were selected in DMEM containing 600 μg/ml Geneticin (Invitrogen) for LN229, 200 μg/ml for U87MG and 300 μg/ml for LNZ308.
Treatment with BCNU and cisplatin
Dose–response studies (data not shown) were performed in each cell line to identify the IC50 concentrations of 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU; Sigma–Aldrich, St. Louis, MO) and cis-dichlorodiammine platinum II (cisplatin; Sigma–Aldrich). Chemotherapy treatments, when used, were generally given to tumor cells 24 h after siRNA treatment. BCNU was dissolved in ethanol and cisplatin was dissolved in DMSO. Final concentrations of ethanol and DMSO were respectively less than 0.5 or 0.1%. These solvent concentrations were determined to have no effect on viability (data not shown).
Assays to measure cytotoxicity and viability
Effects of manipulations, such as treatment with chemotherapeutic drugs or siRNA on the viability of cells, were assessed with the Cell-titer Glo kit (Promega, Madison, WI), an assay that measures the amount of ATP. Briefly, 2 × 104 cells were plated in 96 well plates and then treated with siRNA on day 1 and, for selected experiments, treated with chemotherapeutic drugs on day 2. For caspase inhibition experiments, 20 μM carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone (ZVAD.fmk, Calbiochem, San Diego, CA) was applied 30 min prior to the putative cellular insult. After a period of 4–7 days, ATP assays were performed per the manufacturer's instructions. Fluorescence was quantified using a Fluostar plate reader (BMG Labtech, Offenburg, Germany). In an additional experiment (data not shown), the relationship between ATP and cell count was found to be highly linear (r2 = 0.98) and constant despite treatment with siRNA. All experiments were performed in replicates of at least nine.
RNA extraction and processing
RNA was isolated from plated cells using the RNeasy mini Kit (Qiagen, Hilden, Germany) per the manufacturer's instructions. The isolate was sent for quantitative reverse transcriptase PCR analysis at the Virginia Commonwealth University's nucleic acids facility. In brief, experiments were performed in the ABI Prism 7900 Sequence Detection System (Applied Biosystems, Foster City, CA) using the TaqMan One Step PCR Master Mix Reagents Kit (Applied Biosystems). Samples were tested in triplicate under the conditions recommended by the fabricant. The cycling conditions were: 48°C/30 min; 95°C/10 min; and 40 cycles of 95°C/15 s and 60°C/1 min. The cycle threshold was determined to provide the optimal standard curve values. The probes (5′FAM, 3′TAMRA) and primers were designed using the Primer Express version 2.0. Ribosomal 18S RNA processed using TaqMan pre-developed assay reagents (Applied Biosystems) was used as endogenous control. For WT1, the respective sequences of the forward primer, reverse primer and TaqMan probe were “AGGACTGTGA ACGAAGGTTTTCTC”, “GACAAGTTTTACACTGGA ATGGTTTC”, and “CACCTGTATGTCTCCTTTGGTGT CTTTTGAGCT”. For Bcl-2, the respective sequences of the forward primer, reverse primer and TaqMan probe were “CATGTGTGTGGAGAGCGTCAA”, “GCCGGTTCAG GTACTCAGTCAT”, and “CCTGGTGGACAACATCGC CCTGT”. For Bcl-Xl, the respective sequences of the forward primer, reverse primer and TaqMan probe were “AAT GACCACCTAGAGCCTTGG”, “GCCGGTTCAGGTACTC AGTCAT”, and “CCTGGTGGACAACATCGCCCTGT”. Lastly, for Bad the respective sequences of the forward primer, reverse primer and TaqMan probe were “GGATG AGTGACGAGTTTGTGGAC”, “TCTGCGTTGCTGTGC CC”, and “CTTTAAGAAGGGACTTCCTCGCCCGAAG”.
Protein isolation and Western blot
WT1, IGF1R, H2AX, Beclin 1, Bax and Bak were evaluated by Western blot analysis. Depending on the experiment, protein was extracted from 1 to 7 days after siRNA treatment. A lysis buffer consisting of 50 mM Tris–Cl (ph 6.8), 1% sodium dodecyl sulfate (SDS), 10% glycerol, and 100 ul/10 ml of protease inhibitor (Calbiochem) was applied to trypsinized tumor cells. Concentrations were determined using the Bio-Rad protein assay (Hercules, CA) on a Fluostar plate reader (BMG Labtech).
