

Abstract
Suppressors of cytokine signalling (SOCS) proteins are induced in responses to many stimuli and by binding to cytokine receptors and associated janus kinase (JAK) proteins, directly regulate the activation of the signal transducers and activators of transcription (STATs). STAT proteins regulate the expression of many genes required for the differentiation of various CD4+ T helper cell lineages, and there is now accumulating evidence that SOCS also play essential roles in the regulation and maintenance of CD4+ T-cell polarization. As it is now clear that CD4+ T cells are more plastic than initially thought, it is of particular importance to understand the molecular mechanisms regulating CD4+ T-cell differentiation. Here we review the current understanding of how STATs and SOCS act in concert to influence the polarization of CD4+ T cells and highlight the relevance of this in disease.
Keywords: CD4+ T cells, plasticity, polarization, signal transducer and activator of transcription, suppressors of cytokine signalling
Introduction
After interaction with their cognate antigen, naive CD4+ T cells proliferate and, depending on the cytokine micro-environment, polarize towards different CD4+ lineages, which then shape the immune response. CD4+ lineages include T helper type 1 (Th1), which drives the immune response against intracellular pathogens, Th2, which promotes humoral responses, Th17 cells, which contribute to the elimination of extracellular pathogens, and Foxp3+ regulatory T (Treg) cells, which prevent the development of autoimmunity (Fig. 1a). The differentiation towards each lineage is associated with the up-regulation of specific transcription factors that act as master regulators by controlling the expression of a panel of genes, conferring a specific phenotype1 (Fig. 1a). There is accumulating evidence that CD4+ T-cell lineages are not as stable as initially thought, but rather, in specific environments, secrete cytokines and co-express master regulators specific for other lineages.2 The factors that control CD4+ T-cell stability versus plasticity are currently poorly understood, but it is clear that signal transducer and activator of transcription (STAT) proteins play key roles in this process.
Figure 1.
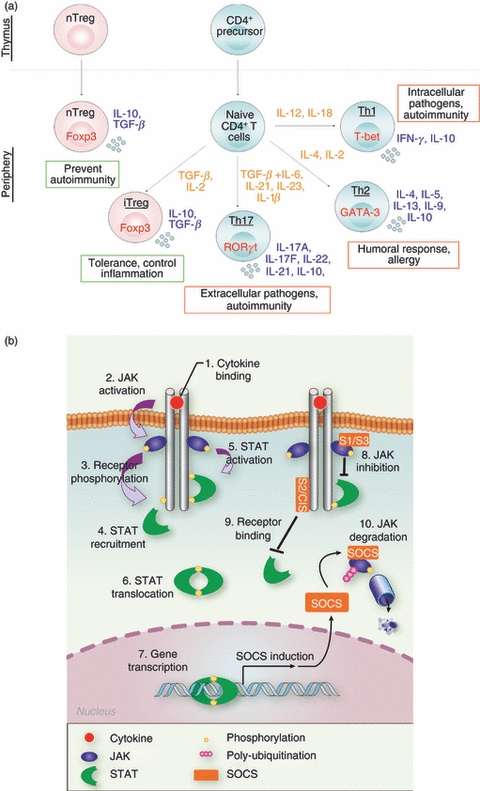
Importance of cytokine signalling for CD4+ T-cell differentiation. (a) Diagrammatic representation of polarization of naive CD4+ T cells towards some of the CD4+ T-cell lineages identified in the mouse. Master regulators are represented in red, polarizing cytokines in orange and cytokines associated with each subset in blue. Red and green boxes identify pro-inflammatory and anti-inflammatory subsets, respectively. (b) Regulation of janus kinase–signal transducer and activator of transcription (JAK-STAT) pathway by suppressor of cytokine signalling (SOCS) proteins. Upon cytokine binding, receptor hetero-dimerization (1) induces JAKs auto-activation (2) which phosphorylate STATs docking sites on the receptor (3). After their recruitment (4), STATs are subsequently phosphorylated by JAKs (5), which promotes their dimerization and translocation to the nucleus (6). SOCS genes are induced following STAT activation (7), and prevent STAT activation either by inhibiting JAK (8), or by competing for STAT docking sites (9). SOCS also mediate JAK ubiquitination, and so target them for proteasomal degradation (10).
Indeed STAT proteins not only control the expression of master regulators, but also directly modulate both permissive and repressive epigenetic events on genes characteristic of each lineage, independently of master regulators.3,4 In particular, STAT4 and STAT6 appear to have opposing effects on several genes, with STAT6 repressing in Th2 cells, the expression of genes characteristic of the Th1 phenotype, such as interleukin-18 receptor 1 (IL-18R1), and STAT4 acting to promote their expression in Th1 cells.5 Therefore STAT proteins directly contribute to the stabilization of CD4+ cell phenotypes.
The suppressor of cytokine signalling (SOCS) proteins are key physiological inhibitors of STAT proteins that are induced following cytokine stimulation. SOCS interact with cytokine receptors or the janus kinases (JAK) and prevent the subsequent activation of STATs.6 Therefore, SOCS govern the magnitude and duration of cytokine responses and not surprisingly, a number of studies have now shown that SOCS also play a key role in CD4+ T-cell polarization and plasticity.7 Here we review what is currently understood about how the SOCS proteins modulate the activation of STAT proteins and consequently influence CD4+ T-cell commitment.
Regulation of STAT activation by SOCS proteins
The activation of STAT proteins following cytokine stimulation is mediated by the JAK family of protein tyrosine kinases that associate with type I and type II cytokine receptors. After cytokine binding, receptor chains cluster and trigger JAK auto-phosphorylation or trans-phosphorylation and consequent activation (Fig. 1b). In turn, JAKs phosphorylate specific tyrosine residues on the receptor cytoplasmic tail that serve as docking sites for STATs. The subsequent STAT tyrosine phosphorylation leads to their dimerization and tetramerization, which facilitate nuclear translocation and binding to specific promoter elements.8
The eight members of the SOCS family (SOCS1 to SOCS7 and CIS) are induced following STAT activation and down-regulate the JAK–STAT cascade in a classic negative feedback loop. SOCS proteins are characterized by an Src-homology type 2 (SH2) domain, which facilitates SOCS binding to JAKs and cytokine receptors and a highly conserved 40-amino-acid C-terminal motif termed the SOCS box. The SOCS box recruits an E3 ubiquitin ligase complex containing elongin-B, elongin-C, Cullin 2 or 5 and the ring finger proteins Rbx1 or Rbx2,6,7,9, which allows SOCS proteins to target cytokine receptors and JAKs for lysosomal or proteasomal degradation. Some SOCS also have additional modes of action, as CIS and SOCS2 may prevent STAT5 binding to the Erythropoietin (EPO) and growth hormone (GH) receptors, respectively, by competing for the tyrosine residues used as docking sites,10,11 and SOCS1, SOCS3 and SOCS5 contain a kinase inhibitory region that inhibits JAK catalytic activity.12,13 Therefore, SOCS proteins prevent STAT activation by blocking their recruitment to the cytokine receptor or by inhibiting their phosphorylation by JAKs. They consequently control the duration and strength of cytokine responses,7 and as discussed herein, play an essential role in the regulation of CD4+ T-cell polarization.
