

Summary
The ESX-1 secretion system is required for pathogenicity of Mycobacterium tuberculosis (Mtb). Despite considerable research, little is known about the structural components of ESX-1, or how these proteins are assembled into the active secretion apparatus. Here, we exploit the functionally related ESX-1 apparatus of Mycobacterium smegmatis (Ms) to show that fluorescently tagged proteins required for ESX-1 activity consistently localize to the cell pole, identified by time-lapse fluoro-microscopy as the non-septal (old) pole. Deletions in Msesx1 prevented polar localization of tagged proteins, indicating the need for specific protein-protein interactions in polar trafficking. Remarkably, expression of the Mtbesx1 locus in Msesx1 mutants restored polar localization of tagged proteins, indicating establishment of the MtbESX-1 apparatus in M. smegmatis. This observation illustrates the cross-species conservation of protein interactions governing assembly of ESX-1, as well as polar localization. Importantly, we describe novel non-esx1 encoded proteins that affect ESX-1 activity, that co-localize with ESX-1, and that are required for ESX-1 recruitment and assembly. This analysis provides new insights into the molecular assembly of this important determinant of Mtb virulence.
Keywords: ESX-1, Mycobacteria, Protein localization, Secretion
Introduction
Bacteria utilize secretion systems to move substrates across the barriers presented by the cell membrane and/or cell wall (Christie et al., 2005, Papanikou et al., 2007, Hayes et al., 2010). The secretion of protein substrates is required for bacteria to decorate their surfaces with receptors and ligands, and to secrete effectors into their environment. The target of these effectors can either be other bacteria or eukaryotic cells, depending on the bacterium’s lifestyle. For bacterial pathogens, secretion systems have become essential for their ability to survive, replicate and disseminate in their hosts. For example, the Legionella pneumophila Icm/Dot Type IV section system secretes over 250 substrates, some of which contribute to establishment of a successful infection and subsequent pathogenesis (Zhu et al., 2011).
Seven secretion systems have been described thus far, each distinguished by hallmarks in their structures or functions (Tseng et al., 2009). The Type VII Secretion Systems (T7SS) were originally described in the pathogen Mycobacterium tuberculosis (Mtb), which encodes five related, but functionally distinct, T7SS (Gey Van Pittius et al., 2001, Pallen, 2002). Each apparatus is encoded by a large multigene locus named esx1-5, which expresses related sets of proteins, although they do not cross complement (Abdallah et al., 2007, DiGiuseppe Champion and Cox, 2007). The archetypal T7SS is the ESX-1 secretory apparatus of Mtb (Bitter et al., 2009). Interest in Mtb ESX-1 was further intensified by the finding that a deletion of seven esx1 genes is the primary attenuating mutation in the avirulent M. bovis vaccine strain, BCG (Fig. 1B; (Behr et al., 1999, Mahairas et al., 1996). This conclusion has been widely corroborated, with a role for ESX-1 being linked to bacterial survival in host macrophages (Behr et al., 1999, Hsu et al., 2003, Lewis et al., 2003, Mahairas et al., 1996, Pym et al., 2002).
Fig. 1.

M. smegmatis and M. tuberculosis Esx-1 proteins localize to a cell pole in M. smegmatis cells. (A) Model of the ESX-1 secretion apparatus identifying key proteins discussed in this work. EccCab is an ATPase thought to deliver the EsxA and EsxB heterodimer, and associated proteins, to the pore (EccD). The pore components of the outer membrane are unknown. For simplicity, only one (EspE) of the many proteins co-secreted with EsxAB is shown. (B) Comparative genetic map of the esx1 operons of M. smegmatis and Mtb, highlighting key genes. Gene orthologs and paralogs are colour coded. The bar below the Mtb map indicates the deletion found in M. bovis(BCG) resulting in its attenuation. (C–E) Differential interference contrast (DIC, left panel), FITC (center), and merged (right) images of M. smegmatis showing that YFP is distributed throughout the cells (C), while YFP-tagged EspEms or EspEmt (D), and the ESX-1-associated ATPase, EccCbms, and it’s Mtb orthologue, EccCbmt (E), localize to the polar regions of M. smegmatis cells. 630X total magnification.
Exactly how Mtb ESX-1 promotes pathogenicity is unknown, although most hypotheses focus on the potential functions of the proteins encoded by genes within the esx1 locus, which are required for the survival and dissemination of Mtb in the host (Simeone et al., 2009). The primary substrate secreted by ESX-1 is a heterodimeric product of the tandem esxB and esxA genes (Berthet et al., 1995, Renshaw et al., 2002). Efforts to comprehensively identify all ESX-1 substrates are complicated by the apparent absence of a distinct secretion signal, and the mutually dependent secretion of the majority of known secreted proteins: EsxAB secretion is dependent on the co-secretion of proteins encoded from both esx1 (eg., EspE) and non-esx1 genes (eg., EspA; (Fortune et al., 2005, MacGurn et al., 2005).