Proteins were resolved using NuPAGE 4–12% Bis–Tris mini-gels (Invitrogen), except in the case of WT1 immunoblotting, for which SDS–PAGE was performed. After separation, protein was transferred electrophoretically to a 0.2 μm nitrocellulose membrane (Invitrogen) and blocked in 5% nonfat milk. The blots were incubated with primary antibodies overnight at 4°C. Antibodies were applied using the following dilutions: 1:1,000 mouse anti-phospho-H2AX (Millipore/Upstate, Billerica, MA), 1:5,000 mouse anti–β-actin (Sigma–Aldrich), 1:1,000 rabbit anti-beclin 1 (Santa Cruz, Santa Cruz, CA), 1:2,000 rabbit anti-cyclophilin A (Millipore/Upstate, Billerica, MA), rabbit anti-IGF1R-beta (Cell Signaling Technology, Danvers, MA) and 1:200 mouse anti-WT1 antibodies (Dako, Carpinteria, CA). Subsequently, the blots were washed six times in Tris buffered saline containing 0.05% Tween-20 before and after 1 h of incubation at room temperature with horseradish peroxidase-conjugated secondary antibodies. Enzyme-linked chemiluminescence was performed according to the manufacturer's protocol (Amersham, Buckinghamshire, England). Blots were performed in triplicate. Cyclophilin A and beta-actin immunoblots were used to control for protein loading due molecular weight considerations, and were found to vary coordinately in the studies carried out (data not shown).
Terminal deoxynucleotidyltransferase dUTP nick end labeling (TUNEL)
The Apo-Direct assay (BD Biosciences, Rockville, MD) was used to detect apoptosis in the treated tumor cells according to the manufacturer's instructions. Briefly, the 3′ hydroxyl ends of fragmented DNA were labeled in a single-step reaction with a FITC-deoxyuridine triphosphates (dUTP) conjugate. Incorporation of the fluorescent conjugate was detected using a BD FACS Canto flow cytometer (BD Biosciences). Forward and side scatter was adjusted to omit cellular debris and 10,000 events were analyzed in the Fl2 channel.
Cell cycle analysis
For cell cycle analysis, tumor cells were detached with trypsin-EDTA, washed and collected. Cells were then stained overnight in a cold propidium iodide staining solution consisting of 3.8 × 103 M Na citrate, 0.05 mg/ml propidium iodide, 0.11% triton X-100 and 7 Kunits/ml RNase B (Sigma–Aldrich). DNA content was analyzed with a FACScan flow cytometer. Data were analyzed using Cell Quest (Becton–Dickinson, San Jose, CA, USA) and ModFit LT (Verity, Topsham, ME) software packages.
Senescence-associated beta-galactosidase staining
To examine senescence, beta-galactosidase (ß-Gal) staining was performed using 5-bromo-4-chloro-3-indoly A-D-galactopyranoside (X-Gal; Gold Biotechnology, Inc., St. Louis, MO). Cells were seeded at a density of 1 × 105 cells/well in 6 well plates. As described, on days 1 and 2 after plating, the cells were subjected to siRNA treatments with or without BCNU and cisplatin. Fixation was performed with 3% paraformaldehyde on day 5 at room temperature for 30 min. After a PBS wash, cells were incubated overnight at 37.8°C without CO2 in staining solution consisting of 1 mg/ml X-Gal, 40 mM citric acid/sodium phosphate buffer (pH = 6.0), 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 2 mM MgCl2, and 150 mM NaCl.
Caspase 3/7 activation
Activity of effector caspases 3 and 7 were measured using the Caspase Glo 3/7 assay (Promega). Tumor cells were seeded on 96 well plates and then treated with siRNA with or without the IC50 dose of BCNU and cisplatin. Luminescence resulting from the interaction of caspase 3/7 with Z-DEVD-aminoluciferin was determined per the manufacturer's instructions. A Fluostar plate reader (BMG Labtech) was used to quantify fluorescence.
To account for the effect of BCNU and cisplatin on cell viability, caspase 3/7 activity was normalized to cell number. Cell number was calculated in a two-step manner. First, we determined that the ATP (RLU)/cell (3.96 RLU/cell) was not significantly different in the treated versus control groups (data not shown). Then, we determined the amount of ATP in the wells by concurrently performing ATP and caspase 3/7 assays. Thus, the normalized caspase activity was calculated by the formula: caspase/(ATP/3.96).