Control of Th1 cell commitment
The Th1 cells secrete high levels of interferon-γ (IFN-γ) and IL-2, and drive immunity against intracellular pathogens but also promote autoimmunity. Interleukin-12, in synergy with IL-18, drives Th1 differentiation, in large part via induction of T-bet (T-Box expressed in T cells), a master regulator transcription factor that controls the expression of IFN-γ.14 Interleukin-12 signals through JAK2 and Tyk2, and activates mainly STAT4, also a key transcription factor for Th1 commitment4 (Fig. 2). Indeed, STAT4-deficient CD4+ T cells do not produce IFN-γ following IL-12 or Listeria monocytogenes stimulation,15,16 and STAT4-deficient mice fail to secrete IFN-γ in response to Toxoplasma gondii and therefore die as the result of an uncontrolled parasite burden.17 It later emerged that STAT4 controls T-bet expression,18,19 with which it then collaborates for efficient binding to the Ifng promoter1 and to induce both IL-18Rα and IL-12Rβ2.3 The STAT4 also induces tumour progression locus 2 (Tpl-2), a serine threonine kinase essential for T-bet and STAT4 up-regulation and so essential for optimal IFN-γ secretion.20 Therefore STAT4 not only promotes the expression of IFN-γ and T-bet, but also of other genes that consolidate the Th1 phenotype (Fig. 2), as summarized in Table 1.
Figure 2.
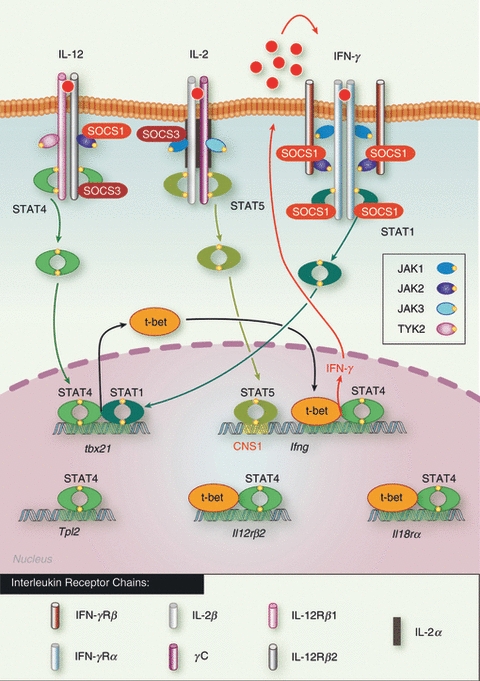
Schematic representation of the control of T helper type 1 (Th1) differentiation by signal transducer and activator of transcription (STAT) and suppressor of cytokine signalling (SOCS) proteins. The first event of Th1 polarization is the activation of STAT4 following IL-12 signalling which results in T-bet induction. STAT4 and T-bet then synergize to induce interferon-γ (IFN-γ), interleukin-12 receptor β2 (IL-12Rβ2) and IL-18Rα. IFN-γ acts in an autocrine positive feedback loop through STAT1 activation, by further inducing T-bet expression, whereas IL-2-mediated STAT5 activation directly contributes to enhance IFN-γ secretion. SOCS1 strongly inhibits IFN-γ signalling not only by inhibiting janus kinase 2 (JAK2) activity but also by preventing STAT1 binding to IFN-γRα chain, whereas SOCS3 targets IL-12Rβ so preventing STAT4 activation, and consequently both SOCS1 and SOCS3 inhibit the development of Th1 cells.
Table 1.
Summary of genes regulated by, and effect of signal transducer and activator of transcription (STAT) proteins during CD4+ T-cell polarization
| Effect | |||||
|---|---|---|---|---|---|
| CD4+ lineage | STAT protein (cytokine) | Gene regulated | Gene | Polarization | References |
| Th1 | STAT4 (IL-12) | Ifng | Induction | + | 18 |
| tbx21 | Induction | + | 19 | ||
| Il12rB2 | Induction | + | 3,19 | ||
| Il18r | Induction | + | 3 | ||
| Tpl2 | Induction | + | 20 | ||
| STAT1 (IFN-γ) | tbx21 | Expression enhancement | NC | 22 | |
| STAT5 (IL-2) | Ifng | Expression enhancement | + | 23 | |
| Th2 | STAT6 (IL-4) | Gata3 | Induction | + | 42 |
| RAD50 | Chromatin modification | + | 49 | ||
| STAT5 (IL-2, TSLP) | Il4 | Induction | + | 52,54,55 | |
| Il4rα | Induction | + | 56 | ||
| Th17 | STAT3 (IL-6, IL-21, IL-23) | Il17a | Induction | + | 65,67 |
| Il17f | Induction | + | 65,67 | ||
| Rorc | Induction | + | 65,67 | ||
| Rora | Induction | + | 67 | ||
| Il6r | Induction | + | 65,67,68 | ||
| Il21 | Induction | + | 65,67,68 | ||
| Foxp3 | Repression | + | 65 | ||
| STAT5 (IL-2) | Il17a | Repression | − | 71 | |
| nTreg | STAT5 (IL-2) | Foxp3 | Induction | + | 76,77 |
| Foxp3+ iTreg | STAT5 (IL-2) | Foxp3 | Induction | + | 76 |
| STAT3 (IL-6) | Foxp3 | Repression | − | 71 | |
| STAT6 (IL-4) | Foxp3 | Repression | − | 79 | |
IFN, interferon; IL-12, interleukin-12; NC, not clear; Th1, T helper type 1; Treg, regulatory T; TSLP, thymic stromal lymphopoietin.
Importantly, IFN-γ also facilitates the development of Th1 cells in a positive autocrine feedback loop,21 and STAT1-deficient T cells have reduced T-bet levels following infection,22 although IFN-γ secretion does not seem to be affected. Moreover, several studies have shown that JAK3 and STAT5 activation by IL-2 enables optimal IFN-γ secretion.23,24 Indeed, JAK3-deficient T cells fail to secrete IFN-γ,23 whereas IL-2-mediated STAT5 activation is required for optimal IFN-γ secretion.23,24 STAT5 binds the first conserved non-coding sequence upstream of the Ifng promoter, which suggests that it might permit T-bet access.23,25 Therefore, STAT1 and STAT5 contribute to Th1 differentiation by enhancing T-bet and IFN-γ expression, respectively (Fig. 2).
SOCS1 is a key inhibitor of IFN-γ signalling26,27 and blocks IFN-γ-mediated STAT1 activation by targeting JAK2 and IFN-γRα chain28 (Fig. 2). The SOCS1-deficient mice also have enhanced type 1 IFN responses, which render them more resistant to viral infection.27 Importantly, SOCS1 is up-regulated during Th1 commitment29 and not surprisingly, SOCS1-deficient T cells proliferate strongly in response to IL-12,30 which enhances their polarization towards the Th1 lineage.31 However, these cells also secrete elevated levels of IL-4, and exhibit heightened IL-4-mediated STAT6 phosphorylation, suggesting that SOCS1 could also be an important regulator of Th2 differentiation. However, IL-6 can induce SOCS1 and subsequently block IFN-γ secretion by T cells without affecting IL-4 production,32 which suggests that the main role of SOCS1 may be to limit Th1 differentiation.
Despite comparably low levels in Th1 cells, SOCS3 and SOCS5 also regulate Th1 differentiation. Indeed through binding to the IL-12Rβ2 chain, SOCS3 prevents STAT4 activation (Fig. 2) and constitutive expression of SOCS3 in CD4+ T cells was shown to hinder Th1 polarization.33 Consistent with these findings, up-regulation of SOCS3 by IL-2 was found to prevent acute graft-versus-host disease by inhibiting the Th1 response.34 However, SOCS3 deletion in T cells also resulted in decreased Th1 differentiation, although this was proposed to be indirect. Indeed, increased IL-10 and transforming growth factor (TGF-β) secretion was also observed in these cells, perhaps suggesting that SOCS3 may limit Treg cell development.35
The role of SOCS5 is more controversial. Indeed, despite being highly expressed in Th1 cells,36 disruption of the socs5 gene does not affect the ability of cells to differentiate either towards Th1 or Th2.37 Over-expression of SOCS5 in T cells is associated with increased levels of IL-12, IFN-γ and tumour necrosis factor-α in a mouse model of septic peritonitis,38 but this could be indirectly the result of enhanced macrophage activity, possibly through increased IFN-γ secretion by T cells.36,39 Finally, Th1 differentiation does not seem to be affected by higher levels of SOCS5,36 and so the exact role of SOCS5 in Th1 differentiation remains unclear. By regulating IL-12-mediated STAT4 activation and IFN-γ-mediated STAT1 signals, SOCS1, SOCS3 and SOCS5 certainly modulate the development of Th1 cells, although the role of individual SOCS is, even at this point, far from clear. Our current understanding is summarized in Table 2.