The structure of the secretory apparatus itself is also poorly defined. Current models of ESX-1 structure have largely been inferred by determining which genes are conserved within homologous esx loci (called Ecc for Esx conserved component), followed by analyses of the encoded proteins to identify features such as transmembrane domains and protein-protein interactions (Fig. 1A; Bitter et al., 2009). Briefly, the trans-membrane protein EccD is thought to form the inner-membrane channel through which EsxAB are secreted. Substrate translocation through the channel is thought to be powered by the ATPase, EccC, which is often encoded by split genes eccCa and eccCb. It is difficult to directly interrogate how, and where, the apparatus components are assembled because of the thick, waxy layer of mycolic acids characteristic of mycobacteria outer-membranes. Thus far, the only in situ characterization of the assembled apparatus has been performed using an M. marinum ΔkasB strain defective in mycolic acid synthesis, which results in a more permeable cell wall, allowing an initial antibody-based approach to localizing exposed ESX-1 epitopes (Carlsson et al., 2009). These studies suggested that the ESX-1 apparatus is localized to a bacterial pole. However, this immuno-fluorescence-based approach was limited to recognition of surface exposed epitopes and by having to use a mutant mycobacteria with a defective cell wall. Consequently, the majority of cytoplasmic components of the secretion apparatus cannot be visualized in their native context by this method.
In order to visualize the localization and assembly of the ESX-1 secretion apparatus, we have systematically expressed fluorescent-protein fusions of putative and candidate ESX-1 components in M. smegmatis: esx1ms is genetically and functionally conserved with that of Mtb (Fig. 1B; Coros et al., 2008, Flint et al., 2004, Converse and Cox, 2005). Analogous to its Mtb counterpart, ESX-1ms secretes a heterodimer of EsxABms (71% and 62% identical to the same proteins in Mtb). Here, we have used the M. smegmatis model system to track the intracellular localization (and co-localization) of ESX1 proteins in live cells and defined the role that individual proteins, implicated in ESX-1 function, play in localization of ESX-1 components.
In M. smegmatis, ESX-1ms also regulates a novel type of conjugative DNA transfer, which we have previously exploited to identify genes required for ESX-1 activity (Coros et al., 2008, Flint et al., 2004). Here we demonstrate that a non-esx1 operon (Msmeg0044-0046, which we rename saeABC), previously shown to be essential for ESX-1ms mediated secretion of EsxAB and for conjugal DNA transfer, is required for appropriate localization of core components of the apparatus. The discovery of a new locus necessary for ESX-1 recruitment to the pole will lead to a more comprehensive understanding of the structure and function of T7SS, and help to build an empirically determined model of the assembly and structure of ESX-1.
Results
ESX-1 associated proteins localize to the polar regions of M. smegmatis cells
In order to determine the cellular location of components associated with the ESX-1 secretion apparatus, esx1-associated genes were PCR amplified from the M. smegmatis mc2155 donor genome and fused, in frame, to a gene encoding a yellow fluorescent protein, YFP-Venus (Nagai et al., 2002). A penta-glycine chain was used as a linker joining the two proteins to reduce steric interference and allow native folding of the N-terminal ESX-1 protein. The genes were introduced into M. smegmatis on a plasmid and expressed from either the Hsp60 or the inducible Ptet promoter (Ehrt et al., 2005, Stover et al., 1991). In preliminary experiments, we established that expression of native YFP resulted in fluorescence distributed throughout the cell (Fig. 1C).
A previous study had shown that the M. marimum protein Mh3864 (EspEmm) is secreted and remains partially associated with the cell pole of the bacterium (Carlsson et al., 2009). We therefore investigated the cellular location of the M. smegmatis ortholog Msmeg0055 (EspEms). Ectopic expression of EspEms tagged at its C-terminus with YFP revealed that it, too, was localized to a pole of the mc2155 cell: cells had foci located at one or both cell poles (Fig. 1D). Our preliminary studies indicate EspEms is not secreted, unlike its M. marinum counterpart, but its polar localization is consistent with the association of EspEms with the ESX-1 apparatus.
We have established that ESX-1 activity is essential for DNA transfer and EsxAB secretion in the M. smegmatis recipient strain, MKD8 (Coros et al., 2008). We sought to determine whether polar localization also occurred in the recipient strain, or if it was a uniquely donor-encoded property. As with the donor strain, EspEms-YFP was localized to the cell poles: 133/207 recipient cells examined contained polar foci and 86/133 had a single polar focus (Fig. S1A). We have since shown that all the fusion proteins described below localized to a pole in both donor and recipient and, therefore, we will focus on localization of ESX-1 in the donor strain (data for the recipient are shown in Fig. S2). Throughout these studies, we noted some variability in the percentage of cells containing fluorescent foci of a particular protein. For example, a higher percentage of recipient cells (41%) contained a single polar focus of EspE compared with the donor (24%). This implies there are growth, environmental and expression factors influencing protein production and assembly that we do not, as yet, understand. The precise location of the foci at the pole of the cell varied between a location at the tip apex to one near the onset of tip curvature (peripolar). We therefore use the term polar here to indicate at, or very close to, the cell pole.