Detection and quantification of acidic vesicular organelles with acridine orange
Autophagy is characterized by the sequestration of cytoplasmic components into acidic vesicular organelles (AVOs) that fluoresce bright red when stained with acridine orange (Sigma–Aldrich) [79, 80]. To detect acridine orange staining, AVOs were visualized by epifluorescent microscopy and quantified with flow cytometry. Acridine orange staining (1 μg/ml for 15 min) was conducted 3 days after treatment with siRNA and the chemotherapeutic agents. For microscopy, coverslips were wet-mounted on glass slides without fixation. Fluorescent pictures were obtained with a Nikon Eclipse E800 M (Tokyo, Japan) and recorded using the Spot version 3.5.6 software package (Diagnostic Instruments, Sterling Heights, MI). For flow cytometry, cells were washed in PBS, detached with 0.05% trypsin–EDTA (Invitrogen), collected and centrifuged, and finally, resuspended in PBS. Analysis was performed on a BD FACSCanto flow cytometer (BD Biosciences, San Jose, CA) using the BDFACS Diva software package (BD Biosciences). Cells underwent excitation with blue (488 nm) laser and the resultant green (Fl2 channel) and red (Fl4 channel) emission data of 104 cells were analyzed. Cells that were double positive were considered to be autophagic. Thresholds were set so that 1–2% of negative control cells were positive.
Phospho-53BP1 immunofluorescence staining
After treatment with WT1 siRNA, tumor cells were cultured for 3 days on sterile 10 mm coverslips placed in a 6 well plate. The samples were then fixed with 4% paraformaldehyde, permeabilized with 0.25% Triton × 100 in PBS, and blocked with 1:10 Donkey serum in 0.01% Triton ×100/PBS. Staining was performed by overnight incubation of the phospho-53BP1 primary antibody (Cell Signaling Technology) at a dilution of 1:400, followed with 0.01% Triton ×100/PBS washes, and overnight incubation of the secondary antibody, goat anti-rabbit IgG conjugated with Alexa 488 fluorochromes (Molecular Probe), at a dilution of 1:200. Coverslips were then mounted on slides using the Prolong antifade kit (Molecular Probe) and examined on a Leica DM IRE2 confocal microscope using the Leica confocal software package (Heidelberg, Germany).
Synergy calculations
Using a procedure described by Hata et al. [42] and Zhang et al. [8] synergy was determined by comparing predicted versus actual survival values. Predicted survival (c) was calculated from the equation c = (a × b)/100, where a and b represent survival values for a single agent. Drug interactions were considered synergistic if the actual survival values were less than 70% of the predicted survival values.
Statistical analysis
The data were expressed as means ± the standard deviation. Microsoft Excel 2002 (Redmond, WA) and Analyze-It (Leeds, England) programs were used to perform two-tailed T-test or one-way analysis of variance. If analysis of variance indicated a significant difference amongst means, post-hoc analysis was executed with the Bonferroni correction. A P-value of equal or less than 0.05 was deemed statistically significant.
Acknowledgments
We were granted invaluable assistance on this project from a number of people, for which we are deeply appreciative. Specifically, we would like to thank Dr. Lynne Elmore for her advice on immunoblotting and immunofluorescence of H2AX and 53BP1. We are also indebted to Frances White and Julie Farnsworth at the VCU Flow Cytometry Core Facility, supported in part by NIH Grant P30 CA16058, for their many hours that they committed to this project. Further, the help of Dr. Scott Henderson was critical to the epifluorescent and confocal microscopy that were both performed at the VCU Department of Neurobiology & Anatomy Microscopy Facility, supported, in part, with funding from NIH-NINDS Center core grant 5P30NS047463. This work was also supported by the F. Norton Hord, Jr. fund of the Medical College of Virginia Foundation.
Contributor Information
Mike Y. Chen, Department of Neurosurgery, Virginia Commonwealth University, Medical College of Virginia Hospitals, Virginia Commonwealth University Health System, P.O. Box 980631, Richmond, VA 23298-0631, USA; Department of Anatomy and Neurobiology, Virginia Commonwealth University Hospitals, Richmond, VA 23298, USA
Aaron J. Clark, Department of Neurosurgery, Virginia Commonwealth University, Medical College of Virginia Hospitals, Virginia Commonwealth University Health System, P.O. Box 980631, Richmond, VA 23298-0631, USA; Department of Anatomy and Neurobiology, Virginia Commonwealth University Hospitals, Richmond, VA 23298, USA
Dana C. Chan, Department of Neurosurgery, Virginia Commonwealth University, Medical College of Virginia Hospitals, Virginia Commonwealth University Health System, P.O. Box 980631, Richmond, VA 23298-0631, USA; Department of Anatomy and Neurobiology, Virginia Commonwealth University Hospitals, Richmond, VA 23298, USA
Joy L. Ware, Department of Human Genetics and Pathology, Virginia Commonwealth University Hospitals, Richmond, VA 23298, USA
Shawn E. Holt, Department of Human Genetics and Pathology, Virginia Commonwealth University Hospitals, Richmond, VA 23298, USA
Archana Chidambaram, Department of Anatomy and Neurobiology, Virginia Commonwealth University Hospitals, Richmond, VA 23298, USA.