Table 2.
Summary of signals regulated by, and effect of suppressor of cytokine signalling (SOCS) proteins during CD4+ T-cell polarization
| Effect | |||||
|---|---|---|---|---|---|
| CD4+ lineage | SOCS protein | Signal regulated | Signal | Polarization | References |
| Thl | SOCS1 | IFN-γ/STAT1 | Inhibition | − | 30,31 |
| SOCS3 | IL-12/STAT4 | Inhibition | − | 33 | |
| Th2 | SOCS1 | IL-4/STAT6 | Inhibition | − | 31 |
| SOCS5 | IL-4/STAT6 | Inhibition | −/NC | 37,39 | |
| SOCS3 | IL-12/STAT4 | Inhibition | + | 33,39 | |
| SOCS2 | IL-2/STAT5 | Inhibition | − | 59 | |
| IL-6/STAT3 | Enhancement | − | 59 | ||
| Thl7 | SOCS3 | IL-6/STAT3 | Inhibition | − | 72 |
| IL-23/STAT3 | Inhibition | − | 62 | ||
| SOCS1 | IFN-γ/STAT1 | Inhibition | + | 61 | |
| IL-6/STAT3 | Enhancement | + | 61 | ||
| SOCS2 | IL-6/STAT3 | Enhancement | + | 59 | |
| Foxp3+ Treg | SOCS1 | IL-2/STAT5 | Inhibition | −/+ | 82,83 |
IFN, interferon; IL-12, interleukin-12; NC, not clear; STAT, signal transducer and activator of transcription; Th1, T helper type 1.
Control of Th2 cell commitment
The Th2 cells secrete large amounts IL-4, IL-5, IL-9 and IL-13, and consequently promote the humoral response but also drive IgE class switching and allergic disease.40 The commitment of Th2 cells is essentially driven by IL-4, which activates both JAK1 and JAK3 and the transcription factor STAT641 (Fig. 3). Not surprisingly, STAT6 plays a key role in the acquisition of the Th2 phenotype. In particular STAT6 directly controls the expression of Th2 lineage master regulator, GATA3,42 and enforced expression of STAT6 in Th1 cells re-establishes their ability to secrete IL-4 and IL-5, while repressing IFN-γ and IL-12Rβ2 expression.42 STAT6-deficient T cells fail to polarize towards Th2 in vitro and in vivo,43–45 but the absence of STAT6 does not affect the emergence of Th2 cells in response to Nippostrongylus brasiliensis or Schistosoma mansoni challenge,46–48 which probably reflects the fact that STAT6 does not directly regulate the il4 gene. Instead, induction of IL-4 is controlled by GATA-3, which suggests that STAT6 essentially acts by up-regulating GATA-3 levels, although STAT6 seems to modify the chromatin structure of the Rad50 gene, which may allow optimal transcription of the il4 and il13 genes.49 Importantly, STAT6 also induces growth factor independent-1 (Gfi-1),50 a transcriptional repressor of ifng and Il17, which inhibits TGF-β mediated Foxp3 induction. Therefore STAT6 not only is a key regulator of GATA-3 expression, but further contributes to Th2 commitment by preventing the acquisition of the Th1, Th17 or Foxp3+ Treg cell phenotypes.51
Figure 3.
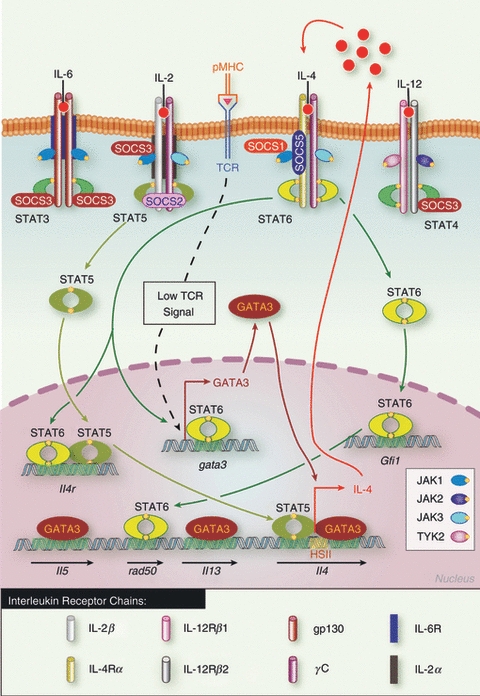
Schematic representation of the control of Th2 differentiation by signal transducer and activator of transcription (STAT) and suppressor of cytokine signalling (SOCS) proteins. Low T-cell receptor activation leads to GATA3 up-regulation, which together with interleukin-2 (IL-2) -mediated STAT5 activation induces early IL-4 secretion. IL-4 then signals through STAT6, which mediates a positive autocrine loop by further up-regulating GATA3 and IL-4Rα expression. SOCS1 and SOCS5 inhibit IL-4-mediated STAT6 activation and therefore negatively regulate Th2 differentiation. SOCS3 positively regulates Th2 differentiation by blocking IL-12 signalling and subsequent Th1 polarization, while SOCS2 regulates IL-2 signalling and so inhibits Th2 differentiation. Adapted from ref. 40.
It is now clear that not only STAT6, but also STAT5 plays an essential role in the initial steps of Th2 differentiation. Indeed, expression of constitutively active STAT5 is sufficient to induce IL-4 expression in cells lacking STAT6 or cultured under Th1 polarizing conditions,52 whereas IL-2 neutralization or STAT5 deletion prevents IL-4 secretion.53 Both STAT5 and GATA3, target the hypersensitivity enhancer region HSII located in the second intron of the il4 gene,52,54,55 and synergize to promote IL-4 secretion. Finally, STAT5 also regulates il4rα expression56 (Fig. 3). This suggests that not only IL-2 but also other cytokines signalling through STAT5, such as thymic stromal lymphopoietin, may be as important as IL-4 in driving Th2 development, as summarized in Table 1.
Both SOCS1 and SOCS5 inhibit IL-4 signalling36,57 (Fig. 3); indeed, SOCS1-deficient T cells secrete increased levels of IL-4.29,31 SOCS5 also inhibits Th2 differentiation,39 but the relevance of this remains controversial because SOCS5-deficient mice do not have increased susceptibility to atopy, perhaps reflecting the close homology and likely redundancy between SOCS4 and SOCS5.37
Interestingly, SOCS3 and SOCS2 also regulate Th2 polarization, positively and negatively, respectively. Indeed, constitutive expression of SOCS3 in T cells confers increased susceptibility in atopic models,33,39,58 while SOCS2-deficient mice develop exacerbated disease because of enhanced Th2 polarization.59 Surprisingly, neither SOCS3 nor SOCS2 seem to directly regulate IL-4 signalling. Instead, SOCS3 is a key regulator of IL-6-mediated or IL-23-mediated STAT360–62 and of IL-12-mediated STAT4 activation33 (Fig. 3), suggesting that SOCS3 may indirectly promote Th2 differentiation by preventing the development of Th1 and Th17 cells. Similarly, SOCS2-deficient CD4+ T cells display reduced STAT3 activation and enhanced STAT5 phosphorylation and so SOCS2 probably inhibits Th2 differentiation by inhibiting IL-2 signalling, while favouring the development of Th17 cells.59 Therefore, SOCS proteins control Th2 differentiation not only by inhibiting the activation of STAT6 and STAT5, but also by regulating the polarization of naive CD4+ T cells towards the other CD4+ lineages (Fig. 3). This is summarized in Table 2.