As EspEms was localized to the cell pole, we investigated whether other ESX-1-associated proteins would be similarly localized. EccCbms (Msmeg0062) encodes an ftsk/spoIIIE family ATPase that is central to ESX-1 function and, presumably, integral to its structure (Fig. 1A). EccCbms was tagged with YFP-Venus and ectopically expressed in M. smegmatis. Localization of this ESX-1-associated protein was very similar to that of EspE: 47% (29/62) of fluorescent cells contained a single EccCbms focus, localized to the polar region of the cell (Figs. 1E, S2E). Not all the ESX-1 proteins we tested were localized. For example, we examined N- and C-terminal fusions of YFP to secreted proteins EsxBms and EsxAms, respectively. In each case, and in both donor and recipient, the EsxA and EsxB fusion proteins were not localized (data not shown). This is not unexpected, as we would predict localization would be a property of structural components of the apparatus.
We next determined whether ESX-1 components from Mtb showed similar localization when heterologously expressed in M. smegmatis. EspEmt and EccCbmt, were similarly tagged and expressed in mc2155. In both cases, cells showed a polar focus of fluorescence (Figs. 1D, E, S3). This is consistent with the previous antibody studies, which indicated that EspEmm was localized to polar regions of the cell surface (Carlsson et al., 2009). These results suggest that the cellular proteins and signals necessary for determining the site of the ESX-1 apparatus are conserved among M. marinum, M. smegmatis and M. tuberculosis.
Localization of EccCbms is dependent on other ESX-1 proteins
We reasoned that protein localization of EccCb or other ESX-1 components reflects their incorporation into a macromolecular complex, and that in the absence of an intact apparatus these proteins would not localize appropriately. Therefore, proteins that showed distinct localization in wild-type M. smegmatis were used as reporters for assembly and localization of the apparatus in ESX-1 mutant backgrounds. As shown above, EccCbms-YFP (as well as EccCbmt-YFP, Fig. 1E) primarily localized to a single cell pole when expressed in wild-type M. smegmatis. When EccCbms-YFP was expressed in a mutant strain containing a large esx1 deletion (Δ0056-0082; Fig. 1B), this ESX-1 protein no longer localized to the cell pole (Fig. 2A). These data confirm that the localization of ESX-1-associated proteins is specific and not an artifact of protein aggregation or expression. EccCbms-YFP must make specific contacts with esx1-encoded proteins that recruit it to the polar region. Current models of the ESX-1 apparatus posit that EccCb may be tethered through interactions with an integral membrane protein, EccCa (Fig. 1A; Abdallah et al., 2007). However, EccCb-YFP did not show polar localization in a background with a smaller deletion (ΔeccCb-esxA) encoding an intact EccCams (Fig. 2B). This observation indicates that the recruitment of EccCbms-YFP to the cell pole requires other esx1-encoded proteins in addition to EccCams.
Fig. 2.
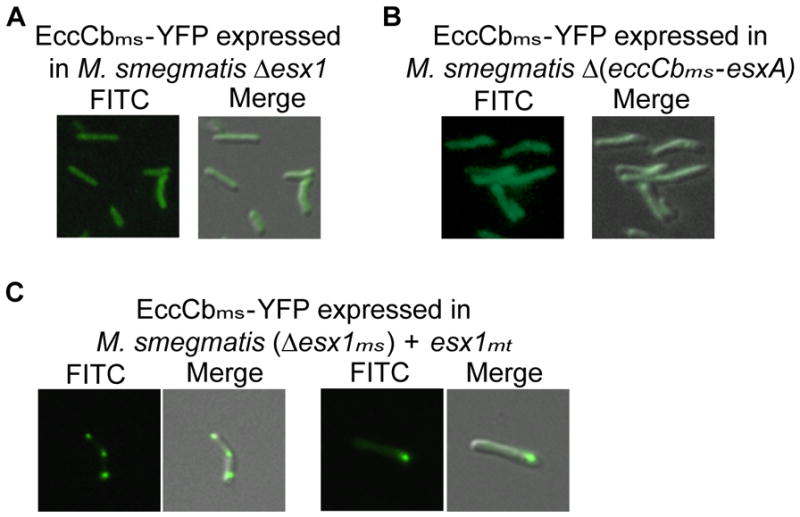
Localization of EccCbms is dependent on other ESX-1-associated proteins. In an M. smegmatis Δesx1 mutant, (A), or a ΔeccCb-esxA mutant (B), EccCbms-YFP no longer assembled at the cell pole and appeared diffuse throughout the cell. Expression of ESX-1mt completely restores polar localization of EccCbms-YFP in the absence of ESX-1ms (C).