Helen L. Fillmore, Department of Neurosurgery, Virginia Commonwealth University, Medical College of Virginia Hospitals, Virginia Commonwealth University Health System, P.O. Box 980631, Richmond, VA 23298-0631, USA; Department of Anatomy and Neurobiology, Virginia Commonwealth University Hospitals, Richmond, VA 23298, USA
William C. Broaddus, Email: wcbroadd@vcu.edu, Department of Neurosurgery, Virginia Commonwealth University, Medical College of Virginia Hospitals, Virginia Commonwealth University Health System, P.O. Box 980631, Richmond, VA 23298-0631, USA; Department of Anatomy and Neurobiology, Virginia Commonwealth University Hospitals, Richmond, VA 23298, USA.
References
- 1.Surawicz TS, McCarthy BJ, Kupelian V, et al. Descriptive epidemiology of primary brain and CNS tumors: results from the Central Brain Tumor Registry of the United States, 1990–1994. Neuro Oncol. 1999;1:14–25. doi: 10.1093/neuonc/1.1.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ballman KV, Buckner JC, Brown PD, et al. The relationship between six-month progression-free survival and 12-month overall survival end points for phase II trials in patients with glioblastoma multiforme. Neuro Oncol. 2007;9(1):29–38. doi: 10.1215/15228517-2006-025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Surawicz TS, Davis F, Freels S, et al. Brain tumor survival: results from the national cancer data base. J Neurooncol. 1998;40:151–160. doi: 10.1023/a:1006091608586. [DOI] [PubMed] [Google Scholar]
- 4.Loeb DM, Evron E, Patel CB, et al. Wilms' tumor suppressor gene (WT1) is expressed in primary breast tumors despite tumor-specific promoter methylation. Cancer Res. 2001;61:921–925. [PubMed] [Google Scholar]
- 5.Miwa H, Beran M, Saunders GF. Expression of the Wilms' tumor gene (WT1) in human leukemias. Leukemia. 1992;6(5):405–409. Abstract. [PubMed] [Google Scholar]
- 6.Nakahara Y, Okamoto H, Mineta T, et al. Expression of the Wilms' tumor gene product WT1 in glioblastomas and medulloblastomas. Brain Tumor Pathol. 2004;21:113–116. doi: 10.1007/BF02482185. [DOI] [PubMed] [Google Scholar]
- 7.Oji Y, Suzuki T, Nakano Y, et al. Overexpression of the Wilms' tumor gene WT1 in primary astrocytic tumors. Cancer Sci. 2004;95:822–827. doi: 10.1111/j.1349-7006.2004.tb02188.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang L, Lau YK, Xia W, et al. Tyrosine kinase inhibitor emodin suppresses growth of HER-2/neu-overexpressing breast cancer cells in athymic mice and sensitizes these cells to the inhibitory effect of paclitaxel. Clin Cancer Res. 1999;5:343–353. [PubMed] [Google Scholar]
- 9.Clark AJ, Dos Santos WG, McCready J, et al. Wilms tumor 1 expression in malignant gliomas and correlation of +KTS isoforms with p53 status. J Neurosurg. 2007;107:586–592. doi: 10.3171/JNS-07/09/0586. [DOI] [PubMed] [Google Scholar]
- 10.Armstrong JF, Pritchard-Jones K, Bickmore WA, et al. The expression of the Wilms' tumour gene, WT1, in the developing mammalian embryo. Mech Dev. 1993;40:85–97. doi: 10.1016/0925-4773(93)90090-k. [DOI] [PubMed] [Google Scholar]
- 11.Pritchard-Jones K, Fleming S, Davidson D, et al. The candidate Wilms' tumour gene is involved in genitourinary development. Nature. 1990;346:194–197. doi: 10.1038/346194a0. [DOI] [PubMed] [Google Scholar]
- 12.Haber DA, Sohn RL, Buckler AJ, et al. Alternative splicing and genomic structure of the Wilms tumor gene WT1. Proc Natl Acad Sci USA. 1991;88:9618–9622. doi: 10.1073/pnas.88.21.9618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scharnhorst V, Dekker P, van der Eb AJ, et al. Internal translation initiation generates novel WT1 protein isoforms with distinct biological properties. J Biol Chem. 1999;274:23456–23462. doi: 10.1074/jbc.274.33.23456. [DOI] [PubMed] [Google Scholar]
- 14.Sharma PM, Bowman M, Madden SL, et al. RNA editing in the Wilms' tumor susceptibility gene, WT1. Genes Dev. 1994;8:720–731. doi: 10.1101/gad.8.6.720. [DOI] [PubMed] [Google Scholar]