Control of Th17 cell commitment
T helper type 17 cells secrete high levels of IL-17A, IL-17F and IL-22 and play a key role at mucosal surfaces where they combat infection by extracellular bacteria. The Th17 cells are highly pro-inflammatory, and an alteration of the Th17 versus Treg cell balance is proposed as a potential mechanism that may induce autoimmunity.63
Commitment of Th17 cells is complex and involves many cytokines and other molecules, which lead to the up-regulation of the RAR-related orphan receptor gamma (RORγt), the master regulator of this lineage.64 Amongst these cytokines, IL-6, IL-21 and IL-23 all signal through STAT3, and not surprisingly, STAT3 is essential for Th17 development. Indeed, disrupted STAT3 expression in T cells blocks Th17 differentiation,65 and confers resistance to experimental autoimmune encephalomyelitis (EAE) and colitis.66,67 STAT3 controls the expression of several key Th17 genes such as il17a, il17f, rora, il6r and il2167–69 but also promotes RORγt while repressing Foxp3 expression,65 so STAT3 is key at all stages of Th17 commitment (Fig. 4).
Figure 4.
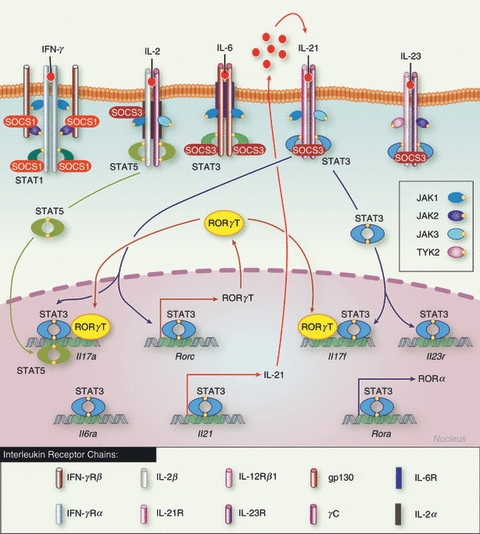
Schematic representation of the control of T helper type 17 (Th17) differentiation by signal transducer and activator of transcription (STAT) and suppressor of cytokine signalling (SOCS) proteins. Interleukin-6 (IL-6) -mediated STAT3 activation initiates the induction of RORγt, IL-17 and IL-21 expression. IL-21 then acts as a positive autocrine loop to further activate STAT3 and up-regulated IL-23R, the expression of which would allow cell responsiveness to IL-23 and its subsequent commitment. IL-2-mediated STAT5 activation inhibits Th17 polarization by direct inhibition of STAT3 transcriptional activity. SOCS3 strongly inhibits STAT3 activation and so prevents the development of Th17 cells, whereas SOCS1 positively regulates Th17 differentiation by blocking interferon-γ signal, and the subsequent polarization towards Th1.
Interestingly, the activation of STAT5 by IL-2 is required for optimal differentiation of Th1, Th2 and Foxp3+ Treg cells, but inhibits the development of Th17 cells.70 Indeed, STAT5 binds several sites on the il17 promoter and directly antagonizes STAT3 transcriptional activity,71 showing that STAT3 and STAT5 exert polar opposite effects on IL-17 expression in the context of Th17 differentiation (Fig. 4). This suggests that STAT5 is an essential regulator of CD4+ T-cell plasticity because IL-2 promotes Th1 and Th2 responses, whereas the absence of IL-2 favours the emergence of Th17 cells, as summarized in Table 1.
The SOCS3 protein is a well known inhibitor of STAT3 activation in various cell types, and in particular inhibits IL-6 and IL-23 signalling in CD4+ T cells60–62 (Fig. 4). As might have been expected, SOCS3 deletion in T cells favours IL-17 secretion in vitro62 and in vivo,72 whereas enforced expression of SOCS3 inhibits polarization towards Th17 and delays the onset of EAE.61 Moreover, mutation of the SOCS3 binding site on gp130 results in increased IL-17 secretion60 and spontaneous arthritis.73 Finally, it has been proposed that TGF-β inhibits SOCS3 expression, and subsequently prolongs STAT3 activation, which perhaps explains how TGF-β enhances Th17 differentiation.74
Therefore, SOCS3 clearly inhibits the development of Th17 cells, but SOCS1 and SOCS2 appear to have the opposite effect. Indeed, disruption of SOCS1 expression in T cells strongly inhibits Th17 differentiation and diminishes disease in EAE models.61 This is associated with increased IFN-γ-mediated STAT1 activation, enhanced SOCS3 levels, attenuated STAT3 phosphorylation and reduced TGF-β transcriptional activity. These observations indicate that SOCS1 promotes Th17 differentiation possibly by modulating TGF-β signalling, but also indirectly by preventing Th1 lineage polarization and by regulating SOCS3 levels. Interestingly, SOCS2-deficient CD4+ T cells also have impaired IL-17 secretion, consistent with reduced STAT3 activation and elevated SOCS3 levels.59 Therefore the positive effect of SOCS1 and SOCS2 on Th17 differentiation might well be simply the consequence of increased SOCS3 levels, which confirms that the regulation of STAT3 activation by SOCS3 is an essential mechanism to limit Th17 development. Our current understanding is summarized in Table 2.
Control of Foxp3+ Treg cell function
Foxp3 is the master regulator of two lineages of Treg cells: natural Treg (nTreg) cells, which mature in the thymus, and Foxp3+-inducible Treg (iTreg) cells, which arise in the periphery from naive CD4+ T cells. These cells play a key role in the prevention of autoimmunity, because immunodysregulation polyendocrinopathy enteropathy X-linked (IPEX) syndrome and the scurfy phenotype, two severe autoimmune conditions in human and mouse, respectively, are the result of mutations within the foxp3 gene. Induction of Foxp3 is associated with the acquisition of a suppressive phenotype, which allows these cells to limit inflammatory responses. One key cytokine for Foxp3+ lineages is IL-2, which is essential for nTreg cell development in the thymus, Foxp3 induction in the periphery and the maintenance of Foxp3+ T-cell homeostasis.75–77 Indeed, disruption of the jak3 or stat5 genes abrogates Foxp3 expression,77 while constitutive activation of STAT5 restores the ability of IL-2Rβ-deficient mice to induce Foxp3.76 STAT5 binds the Foxp3 promoter76,77 but whether STAT5 regulates the expression of other genes contributing to the Foxp3+ Treg cell phenotype is not known. Induction of Foxp3 in naive CD4+ T cells is driven by TGF-β, a process inhibited by IL-6-mediated STAT378 and IL-4-mediated STAT6 activation.79 STAT4 activation following IL-12 stimulation has also been proposed to antagonize Foxp3 expression, as STAT4-deficient mice have elevated Foxp3+ Treg cells in the lung in an ovalbumin-induced asthma model80 but whether this is a direct effect has not yet been assessed.
To date, how SOCS proteins regulate Foxp3 expression is poorly understood. Deletion of SOCS1 in T cells results in increased Foxp3+ T-cell numbers in thymus, whereas its forced expression has the opposite effect.81,82 Mice lacking SOCS1 specifically in Foxp3+ Treg cells also presented with increased Foxp3+ Treg cell populations in the thymus and in the periphery, possibly as the result of a lack of IL-2 signalling.83 Interestingly, these mice spontaneously developed clinical signs of conjunctivitis and dermatitis associated with increased IFN-γ secretion by T cells in vivo.83 Therefore, SOCS1 clearly affected Foxp3+ Treg cell development and stability but the mechanism involved is still unclear. Finally, constitutive expression of SOCS3 seemed to affect the ability of Foxp3+ Treg cells to proliferate and to inhibit the proliferation of conventional T cells in vitro,84 whereas SOCS3 deletion in dendritic cells favours the expansion of Foxp3+ Treg cells.85 Foxp3 may regulate SOCS2 and SOCS3 expression86,87 and high levels of SOCS2 mRNA are found in both Foxp3+ CD4+ T-cell lineages.87–89 Therefore, SOCS3 and SOCS2 might also be important regulators of Foxp3+ Treg cell function, but this needs to be further investigated. These finding are summarized in Tables 1 and 2.