Functional conservation between the components of M. smegmatis and MtbESX-1 has been repeatedly observed. For example, inactivating transposon insertions in M. smegmatis esx1 genes affect DNA transfer and can be ameliorated by complementation with a cosmid carrying Mtb genes (Rv3861-Rv3885), which encompasses the entire Mtbesx1 locus (Rv3864-Rv3883; Fig. 1B; Flint et al., 2004). Therefore, we assessed the ability of this esx1mt cosmid to complement the EccCbms localization defect of the Δesx1ms. Expression of ESX-1mt restored polar localization of EccCbms in the absence of ESX-1ms (Fig. 2C). This result further underscores the functional conservation of the ESX-1 apparatus between M. smegmatis and Mtb, and suggests that the MtbESX-1 apparatus is also recruited to, and assembled at, the cell pole, while incorporating EccCbms-YFP.
In addition to the above experiments, we also assessed the ability of the EccCb-YFP-fusion to rescue conjugal DNA transfer to further demonstrate that the protein is expressed in a functional form. A transposon insertion in eccCbms reduces transfer to undetectable levels (<1 transfer event per 109 donor cells). Expression of eccCbms-YFP in the mutant background restored DNA transfer over 500-fold (5.5 × 10−7 events per donor). Western analyses confirmed that the fusion protein was expressed at similar levels in both wild-type and mutant backgrounds, and that there was minimal proteolytic cleavage of the full-length fusion protein (Fig S4). Together, these data suggest that the EccCb-YFP-fusion protein is functional and that fluorescent foci are sites of functional ESX-1 complexes containing intact EccCb-YFP.
Non-esx1 encoded proteins also localize to the cell poles of M. smegmatis
Transposon mutagenesis screens have identified genes that are not encoded from within the esx1ms genetic locus, yet are required for both ESX-1-dependent conjugal DNA transfer and EsxAB secretion (Coros et al., 2008, and this work). Non-esx1-encoded proteins may directly affect the structure of the ESX-1 apparatus itself, or through indirect means, as might be expected from a transcription factor or protein modification enzyme. In particular, we isolated multiple insertions in a three-gene operon, Msmeg0044-0046 (which we will refer to as saeA-C ), located 7-kb upstream from the start of the esx1ms locus (Fig. 3A). Ectopic expression of the last gene in this three-gene operon, SaeC, as a YFP-Venus fusion resulted in its discrete localization to a single polar region in the majority of fluorescing donor cells (Figs. 3B, C and S2H). This uni-polar localization is consistent with the localization of the esx1-encoded proteins described above, and suggests that proteins encoded in this non-esx1 operon are also integral components of the ESX-1 secretory apparatus.
Fig. 3.

Proteins encoded by the non-esx1 genes saeA-C are essential for ESX-1 function and localize to discrete foci at the cell pole. (A) Genetic map of the sae operon and flanking genes. (B) A wide field FITC image (left panel) showing polar localization of YFP-tagged SaeC, and enlarged images of wild-type bacteria featured in the boxed areas. (C) Merged images of a single cell showing YFP-tagged SaeC fluorescing at a cell pole. (D) YFP-tagged SaeA is also localized to cell poles. 630× total magnification.
A precise deletion of saeC was created, and this mutation abolished EsxAB secretion (Fig 4), consistent with the known phenotype of saeA and saeB mutants (Coros et al., 2008). Deletion of saeC in the recipient strain abolished conjugal DNA transfer, but transfer was rescued over a 1000-fold by ectopic expression of either the wild-type or the fluorescently tagged SaeC to within 10-fold of wild-type levels (Table 1). This complementation is consistent with expression of a functional fluorescently tagged protein that is localized to a cell pole.
Fig. 4.
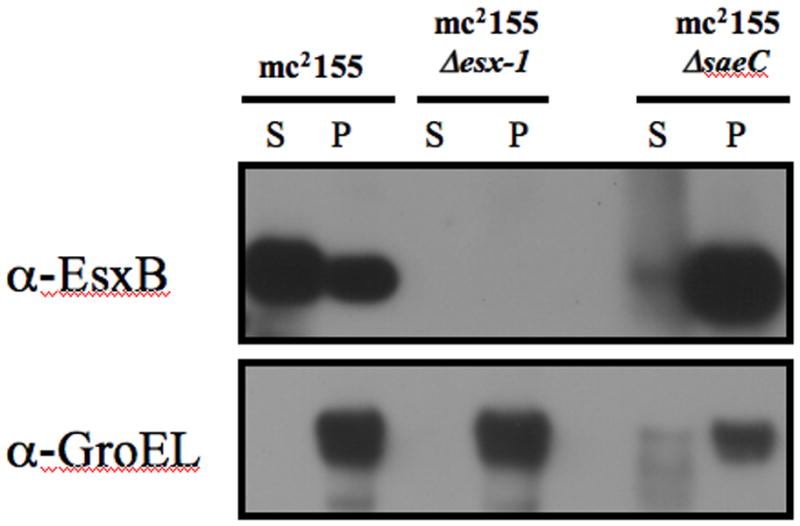
SaeC is required for EsxB secretion. The top panel shows that mc2155 expresses and secretes EsxB into the culture supernatant (S). By contrast, in the mc2155ΔsaeC deletion strain, EsxB accumulates in the cell pellet (P), indicating SaeC is required for EsxB secretion. The esx1 deletion strain does not express EsxB. The lower panel demonstrates that GroEL (a marker for cytoplasmic proteins) was detected primarily in the cell pellet, indicating very little cell leakage occurred into the culture filtrate.