- 15.Call KM, Glaser T, Ito CY, et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms' tumor locus. Cell. 1990;60:509–520. doi: 10.1016/0092-8674(90)90601-a. [DOI] [PubMed] [Google Scholar]
- 16.Keilholz U, Menssen HD, Gaiger A, et al. Wilms' tumour gene 1 (WT1) in human neoplasia. Leukemia. 2005;19(8):1318–1323. doi: 10.1038/sj.leu.2403817. [DOI] [PubMed] [Google Scholar]
- 17.Wagner KJ, Roberts SG. Transcriptional regulation by the Wilms' tumour suppressor protein WT1. Biochem Soc Trans. 2004;32:932–935. doi: 10.1042/BST0320932. [DOI] [PubMed] [Google Scholar]
- 18.Davies RC, Calvio C, Bratt E, et al. WT1 interacts with the splicing factor U2AF65 in an isoform-dependent manner and can be incorporated into spliceosomes. Genes Dev. 1998;12:3217–3225. doi: 10.1101/gad.12.20.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caricasole A, Duarte A, Larsson SH, et al. RNA binding by the Wilms tumor suppressor zinc finger proteins. Proc Natl Acad Sci USA. 1996;93:7562–7566. doi: 10.1073/pnas.93.15.7562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spraggon L, Dudnakova T, Slight J, et al. hnRNP-U directly interacts with WT1 and modulates WT1 transcriptional activation. Oncogene. 2007;26(10):1484–1491. doi: 10.1038/sj.onc.1209922. [DOI] [PubMed] [Google Scholar]
- 21.Maheswaran S, Englert C, Bennett P, et al. The WT1 gene product stabilizes p53 and inhibits p53-mediated apoptosis. Genes Dev. 1995;9:2143–2156. doi: 10.1101/gad.9.17.2143. [DOI] [PubMed] [Google Scholar]
- 22.Johnstone RW, See RH, Sells SF, et al. A novel repressor, par-4, modulates transcription and growth suppression functions of the Wilms' tumor suppressor WT1. Mol Cell Biol. 1996;16:6945–6956. doi: 10.1128/mcb.16.12.6945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scharnhorst V, Dekker P, van der Eb AJ, et al. Physical interaction between Wilms tumor 1 and p73 proteins modulates their functions. J Biol Chem. 2000;275:10202–10211. doi: 10.1074/jbc.275.14.10202. [DOI] [PubMed] [Google Scholar]
- 24.Algar EM, Khromykh T, Smith SI, et al. A WT1 antisense oligonucleotide inhibits proliferation and induces apoptosis in myeloid leukaemia cell lines. Oncogene. 1996;12:1005–1014. [PubMed] [Google Scholar]
- 25.Oji Y, Nakamori S, Fujikawa M, et al. Overexpression of the Wilms' tumor gene WT1 in pancreatic ductal adenocarcinoma. Cancer Sci. 2004;95:583–587. doi: 10.1111/j.1349-7006.2004.tb02490.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tuna M, Chavez-Reyes A, Tari AM. HER2/neu increases the expression of Wilms' tumor 1 (WT1) protein to stimulate S-phase proliferation and inhibit apoptosis in breast cancer cells. Oncogene. 2005;24:1648–1652. doi: 10.1038/sj.onc.1208345. [DOI] [PubMed] [Google Scholar]
- 27.Zapata-Benavides P, Tuna M, Lopez-Berestein G, et al. Downregulation of Wilms' tumor 1 protein inhibits breast cancer proliferation. Biochem Biophys Res Commun. 2002;295:784–790. doi: 10.1016/s0006-291x(02)00751-9. [DOI] [PubMed] [Google Scholar]
- 28.Oji Y, Ogawa H, Tamaki H, et al. Expression of the Wilms' tumor gene WT1 in solid tumors and its involvement in tumor cell growth. Jpn J Cancer Res. 1999;90:194–204. doi: 10.1111/j.1349-7006.1999.tb00733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ware j, Roberts C, Richardson A, London C, Kroecher A, Amantana A, Devi G. WT1: a novel target for antisense mediated prostate tumor therapy (Abstract #594). Poster presentation American Association of Cancer Research 2005; Anaheim CA. 16/04/2005.2005. [Google Scholar]
- 30.Mayo MW, Wang CY, Drouin SS, et al. WT1 modulates apoptosis by transcriptionally upregulating the bcl-2 protooncogene. EMBO J. 1999;18:3990–4003. doi: 10.1093/emboj/18.14.3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hewitt SM, Hamada S, McDonnell TJ, et al. Regulation of the protooncogenes bcl-2 and c-myc by the Wilms' tumor suppressor gene WT1. Cancer Res. 1995;55:5386–5389. [PubMed] [Google Scholar]