SOCS and STAT in human disorders
The close relationship between STAT mutations and diseases was recently reviewed.4 Briefly, STAT1 mutation confers increased susceptibility to viral and mycobacterial infections,90,91 whereas STAT5b deficiency is associated with reduced accumulation and impaired function of Treg cells.92 Moreover, polymorphisms in not only STAT3, but also in IL23R and JAK2 loci, correlate with Crohn's disease.93–95 Therefore, appropriate activation of the STAT proteins is clearly required for the development of a healthy immune response. Interestingly, several studies show abnormal expression of SOCS proteins in autoimmune diseases. In particular, SOCS1 mRNA is elevated in patients who present with systemic lupus erythematosus96 and rheumatoid arthritis,97 and single nucleotide polymorphisms in SOCS1 are associated with multiple sclerosis98 and coeliac disease.99 All of these autoimmune pathologies are characterized by increased IL-17 secretion, which would be consistent with the fact that SOCS1 promotes the development of Th17 cells.
Compellingly, the correlation between SOCS3 expression and the severity of atopy is also apparent in patients. Markedly elevated SOCS3 expression is observed in skin samples from patients suffering from severe atopic dermatitis (AD) when compared with individuals with normal skin or with the Th1-mediated condition psoriasis.100 Furthermore, specific haplotypes of the SOCS3 gene have been linked with AD in two independent Swedish childhood cohorts and SOCS3 mRNA is more highly expressed in AD skin.101 The detection of elevated SOCS3 expression in peripheral T cells and in AD skin may be of particular relevance because the SOCS3 gene is located on chromosome 17q25, one of the established AD genetic loci.102 Similarly, SOCS3 expression in T cells positively correlates with the severity of asthma and AD,33 whereas elevated SOCS3 mRNA levels and polymorphisms within the SOCS3 locus are found in patients with AD.101
Asthmatics also present with polymorphisms within the SOCS1 promoter, consistent with the fact that SOCS3 and SOCS1 regulate Th2 differentiation.103 The correlation between elevated SOCS1 expression and asthma severity in patients suggests that SOCS1 may inhibit IFN-γ-dependent Th1 differentiation, thereby enhancing Th2-mediated pathology.104 Of note, disruption of SOCS2 expression increases murine susceptibility to atopy but whether this is of relevance in patients has yet to be determined.59 Taken together, these different studies confirm the importance of SOCS proteins in the regulation of human pathogenic immune responses.
Conclusions
Clearly, both STATs and SOCS are key regulators of lineage commitment and collaborate to tightly regulate CD4+ T-cell polarization. As with STATs, SOCS often exert opposing effects and may cross-regulate one another,59,61,105,106 and although murine null models exemplify this cross-compensation, this may well reflect reality because SOCS proteins are differentially expressed in individual CD4+ lineages.29 The studies summarized here point to an essential role for SOCS proteins in the regulation of CD4+ T-cell polarity because SOCS1 and SOCS3 indirectly favour Th17 and Th2 differentiation, respectively, by limiting the development of Th1 cells,33,61 and SOCS2 favours polarization towards Th17 at the expense of Th2 differentiation.59 Hence, SOCS proteins do not simply regulate CD4+ T-cell commitment by inhibiting specific JAK/STAT responses, but rather, they adjust the balance between each lineage, suggesting that they might play an essential role in the regulation of CD4+ T-cell plasticity. It will be important to determine the relative expression of each SOCS in the context of human CD4+ T-cell polarization and ascertain whether these proteins might represent potential targets to medicate the growing allergy and autoimmune disease burden observed in recent decades.
Disclosures
The authors have no conflicts of interest to disclose.
References
- 1.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*) Annu Rev Immunol. 2010;28:445–89. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murphy KM, Stockinger B. Effector T cell plasticity: flexibility in the face of changing circumstances. Nat Immunol. 2010;11:674–80. doi: 10.1038/ni.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thieu VT, Yu Q, Chang HC, Yeh N, Nguyen ET, Sehra S, Kaplan MH. Signal transducer and activator of transcription 4 is required for the transcription factor T-bet to promote T helper 1 cell-fate determination. Immunity. 2008;29:679–90. doi: 10.1016/j.immuni.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O'Shea JJ, Lahesmaa R, Vahedi G, Laurence A, Kanno Y. Genomic views of STAT function in CD4+ T helper cell differentiation. Nat Rev Immunol. 2011;11:239–50. doi: 10.1038/nri2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wei L, Vahedi G, Sun HW, et al. Discrete roles of STAT4 and STAT6 transcription factors in tuning epigenetic modifications and transcription during T helper cell differentiation. Immunity. 2010;32:840–51. doi: 10.1016/j.immuni.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elliott J, Johnston JA. SOCS: role in inflammation, allergy and homeostasis. Trends Immunol. 2004;25:434–40. doi: 10.1016/j.it.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 7.Palmer DC, Restifo NP. Suppressors of cytokine signaling (SOCS) in T cell differentiation, maturation, and function. Trends Immunol. 2009;30:592–602. doi: 10.1016/j.it.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shuai K, Liu B. Regulation of JAK-STAT signalling in the immune system. Nat Rev Immunol. 2003;3:900–11. doi: 10.1038/nri1226. [DOI] [PubMed] [Google Scholar]
- 9.Babon JJ, Sabo JK, Zhang JG, Nicola NA, Norton RS. The SOCS box encodes a hierarchy of affinities for Cullin5: implications for ubiquitin ligase formation and cytokine signalling suppression. J Mol Biol. 2009;387:162–74. doi: 10.1016/j.jmb.2009.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsumoto A, Masuhara M, Mitsui K, Yokouchi M, Ohtsubo M, Misawa H, Miyajima A, Yoshimura A. CIS, a cytokine inducible SH2 protein, is a target of the JAK-STAT5 pathway and modulates STAT5 activation. Blood. 1997;89:3148–54. [PubMed] [Google Scholar]
- 11.Greenhalgh CJ, Rico-Bautista E, Lorentzon M, et al. SOCS2 negatively regulates growth hormone action in vitro and in vivo. J Clin Invest. 2005;115:397–406. doi: 10.1172/JCI22710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sasaki A, Yasukawa H, Suzuki A, et al. Cytokine-inducible SH2 protein-3 (CIS3/SOCS3) inhibits janus tyrosine kinase by binding through the N-terminal kinase inhibitory region as well as SH2 domain. Genes Cells. 1999;4:339–51. doi: 10.1046/j.1365-2443.1999.00263.x. [DOI] [PubMed] [Google Scholar]
- 13.Yasukawa H, Misawa H, Sakamoto H, et al. The JAK-binding protein JAB inhibits janus tyrosine kinase activity through binding in the activation loop. EMBO J. 1999;18:1309–20. doi: 10.1093/emboj/18.5.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu J, Paul WE. Peripheral CD4+ T-cell differentiation regulated by networks of cytokines and transcription factors. Immunol Rev. 2010;238:247–62. doi: 10.1111/j.1600-065X.2010.00951.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaplan MH, Sun YL, Hoey T, Grusby MJ. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature. 1996;382:174–7. doi: 10.1038/382174a0. [DOI] [PubMed] [Google Scholar]