Table 1.
Results of filter mating assays.
| Conjugation pair (selectable marker) | Transfer frequency of recipient (transconjugants/recipients) * |
|---|---|
| MKD6 (Km) × MKD8 (Sm) | 1.02 × 10−4 ± 0.6 |
| MKD6 (Km) × MKD8ΔsaeC (Hyg) | < 1.00 × 10−9 ± 0 ** |
| MKD6 (Km) × MKD8 ΔsaeC + saeC (Apy) | 7.7 × 10−6 ± 0.6 |
| MKD6 (Km) × MKD8 ΔsaeC + saeC-YFP (Apy) | 9.0 × 10−6 ± 0.5 |
values are the average of at least 3 mating experiments
no transconjugants were detected after 8 separate mating experiments
SaeA (Fig. 3D), but not SaeB (fusions of this protein were toxic to the cell), also localized to a cell pole, though its localization was conditional. When M. smegmatis was cultured in the presence of 0.05% Tween 80, SaeA-YFP did not localize to the poles; instead, cells appeared swollen with punctate fluorescence distributed throughout the cytoplasm (Fig. S1B). By contrast, growth in the absence of Tween 80 resulted in the localization of SaeA-YFP to the pole in both donor (Fig. 3D) and recipient cells (Figs. S2I-L). We examined the effect of Tween 80 on the other fusion proteins, but it had no effect on protein localization (data not shown). We speculate that the detergent properties of Tween 80, combined with overexpression of SaeA-YFP, alters the cell wall structure and perturbs ESX-1 assembly and localization.
ESX-1 proteins co-localize at the cell pole
So far we have presented evidence that independent ESX-1-associated proteins localize to the polar region of M. smegmatis. In order to show that these proteins assemble at a specific and unique cellular location, we co-expressed proteins fused with differently fluorescing proteins. SaeC-YFP was co-expressed with either EspEms-TdTomato or EccCbms-TdTomato from a bi-cistronic episomal expression vector. In all cases (n=49), when dual fluorescence was observed within a cell it occurred at the same pole, as indicated by merging the images (Fig. 5). Importantly, SaeC co-localization with both EspE and EccCb shows that ESX-1 associated proteins, regardless of whether they are encoded within the esx1 locus or elsewhere, traffic to the same location.
Fig. 5.
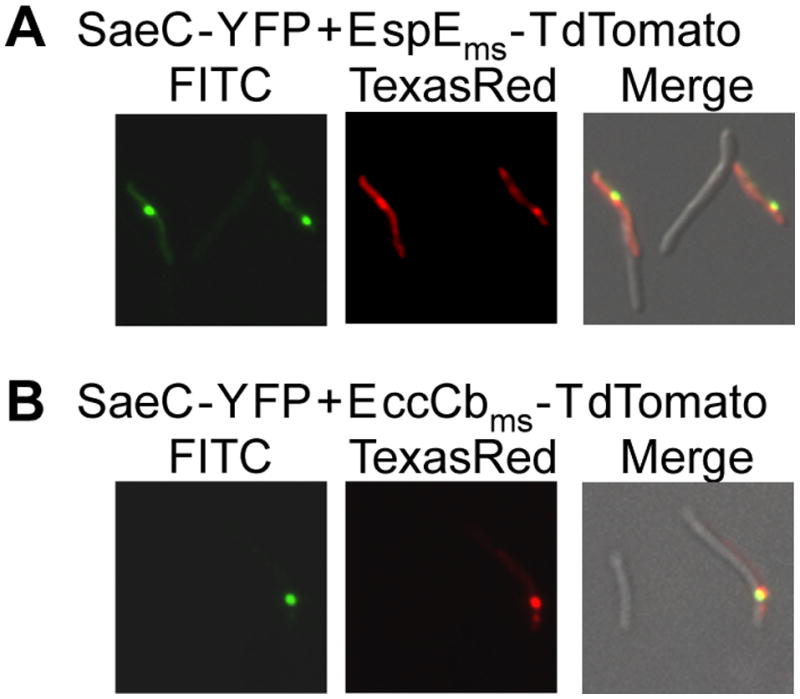
Proteins crucial to ESX-1 function co-localize in the polar region of M. smegmatis cells. (A) The non-esx1-encoded protein, SaeC-YFP, and EspEms-TdTomato co-localize when co-expressed in the same wild-type cell. (B) SaeC-YFP also co-localizes with EccCbms-TdTomato in the polar region. 630× total magnification
ESX-1 primarily associates with the non-septal pole
In the majority of cells, the fluorescent ESX-1 components are observed at a single cell pole, suggesting that the poles are functionally distinct. To unambiguously evaluate polar identity, we compiled time-lapse fluoromicroscopy images of M. smegmatis ectopically expressing SaeC-YFP. We used live-cell imaging to follow the localization of SaeC over time in multiple, independent, clonal lineages. With image capture at 10 minute intervals, each cell division was observed, with a definitive assignment of each resulting “old” and “new” pole (Fig. 6 and Movie S1, Movie S2).