- 32.Heckman C, Mochon E, Arcinas M, et al. The WT1 protein is a negative regulator of the normal bcl-2 allele in t(14;18) lymphomas. J Biol Chem. 1997;272:19609–19614. doi: 10.1074/jbc.272.31.19609. [DOI] [PubMed] [Google Scholar]
- 33.Loeb DM. WT1 influences apoptosis through transcriptional regulation of Bcl-2 family members. Cell Cycle. 2006;5:1249–1253. doi: 10.4161/cc.5.12.2807. [DOI] [PubMed] [Google Scholar]
- 34.Damon SE, Plymate SR, Carroll JM, et al. Transcriptional regulation of insulin-like growth factor-I receptor gene expression in prostate cancer cells. Endocrinology. 2001;142:21–27. doi: 10.1210/endo.142.1.7890. [DOI] [PubMed] [Google Scholar]
- 35.Idelman G, Glaser T, Roberts CT, Jr, et al. WT1–p53 interactions in insulin-like growth factor-I receptor gene regulation. J Biol Chem. 2003;278:3474–3482. doi: 10.1074/jbc.M211606200. [DOI] [PubMed] [Google Scholar]
- 36.Tajinda K, Carroll J, Roberts CT., Jr Regulation of insulin-like growth factor I receptor promoter activity by wild-type and mutant versions of the WT1 tumor suppressor. Endocrinology. 1999;140:4713–4724. doi: 10.1210/endo.140.10.7065. [DOI] [PubMed] [Google Scholar]
- 37.Chen Y, Douglass T, Jeffes EW, et al. Living T9 glioma cells expressing membrane macrophage colony-stimulating factor produce immediate tumor destruction by polymorphonuclear leukocytes and macrophages via a “paraptosis”-induced pathway that promotes systemic immunity against intracranial T9 gliomas. Blood. 2002;100:1373–1380. doi: 10.1182/blood-2002-01-0174. [DOI] [PubMed] [Google Scholar]
- 38.Jadus MR, Chen Y, Boldaji MT, et al. Human U251MG glioma cells expressing the membrane form of macrophage colony-stimulating factor (mM-CSF) are killed by human monocytes in vitro and are rejected within immunodeficient mice via paraptosis that is associated with increased expression of three different heat shock proteins. Cancer Gene Ther. 2003;10:411–420. doi: 10.1038/sj.cgt.7700583. [DOI] [PubMed] [Google Scholar]
- 39.Sperandio S, de BI, Bredesen DE. An alternative, nonapoptotic form of programmed cell death. Proc Natl Acad Sci USA. 2000;97:14376–14381. doi: 10.1073/pnas.97.26.14376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sperandio S, Poksay K, de BI, et al. Paraptosis: mediation by MAP kinases and inhibition by AIP-1/Alix. Cell Death Differ. 2004;11:1066–1075. doi: 10.1038/sj.cdd.4401465. [DOI] [PubMed] [Google Scholar]
- 41.Liu Y, Lehar S, Corvi C, et al. Expression of the insulin-like growth factor I receptor C terminus as a myristylated protein leads to induction of apoptosis in tumor cells. Cancer Res. 1998;58:570–576. [PubMed] [Google Scholar]
- 42.Hata Y, Sandler A, Loehrer PJ, et al. Synergism of taxol and gallium nitrate in human breast carcinoma cells: schedule dependency. Oncol Res. 1994;6:19–24. [PubMed] [Google Scholar]
- 43.Mochan TA, Venere M, DiTullio RA, Jr, et al. 53BP1 and NFBD1/MDC1-Nbs1 function in parallel interacting pathways activating ataxia-telangiectasia mutated (ATM) in response to DNA damage. Cancer Res. 2003;63:8586–8591. [PubMed] [Google Scholar]
- 44.Morrison AJ, Highland J, Krogan NJ, et al. INO80 and gamma-H2AX interaction links ATP-dependent chromatin remodeling to DNA damage repair. Cell. 2004;119:767–775. doi: 10.1016/j.cell.2004.11.037. [DOI] [PubMed] [Google Scholar]
- 45.Ito H, Daido S, Kanzawa T, et al. Radiation-induced autophagy is associated with LC3 and its inhibition sensitizes malignant glioma cells. Int J Oncol. 2005;26:1401–1410. [PubMed] [Google Scholar]
- 46.Kanzawa T, Kondo Y, Ito H, et al. Induction of autophagic cell death in malignant glioma cells by arsenic trioxide. Cancer Res. 2003;63:2103–2108. [PubMed] [Google Scholar]
- 47.Chi S, Kitanaka C, Noguchi K, et al. Oncogenic Ras triggers cell suicide through the activation of a caspase-independent cell death program in human cancer cells. Oncogene. 1999;18:2281–2290. doi: 10.1038/sj.onc.1202538. [DOI] [PubMed] [Google Scholar]