- 16.Thierfelder WE, van Deursen JM, Yamamoto K, et al. Requirement for Stat4 in interleukin-12-mediated responses of natural killer and T cells. Nature. 1996;382:171–4. doi: 10.1038/382171a0. [DOI] [PubMed] [Google Scholar]
- 17.Cai G, Radzanowski T, Villegas EN, Kastelein R, Hunter CA. Identification of STAT4-dependent and independent mechanisms of resistance to Toxoplasma gondii. J Immunol. 2000;165:2619–27. doi: 10.4049/jimmunol.165.5.2619. [DOI] [PubMed] [Google Scholar]
- 18.Usui T, Nishikomori R, Kitani A, Strober W. GATA-3 suppresses Th1 development by downregulation of Stat4 and not through effects on IL-12Rβ2 chain or T-bet. Immunity. 2003;18:415–28. doi: 10.1016/s1074-7613(03)00057-8. [DOI] [PubMed] [Google Scholar]
- 19.Usui T, Preiss JC, Kanno Y, Yao ZJ, Bream JH, O'Shea JJ, Strober W. T-bet regulates Th1 responses through essential effects on GATA-3 function rather than on IFNG gene acetylation and transcription. J Exp Med. 2006;203:755–66. doi: 10.1084/jem.20052165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Watford WT, Hissong BD, Durant LR, et al. Tpl2 kinase regulates T cell interferon-γ production and host resistance to Toxoplasma gondii. J Exp Med. 2008;205:2803–12. doi: 10.1084/jem.20081461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bradley LM, Dalton DK, Croft M. A direct role for IFN-γ in regulation of Th1 cell development. J Immunol. 1996;157:1350–8. [PubMed] [Google Scholar]
- 22.Lieberman LA, Banica M, Reiner SL, Hunter CA. STAT1 plays a critical role in the regulation of antimicrobial effector mechanisms, but not in the development of Th1-type responses during toxoplasmosis. J Immunol. 2004;172:457–63. doi: 10.4049/jimmunol.172.1.457. [DOI] [PubMed] [Google Scholar]
- 23.Shi M, Lin TH, Appell KC, Berg LJ. Janus-kinase-3-dependent signals induce chromatin remodeling at the ifng locus during T helper 1 cell differentiation. Immunity. 2008;28:763–73. doi: 10.1016/j.immuni.2008.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liao W, Lin JX, Wang L, Li P, Leonard WJ. Modulation of cytokine receptors by IL-2 broadly regulates differentiation into helper T cell lineages. Nat Immunol. 2011;12:551–9. doi: 10.1038/ni.2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murphy KM. Permission to proceed: Jak3 and STAT5 signaling molecules give the green light for T helper 1 cell differentiation. Immunity. 2008;28:725–7. doi: 10.1016/j.immuni.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Starr R, Metcalf D, Elefanty AG, Brysha M, Willson TA, Nicola NA, Hilton DJ, Alexander WS. Liver degeneration and lymphoid deficiencies in mice lacking suppressor of cytokine signaling-1. Proc Natl Acad Sci U S A. 1998;95:14395–9. doi: 10.1073/pnas.95.24.14395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alexander WS, Starr R, Fenner JE, et al. SOCS1 is a critical inhibitor of interferon-γ signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell. 1999;98:597–608. doi: 10.1016/s0092-8674(00)80047-1. [DOI] [PubMed] [Google Scholar]
- 28.Sakamoto H, Yasukawa H, Masuhara M, et al. A janus kinase inhibitor, JAB, is an interferon-γ-inducible gene and confers resistance to interferons. Blood. 1998;92:1668–76. [PubMed] [Google Scholar]
- 29.Egwuagu CE, Yu CR, Zhang M, Mahdi RM, Kim SJ, Gery I. Suppressors of cytokine signaling proteins are differentially expressed in Th1 and Th2 cells: implications for Th cell lineage commitment and maintenance. J Immunol. 2002;168:3181–7. doi: 10.4049/jimmunol.168.7.3181. [DOI] [PubMed] [Google Scholar]
- 30.Eyles JL, Metcalf D, Grusby MJ, Hilton DJ, Starr R. Negative regulation of interleukin-12 signaling by suppressor of cytokine signaling-1. J Biol Chem. 2002;277:43735–40. doi: 10.1074/jbc.M208586200. [DOI] [PubMed] [Google Scholar]
- 31.Fujimoto M, Tsutsui H, Yumikura-Futatsugi S, et al. A regulatory role for suppressor of cytokine signaling-1 in T(h) polarization in vivo. Int Immunol. 2002;14:1343–50. doi: 10.1093/intimm/dxf094. [DOI] [PubMed] [Google Scholar]
- 32.Diehl S, Anguita J, Hoffmeyer A, Zapton T, Ihle JN, Fikrig E, Rincon M. Inhibition of Th1 differentiation by IL-6 is mediated by SOCS1. Immunity. 2000;13:805–15. doi: 10.1016/s1074-7613(00)00078-9. [DOI] [PubMed] [Google Scholar]
- 33.Seki Y, Inoue H, Nagata N, et al. SOCS-3 regulates onset and maintenance of TH2-mediated allergic responses. Nat Med. 2003;9:1047–54. doi: 10.1038/nm896. [DOI] [PubMed] [Google Scholar]
- 34.Zhao J, Zhang T, He H, Xie Y. Interleukin-2 inhibits polarization to T helper type 1 cells and prevents mouse acute graft-versus-host disease through up-regulating suppressors of cytokine signalling-3 expression of naive CD4+ T cells. Clin Exp Immunol. 2010;160:479–88. doi: 10.1111/j.1365-2249.2010.04089.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kinjyo I, Inoue H, Hamano S, et al. Loss of SOCS3 in T helper cells resulted in reduced immune responses and hyperproduction of interleukin 10 and transforming growth factor-β1. J Exp Med. 2006;203:1021–31. doi: 10.1084/jem.20052333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seki Y, Hayashi K, Matsumoto A, et al. Expression of the suppressor of cytokine signaling-5 (SOCS5) negatively regulates IL-4-dependent STAT6 activation and Th2 differentiation. Proc Natl Acad Sci U S A. 2002;99:13003–8. doi: 10.1073/pnas.202477099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brender C, Columbus R, Metcalf D, et al. SOCS5 is expressed in primary B and T lymphoid cells but is dispensable for lymphocyte production and function. Mol Cell Biol. 2004;24:6094–103. doi: 10.1128/MCB.24.13.6094-6103.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Watanabe H, Kubo M, Numata K, Takagi K, Mizuta H, Okada S, Ito T, Matsukawa A. Overexpression of suppressor of cytokine signaling-5 in T cells augments innate immunity during septic peritonitis. J Immunol. 2006;177:8650–7. doi: 10.4049/jimmunol.177.12.8650. [DOI] [PubMed] [Google Scholar]
- 39.Ozaki A, Seki Y, Fukushima A, Kubo M. The control of allergic conjunctivitis by suppressor of cytokine signaling (SOCS)3 and SOCS5 in a murine model. J Immunol. 2005;175:5489–97. doi: 10.4049/jimmunol.175.8.5489. [DOI] [PubMed] [Google Scholar]
- 40.Paul WE, Zhu J. How are TH2-type immune responses initiated and amplified? Nat Rev Immunol. 2010;10:225–35. doi: 10.1038/nri2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rochman Y, Spolski R, Leonard WJ. New insights into the regulation of T cells by γ(c) family cytokines. Nat Rev Immunol. 2009;9:480–90. doi: 10.1038/nri2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kurata H, Lee HJ, O'Garra A, Arai N. Ectopic expression of activated Stat6 induces the expression of Th2-specific cytokines and transcription factors in developing Th1 cells. Immunity. 1999;11:677–88. doi: 10.1016/s1074-7613(00)80142-9. [DOI] [PubMed] [Google Scholar]
- 43.Kaplan MH, Schindler U, Smiley ST, Grusby MJ. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity. 1996;4:313–9. doi: 10.1016/s1074-7613(00)80439-2. [DOI] [PubMed] [Google Scholar]