Fig. 6.

ESX-1 primarily associates with the “old” cell pole. SaeC-YFP localized to the old cell pole of M. smegmatis mc2155 cells. Bacteria were loaded into a heated flow-cell growth environment and fluorescent images were captured every 10 minutes (A to D) with a semi-automated imaging system. Black arrows, old pole. Red arrow, site of new (septal) pole.
The fluorescence of the SaeC-YFP fusion is readily discernable in the time-lapse images and localizes to a single pole, in agreement with the conventional fluoromicroscopy images. Fig. 6 shows the division of two cells into four, and the appearance of three new foci. Initially, a single focus is observed at an old cell pole (black arrow) and this localized fluorescence remains associated with that pole following cell division at the septum (red arrow). Here, “old pole” is defined as the pole distal to the divisional septum (“new pole”). New fluorescent foci then appear in the other, previously non-fluorescing, daughter cells: in two of these daughter cells the foci are localized at the old pole (black arrows), while in the other daughter cell the protein is localized to the new pole (where division occurred). In 35/47 cells that had undergone cell division, in 8 independently monitored cell clusters, SaeC-YFP was localized to the old pole. Four foci appeared at the new pole, while the remaining foci (8) could not be properly assigned because there is increasing ambiguity due to cells clumping and moving as the cell number increased.
SaeC is essential for polar localization of EccCbms
As proteins encoded from the saeA-C operon are co-localized with ESX-1 proteins, we examined whether their localization was dependent on a complete ESX-1 apparatus. Surprisingly, SaeC-YFP was localized to the cell pole even in the absence of ESX-1 core components showing that its polar localization is independent of esx1-encoded proteins (Fig. 7A). Conversely, to determine whether esx1-encoded components are dependent on Sae proteins, EccCbms-YFP localization was evaluated in a ΔsaeA-C mutant background. EccCbms-YFP was not localized in this background, even though the esx1 locus was intact (Fig. 7B). A similar phenotype was also observed in a mutant background with a precise deletion of saeC (Fig. 7C). Together, these data suggest that the SaeA-C proteins, and SaeC in particular, initiate an early step in the assembly of the esx1-encoded proteins at the pole. We also predict that the defect in DNA-transfer and EsxA/EsxB secretion in Δsae strains is because ESX-1 is either not assembled, or is mis-localized.
Fig. 7.

Localization of EccCbms is dependent on SaeC, but localization of SaeC is not dependent on ESX-1. SaeC, localizes to the poles in the absence of ESX-1 core components (Δesx1) (A). EccCbms-YFP was not localized in either a ΔsaeA-saeC (B), or a ΔsaeC mutant background (C), 630× total magnification.
Discussion
Despite the focused research on ESX-1 and related T7S systems, the composition of the secretion apparatus and the mechanism of secretion are still unknown. For example, in most cases, it is not known whether genes required for ESX-1 activity perform a structural role or modulate secretion through indirect regulatory contributions, such as chaperone activity. Protein localization will help to differentiate between these possibilities, and inform on how the secretion apparatus is assembled.
The fusion of monomeric fluorescent proteins, primarily YFP-Venus in this study, with proteins known to affect ESX-1 function, provides a non-invasive way to visualize where in the cell the secretion system is assembled. This study addresses fundamental questions concerning the ESX-1 secretion machine. Does a given Esx protein localize within the cell and, if so, does it co-localize with other ESX-1 proteins, suggesting it plays a structural role? If multiple ESX-1 structural (localized) proteins are used as reporters, can mutations in other genes define the order-of-assembly of the apparatus? YFP fusions of two known esx1-encoded proteins, EspEms and EccCbms, revealed that these proteins localize to a single cell pole in the bacillus, corroborating a previous report in M. marinum with immunodetection of EspEmm and EccCamm (Carlsson et al., 2009). Moreover, proteins encoded by a non-esx1 operon, saeA-C, (Msmeg0044-0046), previously reported to affect ESX-1 function, are also polar localized. The use of two-color fluoromicroscopy showed that SaeC-YFP localized to the same pole as the esx1-encoded fusion proteins, consistent with its functional requirement for ESX-1 mediated secretion and DNA transfer activities. When EccCbms was used as a reporter for ESX-1 assembly, we identified mutations that prevented its polar localization. This could be interpreted as the loss of a specific interaction needed to recruit EccCbms to the secretion apparatus, or as dissolution of the apparatus itself. The use of other ESX-1 structural protein reporters will help to distinguish between these possibilities. Notably, deletion of esx1 genes prevented assembly of EccCbms at the cell pole indicating that other esx1-encoded proteins are necessary for localization. These proteins may act as chaperones or as scaffolds for the structural proteins.