- 48.Kanzawa T, Germano IM, Komata T, et al. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004;11:448–457. doi: 10.1038/sj.cdd.4401359. [DOI] [PubMed] [Google Scholar]
- 49.Ito H, Aoki H, Kuhnel F, et al. Autophagic cell death of malignant glioma cells induced by a conditionally replicating adenovirus. J Natl Cancer Inst. 2006;98:625–636. doi: 10.1093/jnci/djj161. [DOI] [PubMed] [Google Scholar]
- 50.Katayama M, Kawaguchi T, Berger MS, et al. DNA damaging agent-induced autophagy produces a cytoprotective adenosine triphosphate surge in malignant glioma cells. Cell Death Differ. 2007;14:548–558. doi: 10.1038/sj.cdd.4402030. [DOI] [PubMed] [Google Scholar]
- 51.Abedin MJ, Wang D, McDonnell MA, et al. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ. 2007;14:500–510. doi: 10.1038/sj.cdd.4402039. [DOI] [PubMed] [Google Scholar]
- 52.Kreidberg JA, Sariola H, Loring JM, et al. WT-1 is required for early kidney development. Cell. 1993;74:679–691. doi: 10.1016/0092-8674(93)90515-r. [DOI] [PubMed] [Google Scholar]
- 53.Izumoto S, Tsuboi A, Oka Y, et al. Phase II clinical trial of Wilms tumor 1 peptide vaccination for patients with recurrent glioblastoma multiforme. J Neurosurg. 2008;108:963–971. doi: 10.3171/JNS/2008/108/5/0963. [DOI] [PubMed] [Google Scholar]
- 54.Englert C, Hou X, Maheswaran S, et al. WT1 suppresses synthesis of the epidermal growth factor receptor and induces apoptosis. EMBO J. 1995;14:4662–4675. doi: 10.1002/j.1460-2075.1995.tb00148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Han Y, San Marina S, Liu J, et al. Transcriptional activation of c-myc proto-oncogene by WT1 protein. Oncogene. 2004;23:6933–6941. doi: 10.1038/sj.onc.1207609. [DOI] [PubMed] [Google Scholar]
- 56.Liu XW, Gong LJ, Guo LY, et al. The Wilms' tumor gene product WT1 mediates the down-regulation of the rat epidermal growth factor receptor by nerve growth factor in PC12 cells. J Biol Chem. 2001;276:5068–5073. doi: 10.1074/jbc.M008776200. [DOI] [PubMed] [Google Scholar]
- 57.Drummond IA, Madden SL, Rohwer-Nutter P, et al. Repression of the insulin-like growth factor II gene by the Wilms tumor suppressor WT1. Science. 1992;257:674–678. doi: 10.1126/science.1323141. [DOI] [PubMed] [Google Scholar]
- 58.Gashler AL, Bonthron DT, Madden SL, et al. Human platelet-derived growth factor A chain is transcriptionally repressed by the Wilms tumor suppressor WT1. Proc Natl Acad Sci USA. 1992;89:10984–10988. doi: 10.1073/pnas.89.22.10984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nichols KE, Re GG, Yan YX, et al. WT1 induces expression of insulin-like growth factor 2 in Wilms' tumor cells. Cancer Res. 1995;55:4540–4543. [PubMed] [Google Scholar]
- 60.Broaddus WC, Liu Y, Steele LL, et al. Enhanced radiosensitivity of malignant glioma cells after adenoviral p53 transduction. J Neurosurg. 1999;91:997–1004. doi: 10.3171/jns.1999.91.6.0997. [DOI] [PubMed] [Google Scholar]
- 61.Ishii N, Maier D, Merlo A, et al. Frequent co-alterations of TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol. 1999;9:469–479. doi: 10.1111/j.1750-3639.1999.tb00536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shahrabani-Gargir L, Pandita TK, Werner H. Ataxia-telangiectasia mutated gene controls insulin-like growth factor I receptor gene expression in a deoxyribonucleic acid damage response pathway via mechanisms involving zinc-finger transcription factors Sp1 and WT1. Endocrinology. 2004;145:5679–5687. doi: 10.1210/en.2004-0613. [DOI] [PubMed] [Google Scholar]
- 63.Morrison DJ, English MA, Licht JD. WT1 induces apoptosis through transcriptional regulation of the proapoptotic Bcl-2 family member Bak. Cancer Res. 2005;65:8174–8182. doi: 10.1158/0008-5472.CAN-04-3657. [DOI] [PubMed] [Google Scholar]
- 64.Rodeck U, Bossler A, Kari C, et al. Expression of the wt1 Wilms' tumor gene by normal and malignant human melanocytes. Int J Cancer. 1994;59:78–82. doi: 10.1002/ijc.2910590116. [DOI] [PubMed] [Google Scholar]