- 44.Shimoda K, van Deursen J, Sangster MY, et al. Lack of IL-4-induced Th2 response and IgE class switching in mice with disrupted Stat6 gene. Nature. 1996;380:630–3. doi: 10.1038/380630a0. [DOI] [PubMed] [Google Scholar]
- 45.Takeda K, Tanaka T, Shi W, et al. Essential role of Stat6 in IL-4 signalling. Nature. 1996;380:627–30. doi: 10.1038/380627a0. [DOI] [PubMed] [Google Scholar]
- 46.Jankovic D, Kullberg MC, Noben-Trauth N, Caspar P, Paul WE, Sher A. Single cell analysis reveals that IL-4 receptor/Stat6 signaling is not required for the in vivo or in vitro development of CD4+ lymphocytes with a Th2 cytokine profile. J Immunol. 2000;164:3047–55. doi: 10.4049/jimmunol.164.6.3047. [DOI] [PubMed] [Google Scholar]
- 47.van Panhuys N, Tang SC, Prout M, et al. In vivo studies fail to reveal a role for IL-4 or STAT6 signaling in Th2 lymphocyte differentiation. Proc Natl Acad Sci U S A. 2008;105:12423–8. doi: 10.1073/pnas.0806372105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Finkelman FD, Morris SC, Orekhova T, et al. Stat6 regulation of in vivo IL-4 responses. J Immunol. 2000;164:2303–10. doi: 10.4049/jimmunol.164.5.2303. [DOI] [PubMed] [Google Scholar]
- 49.Lee DU, Rao A. Molecular analysis of a locus control region in the T helper 2 cytokine gene cluster: a target for STAT6 but not GATA3. Proc Natl Acad Sci U S A. 2004;101:16010–5. doi: 10.1073/pnas.0407031101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu J, Guo L, Min B, Watson CJ, Hu-Li J, Young HA, Tsichlis PN, Paul WE. Growth factor independent-1 induced by IL-4 regulates Th2 cell proliferation. Immunity. 2002;16:733–44. doi: 10.1016/s1074-7613(02)00317-5. [DOI] [PubMed] [Google Scholar]
- 51.Zhu J, Jankovic D, Grinberg A, Guo L, Paul WE. Gfi-1 plays an important role in IL-2-mediated Th2 cell expansion. Proc Natl Acad Sci U S A. 2006;103:18214–9. doi: 10.1073/pnas.0608981103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhu J, Cote-Sierra J, Guo L, Paul WE. Stat5 activation plays a critical role in Th2 differentiation. Immunity. 2003;19:739–48. doi: 10.1016/s1074-7613(03)00292-9. [DOI] [PubMed] [Google Scholar]
- 53.Cote-Sierra J, Foucras G, Guo L, Chiodetti L, Young HA, Hu-Li J, Zhu J, Paul WE. Interleukin 2 plays a central role in Th2 differentiation. Proc Natl Acad Sci U S A. 2004;101:3880–5. doi: 10.1073/pnas.0400339101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hural JA, Kwan M, Henkel G, Hock MB, Brown MA. An intron transcriptional enhancer element regulates IL-4 gene locus accessibility in mast cells. J Immunol. 2000;165:3239–49. doi: 10.4049/jimmunol.165.6.3239. [DOI] [PubMed] [Google Scholar]
- 55.Tanaka S, Motomura Y, Suzuki Y, Yagi R, Inoue H, Miyatake S, Kubo M. The enhancer HS2 critically regulates GATA-3-mediated Il4 transcription in TH2 cells. Nat Immunol. 2011;12:77–85. doi: 10.1038/ni.1966. [DOI] [PubMed] [Google Scholar]
- 56.Liao W, Schones DE, Oh J, Cui Y, Cui K, Roh TY, Zhao K, Leonard WJ. Priming for T helper type 2 differentiation by interleukin 2-mediated induction of interleukin 4 receptor α-chain expression. Nat Immunol. 2008;9:1288–96. doi: 10.1038/ni.1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Losman JA, Chen XP, Hilton D, Rothman P. Cutting edge: SOCS-1 is a potent inhibitor of IL-4 signal transduction. J Immunol. 1999;162:3770–4. [PMC free article] [PubMed] [Google Scholar]
- 58.Nakaya M, Hamano S, Kawasumi M, Yoshida H, Yoshimura A, Kobayashi T. Aberrant IL-4 production by SOCS3-over-expressing T cells during infection with Leishmania major exacerbates disease manifestations. Int Immunol. 2011;23:195–202. doi: 10.1093/intimm/dxq472. [DOI] [PubMed] [Google Scholar]
- 59.Knosp CA, Carroll HP, Elliott J, et al. SOCS2 regulates T helper type 2 differentiation and the generation of type 2 allergic responses. J Exp Med. 2011;208:1523–31. doi: 10.1084/jem.20101167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stumhofer JS, Laurence A, Wilson EH, et al. Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nat Immunol. 2006;7:937–45. doi: 10.1038/ni1376. [DOI] [PubMed] [Google Scholar]
- 61.Tanaka K, Ichiyama K, Hashimoto M, et al. Loss of suppressor of cytokine signaling 1 in helper T cells leads to defective Th17 differentiation by enhancing antagonistic effects of IFN-γ on STAT3 and smads. J Immunol. 2008;180:3746–56. doi: 10.4049/jimmunol.180.6.3746. [DOI] [PubMed] [Google Scholar]
- 62.Chen Z, Laurence A, Kanno Y, et al. Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc Natl Acad Sci U S A. 2006;103:8137–42. doi: 10.1073/pnas.0600666103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bettelli E, Korn T, Oukka M, Kuchroo VK. Induction and effector functions of TH17 cells. Nature. 2008;453:1051–7. doi: 10.1038/nature07036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hirahara K, Ghoreschi K, Laurence A, Yang XP, Kanno Y, O'Shea JJ. Signal transduction pathways and transcriptional regulation in Th17 cell differentiation. Cytokine Growth Factor Rev. 2010;21:425–34. doi: 10.1016/j.cytogfr.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, Dong C. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282:9358–63. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- 66.Harris TJ, Grosso JF, Yen HR, et al. Cutting edge: an in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J Immunol. 2007;179:4313–7. doi: 10.4049/jimmunol.179.7.4313. [DOI] [PubMed] [Google Scholar]
- 67.Durant L, Watford WT, Ramos HL, et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity. 2010;32:605–15. doi: 10.1016/j.immuni.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nurieva R, Yang XO, Martinez G, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–3. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- 69.Zhou L, Ivanov II, Spolski R, et al. IL-6 programs TH-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–74. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 70.Laurence A, Tato CM, Davidson TS, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–81. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 71.Yang XP, Ghoreschi K, Steward-Tharp SM, et al. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat Immunol. 2011;12:247–54. doi: 10.1038/ni.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Taleb S, Romain M, Ramkhelawon B, et al. Loss of SOCS3 expression in T cells reveals a regulatory role for interleukin-17 in atherosclerosis. J Exp Med. 2009;206:2067–77. doi: 10.1084/jem.20090545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ogura H, Murakami M, Okuyama Y, et al. Interleukin-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction. Immunity. 2008;29:628–36. doi: 10.1016/j.immuni.2008.07.018. [DOI] [PubMed] [Google Scholar]
- 74.Qin H, Wang L, Feng T, et al. TGF-β promotes Th17 cell development through inhibition of SOCS3. J Immunol. 2009;183:97–105. doi: 10.4049/jimmunol.0801986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Malek TR, Castro I. Interleukin-2 receptor signaling: at the interface between tolerance and immunity. Immunity. 2010;33:153–65. doi: 10.1016/j.immuni.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL-2 receptor β-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J Immunol. 2007;178:280–90. doi: 10.4049/jimmunol.178.1.280. [DOI] [PubMed] [Google Scholar]