Our demonstration that heterologous expression of the Mtbesx1 operon restored localization of EccCbms in a Δesx1ms background implies that the Mtbesx1 locus recruited EccCbms to the cell pole and further establishes the functional conservation of the ESX-1 apparatus between pathogenic and saprophytic species. This conservation is consistent with the hypothesis that the observed species-specific functional differences (e.g., virulence in Mtb or conjugation in M. smegmatis) are due to differences in secreted proteins, not differences in the secretory apparatus per se. By examining different combinations of YFP-reporter and esx1 mutants, we are beginning to construct a hierarchical interaction map that outlines the potential protein interactions necessary to assemble and form the apparatus.
Localization of EccCbms was also dependent on proteins encoded by saeA-C. This result, combined with the co-localization of SaeC with ESX-1 proteins, suggests that SaeA-C play a role as “keystones” in the ESX-1ms assembly process. Therefore, we propose that this operon be named the sae operon, reflecting the role of the genes (saeABC) in both the scaffolding and assembly of the ESX-1 apparatus. The proteins encoded by this operon have no close homologs in Mtb, suggesting that other proteins might perform the equivalent functional role in Mtb, and these are still to be discovered. Weak Sae homologs exist in more distantly related bacteria such as Rhodococcus and Corynebacterium species, but no function has been assigned to these hypothetical proteins and, as some of these species lack identifiable esx1 genes, the homologs likely have evolved to perform different roles.
The approach used here has limitations in its application and interpretation. Some proteins appear to be refractory to this type of analysis, producing little or no fluorescent signal (Msmeg0071), inconsistent localization patterns, or sick cells (SaeB). While any protein can be tagged and expressed, an ~30 kDa tag may impede protein function or interaction with partner proteins. To address this, we have made both N- and C-terminal fusions, and used a penta-glycine linker joining the fluorescent protein with the target protein to minimize steric interference. Additionally, we have shown that fusion proteins are functionally active and that polar localization of the EccCb-YFP-fusion requires the esx1 operon. Ectopic over-expression of fusion proteins might also contribute to toxicity issues, although we observed little improvement in the apparent health of the cells, or the intensity of the fluorescence using different promoters to drive protein expression (constitutive Hsp60 and inducible Ptet, data not shown). Indeed, the fact that foci were not visible in all cells for a given fusion protein (e.g. for EccCb-YFP, 37 out of 62 cells screened contained polar foci, while the remaining cells exhibited diffuse fluoresence; Fig. S2) indicates inherent heterogeneity in mycobacterial cultures and implies that not all cells have an assembled apparatus.
The non-invasive, real-time assay was exploited to determine the preferred site of ESX-1 assembly. Time-lapse imaging clearly showed that ESX-1 proteins prefer to locate at the “old” cell pole and not at the “new” septal pole. This interpretation differs from a previous report, which suggested that ESX-1 was positioned at the “new” pole (Carlsson et al., 2009). However, the discrepancy is not a disagreement of data, but results from the ambiguous terminology of “old” and “new” pole. Our time-lapse microscopy definitively discriminates between the pre-existing (old) pole, and that generated by septation (new pole). The confusion arises because mycobacteria, like other Actinobacteria, grow apically, i.e. new cell material is added at existing cell tips (old poles) and not at the developing septum (new poles) (Flardh, 2010). It is possible that the apparent peri-polar location of some foci is actually a consequence of the new growth transiently displacing the ESX-1 apparatus from the growing tip.
Unique cellular localization of macromolecular complexes is a common feature of cellular processes such as cell division and DNA replication, and secretion machines are no exception (Burton and Dubnau, 2010). The type IV secretion systems encoded by Agrobacterium tumefacians and Helicobacter pylori are known to assemble at the cell poles (Burton and Dubnau, 2010, Judd et al., 2005, Kutter et al., 2008). Competence proteins responsible for uptake of DNA in Bacillus subtilis are also localized to the cell pole, as is the conjugation machinery necessary for transfer of ICEBs1 in B. subtilis and pSVH1 in Streptomyces (Hahn et al., 2005, Kaufenstein et al., 2011, Reuther et al., 2006). Although the cell pole seems to be a preferred site of DNA translocation in many species and even for attachment of mycobacteriophage (Edgar et al., 2008), so far there is no evidence suggesting that DNA transfer occurs at the growing pole during mycobacterial conjugation. ESX-1 is required for recipient activity, but we do not know whether the ESX-1 apparatus directly mediates DNA uptake. Moreover, esx1 donor mutants are hyper-conjugative indicating ESX-1 suppresses, and does not transport DNA out of the donor cell.