- 65.Wang W, Lee SB, Palmer R, et al. A functional interaction with CBP contributes to transcriptional activation by the Wilms tumor suppressor WT1. J Biol Chem. 2001;276:16810–16816. doi: 10.1074/jbc.M009687200. [DOI] [PubMed] [Google Scholar]
- 66.Simpson LA, Burwell EA, Thompson KA, et al. The antiapoptotic gene A1/BFL1 is a WT1 target gene that mediates granulocytic differentiation and resistance to chemotherapy. Blood. 2006;107:4695–4702. doi: 10.1182/blood-2005-10-4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ito K, Oji Y, Tatsumi N, et al. Antiapoptotic function of 17AA(+)WT1 (Wilms' tumor gene) isoforms on the intrinsic apoptosis pathway. Oncogene. 2006;25:4217–4229. doi: 10.1038/sj.onc.1209455. [DOI] [PubMed] [Google Scholar]
- 68.Daido S, Kanzawa T, Yamamoto A, et al. Pivotal role of the cell death factor BNIP3 in ceramide-induced autophagic cell death in malignant glioma cells. Cancer Res. 2004;64:4286–4293. doi: 10.1158/0008-5472.CAN-03-3084. [DOI] [PubMed] [Google Scholar]
- 69.Takeuchi H, Kondo Y, Fujiwara K, et al. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 2005;65:3336–3346. doi: 10.1158/0008-5472.CAN-04-3640. [DOI] [PubMed] [Google Scholar]
- 70.Loeb DM, Korz D, Katsnelson M, et al. Cyclin E is a target of WT1 transcriptional repression. J Biol Chem. 2002;277:19627–19632. doi: 10.1074/jbc.M201336200. [DOI] [PubMed] [Google Scholar]
- 71.Englert C, Maheswaran S, Garvin AJ, et al. Induction of p21 by the Wilms' tumor suppressor gene WT1. Cancer Res. 1997;57:1429–1434. [PubMed] [Google Scholar]
- 72.Werner H, Roberts CT, Jr, Rauscher FJ, III, et al. Regulation of insulin-like growth factor I receptor gene expression by the Wilms' tumor suppressor WT1. J Mol Neurosci. 1996;7:111–123. doi: 10.1007/BF02736791. [DOI] [PubMed] [Google Scholar]
- 73.Hongo A, Yumet G, Resnicoff M, et al. Inhibition of tumorigenesis and induction of apoptosis in human tumor cells by the stable expression of a myristylated COOH terminus of the insulin-like growth factor I receptor. Cancer Res. 1998;58:2477–2484. [PubMed] [Google Scholar]
- 74.Plymate SR, Bae VL, Maddison L, et al. Reexpression of the type 1 insulin-like growth factor receptor inhibits the malignant phenotype of simian virus 40 T antigen immortalized human prostate epithelial cells. Endocrinology. 1997;138:1728–1735. doi: 10.1210/endo.138.4.5071. [DOI] [PubMed] [Google Scholar]
- 75.Plymate SS, Bae VL, Maddison L, et al. Type-1 insulin-like growth factor receptor reexpression in the malignant phenotype of SV40-T-immortalized human prostate epithelial cells enhances apoptosis. Endocrine. 1997;7:119–124. doi: 10.1007/BF02778078. [DOI] [PubMed] [Google Scholar]
- 76.Meter Van, et al. AKT inhibition enhances BCNU-mediated death in astrocytoma cells independent of PTEN functional status. Poster presentation; Congress of neurological surgeons, Annual meeting and 6th biennial AANS/CNS joint tumor satellite symposium; San Francisco CA. 21/10/2004.2004. [Google Scholar]
- 77.Simpson JR, Horton J, Scott C, et al. Influence of location and extent of surgical resection on survival of patients with glioblastoma multiforme: results of three consecutive Radiation Therapy Oncology Group (RTOG) clinical trials. Int J Radiat Oncol Biol Phys. 1993;26:239–244. doi: 10.1016/0360-3016(93)90203-8. [DOI] [PubMed] [Google Scholar]
- 78.Walker MD, Alexander E, Jr, Hunt WE, et al. Evaluation of BCNU and/or radiotherapy in the treatment of anaplastic gliomas. A cooperative clinical trial. J Neurosurg. 1978;49:333–343. doi: 10.3171/jns.1978.49.3.0333. [DOI] [PubMed] [Google Scholar]
- 79.Mains RE, May V. The role of a low pH intracellular compartment in the processing, storage, and secretion of ACTH and endorphin. J Biol Chem. 1988;263:7887–7894. [PubMed] [Google Scholar]
- 80.Paglin S, Hollister T, Delohery T, et al. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001;61:439–444. [PubMed] [Google Scholar]