- 77.Yao Z, Kanno Y, Kerenyi M, et al. Nonredundant roles for Stat5a/b in directly regulating Foxp3. Blood. 2007;109:4368–75. doi: 10.1182/blood-2006-11-055756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang XO, Nurieva R, Martinez GJ, et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity. 2008;29:44–56. doi: 10.1016/j.immuni.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Takaki H, Ichiyama K, Koga K, et al. STAT6 inhibits TGF-β1-mediated Foxp3 induction through direct binding to the Foxp3 promoter, which is reverted by retinoic acid receptor. J Biol Chem. 2008;283:14955–62. doi: 10.1074/jbc.M801123200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.O'Malley JT, Sehra S, Thieu VT, et al. Signal transducer and activator of transcription 4 limits the development of adaptive regulatory T cells. Immunology. 2009;127:587–95. doi: 10.1111/j.1365-2567.2008.03037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lu LF, Thai TH, Calado DP, et al. Foxp3-dependent microRNA155 confers competitive fitness to regulatory T cells by targeting SOCS1 protein. Immunity. 2009;30:80–91. doi: 10.1016/j.immuni.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhan Y, Davey GM, Graham KL, Kiu H, Dudek NL, Kay TW, Lew AM. SOCS1 negatively regulates the production of Foxp3+ CD4+ T cells in the thymus. Immunol Cell Biol. 2009;87:473–80. doi: 10.1038/icb.2009.23. [DOI] [PubMed] [Google Scholar]
- 83.Lu LF, Boldin MP, Chaudhry A, et al. Function of miR-146a in controlling Treg cell-mediated regulation of Th1 responses. Cell. 2010;142:914–29. doi: 10.1016/j.cell.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pillemer BB, Xu H, Oriss TB, Qi Z, Ray A. Deficient SOCS3 expression in CD4+ CD25+ FoxP3+ regulatory T cells and SOCS3-mediated suppression of Treg function. Eur J Immunol. 2007;37:2082–9. doi: 10.1002/eji.200737193. [DOI] [PubMed] [Google Scholar]
- 85.Matsumura Y, Kobayashi T, Ichiyama K, et al. Selective expansion of foxp3-positive regulatory T cells and immunosuppression by suppressors of cytokine signaling 3-deficient dendritic cells. J Immunol. 2007;179:2170–9. doi: 10.4049/jimmunol.179.4.2170. [DOI] [PubMed] [Google Scholar]
- 86.Muthukumarana P, Chae WJ, Maher S, Rosengard BR, Bothwell AL, Metcalfe SM. Regulatory transplantation tolerance and “stemness”: evidence that Foxp3 may play a regulatory role in SOCS-3 gene transcription. Transplantation. 2007;84:S6–11. doi: 10.1097/01.tp.0000269116.06510.db. [DOI] [PubMed] [Google Scholar]
- 87.Sugimoto N, Oida T, Hirota K, Nakamura K, Nomura T, Uchiyama T, Sakaguchi S. Foxp3-dependent and -independent molecules specific for CD25+ CD4+ natural regulatory T cells revealed by DNA microarray analysis. Int Immunol. 2006;18:1197–209. doi: 10.1093/intimm/dxl060. [DOI] [PubMed] [Google Scholar]
- 88.Hill JA, Feuerer M, Tash K, Haxhinasto S, Perez J, Melamed R, Mathis D, Benoist C. Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity. 2007;27:786–800. doi: 10.1016/j.immuni.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 89.Haribhai D, Lin W, Edwards B, et al. A central role for induced regulatory T cells in tolerance induction in experimental colitis. J Immunol. 2009;182:3461–8. doi: 10.4049/jimmunol.0802535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dupuis S, Jouanguy E, Al-Hajjar S, et al. Impaired response to interferon-α/β and lethal viral disease in human STAT1 deficiency. Nat Genet. 2003;33:388–91. doi: 10.1038/ng1097. [DOI] [PubMed] [Google Scholar]
- 91.Dupuis S, Dargemont C, Fieschi C, et al. Impairment of mycobacterial but not viral immunity by a germline human STAT1 mutation. Science. 2001;293:300–3. doi: 10.1126/science.1061154. [DOI] [PubMed] [Google Scholar]
- 92.Cohen AC, Nadeau KC, Tu W, et al. Cutting edge: decreased accumulation and regulatory function of CD4+ CD25high T cells in human STAT5b deficiency. J Immunol. 2006;177:2770–4. doi: 10.4049/jimmunol.177.5.2770. [DOI] [PubMed] [Google Scholar]
- 93.Franke A, Balschun T, Karlsen TH, et al. Replication of signals from recent studies of Crohn's disease identifies previously unknown disease loci for ulcerative colitis. Nat Genet. 2008;40:713–5. doi: 10.1038/ng.148. [DOI] [PubMed] [Google Scholar]
- 94.Barrett JC, Hansoul S, Nicolae DL, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet. 2008;40:955–62. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Duerr RH, Taylor KD, Brant SR, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–3. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chan HC, Ke LY, Chang LL, et al. Suppressor of cytokine signaling 1 gene expression and polymorphisms in systemic lupus erythematosus. Lupus. 2010;19:696–702. doi: 10.1177/0961203309357437. [DOI] [PubMed] [Google Scholar]
- 97.Chan HC, Ke LY, Liu CC, Chang LL, Tsai WC, Liu HW, Yen JH. Increased expression of suppressor of cytokine signaling 1 mRNA in patients with rheumatoid arthritis. Kaohsiung J Med Sci. 2010;26:290–8. doi: 10.1016/S1607-551X(10)70042-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zuvich RL, McCauley JL, Oksenberg JR, et al. Genetic variation in the IL7RA/IL7 pathway increases multiple sclerosis susceptibility. Hum Genet. 2010;127:525–35. doi: 10.1007/s00439-010-0789-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dubois PC, Trynka G, Franke L, et al. Multiple common variants for celiac disease influencing immune gene expression. Nat Genet. 2010;42:295–302. doi: 10.1038/ng.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Horiuchi Y, Bae SJ, Katayama I. Overexpression of the suppressor of cytokine signalling 3 (SOCS3) in severe atopic dermatitis. Clin Exp Dermatol. 2006;31:100–4. doi: 10.1111/j.1365-2230.2005.01979.x. [DOI] [PubMed] [Google Scholar]
- 101.Ekelund E, Saaf A, Tengvall-Linder M, et al. Elevated expression and genetic association links the SOCS3 gene to atopic dermatitis. Am J Hum Genet. 2006;78:1060–5. doi: 10.1086/504272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cookson WO, Ubhi B, Lawrence R, et al. Genetic linkage of childhood atopic dermatitis to psoriasis susceptibility loci. Nat Genet. 2001;27:372–3. doi: 10.1038/86867. [DOI] [PubMed] [Google Scholar]
- 103.Harada M, Nakashima K, Hirota T, et al. Functional polymorphism in the suppressor of cytokine signaling 1 gene associated with adult asthma. Am J Respir Cell Mol Biol. 2007;36:491–6. doi: 10.1165/rcmb.2006-0090OC. [DOI] [PubMed] [Google Scholar]
- 104.Daegelmann C, Herberth G, Roder S, et al. Association between suppressors of cytokine signalling, T-helper type 1/T-helper type 2 balance and allergic sensitization in children. Clin Exp Allergy. 2008;38:438–48. doi: 10.1111/j.1365-2222.2007.02913.x. [DOI] [PubMed] [Google Scholar]
- 105.Tannahill GM, Elliott J, Barry AC, Hibbert L, Cacalano NA, Johnston JA. SOCS2 can enhance interleukin-2 (IL-2) and IL-3 signaling by accelerating SOCS3 degradation. Mol Cell Biol. 2005;25:9115–26. doi: 10.1128/MCB.25.20.9115-9126.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Piessevaux J, Lavens D, Montoye T, et al. Functional cross-modulation between SOCS proteins can stimulate cytokine signaling. J Biol Chem. 2006;281:32953–66. doi: 10.1074/jbc.M600776200. [DOI] [PubMed] [Google Scholar]