Currently, there is an interesting debate on how a polar site is recognized by a protein. Most recently, localization of several proteins has been suggested to be mediated by their ability to recognize membrane curvature i.e., geometry is the determining factor (Ramamurthi, 2010, Lenarcic et al., 2009, Ramamurthi et al., 2009). However, others have shown that polar-localized proteins needed for DNA uptake in B. subtilis localize to discrete foci even in spheroplasts that have lost their rod shape (Kaufenstein et al., 2011). In the case of mycobacteria, the answer appears to be more complex. The majority of cells contained only one focus implying asymmetry of the poles and indicating localization was not simply determined by membrane curvature. This observation was reinforced by the time-lapse microscopy experiments, which showed that the old pole was the preferred site of assembly. As cell wall synthesis is occurring at this pole, this process, or a component of it, might be the signal that dictates ESX-1 localization. Furthermore, SaeC-YFP localization was independent of esx1-encoded proteins (SaeC-YFP was localized in an esx1 deletion mutant), yet SaeC was critical for localization of major ESX-1 components. Together, these observations suggest that SaeC is both an early recruit and recruiter to the cell pole in ESX-1 assembly. SaeC does not appear to be secreted, as it is not detected in culture filtrates (Fig. S5). Future studies will focus on the role of SaeC (and SaeA,B) in the mechanism and order of ESX-1ms assembly, and whether ESX-1mt assembly requires a comparable activity. The localization of ESX-1 at the site of active cell-wall synthesis suggests that ESX-1 secretion may function in the generation or modification of the mycobacterial cell wall. Such a relationship might explain the myriad effects attributed to ESX-1 in microbe-environment interactions.
Experimental procedures
Bacterial strains, plasmids and media
E. coli DH5α (Invitrogen), E. coli XL10Gold (Stratagene), and ER2925 (New England BioLabs) were used for all experiments to propagate plasmid DNA constructs. E. coli strains were cultured in Terrific Broth or on Luria-Bertani agar at 37°C, supplemented with antibiotics, as necessary, at the following concentrations: Apramycin 50 μg ml−1, Kanamycin 50 μg ml−1, and Hygromycin 100 μg ml−1. M. smegmatis donor strains were derivatives of the laboratory strain, mc2155, and have been described previously, as has the recipient strain MKD8 (Table S3; Parsons et al., 1998). M. smegmatis strains were cultured at 37°C in Trypticase Soy Broth with, or, without 0.05% Tween80 (TSBT and TSB, respectively), or on Trypticase Soy Agar (TSA) plates, supplemented with antibiotics, as necessary, at the following concentrations: Apramycin 50 μg ml−1, Kanamycin 10 μg ml−1, Hygromycin 100 μg ml−1, and Streptomycin 200 μg ml−1.
Creation of fluorescent protein fusions
A yellow fluorescent protein gene, YFPVenus, was PCR amplified using oligonucleotides TGD1536 and TGD1563 (Table S2). YFPVenus was cloned into pMP349 (Consaul and Pavelka, 2004) to create the fluorescent protein expression plasmid, pGD15 (Table S1). Failsafe™ Enzyme Mix (EPICENTRE® Biotechnologies) was used to PCR amplify genes of interest from M. smegmatis mc2155 genomic DNA, or from cell lysates of heat-killed M. tuberculosis H37Rv. PCR products were cloned between the MscI and NheI sites of pGD15 to fuse YFPVenus, in frame, to the 3′ end of the gene. All fusion constructs were sequenced to verify the fusion was in-frame and rule out PCR-induced mutations.
Creation of bi-cistronic expression plasmid for co-localization studies
A red fluorescent protein gene, TdTomato, (Shaner et al., 2004) was PCR amplified and cloned between the BsrGI and EcoRI sites in pGD15, to create pGD15_TdTomato (Table S1). pGD15_TdTomato was then used to co-express TdTomato and YFP fusions.
Microscopy
Fluorescent-protein fusion plasmids were transformed into M. smegmatis and colonies were assessed for fluorescence using a Zeiss Stemi SV6 stereomicroscope with a GFP filter (470nm). Fluorescent colonies were individually inoculated into TSBT and TSB, supplemented with 50 μg ml−1 Apramycin. Bacterial cells were examined with either a Nikon TE 2000 microscope with a Roper HQ 2-shot color cooled CCD camera or a Zeiss Axiovert 200M, with a Lambda 10-2 Filterwheel shutter controler, plus a Hamamatsu Camera and a Hamamatsu ORCA-ER camera controller; all hardware was controlled with OpenLab software. Image analysis was performed using ImageJ (NIH®) or AdobePhotoshop. Images were normalized for brightness, contrast, and resolution. Cells were scored as having 1 focus, multiple foci, diffuse fluorescence, or no fluorescence (Fig. S2), with blinded evaluation for quantitation.
Time lapse microscopy
Cells were imaged in a custom flow-cell microfluidic device made of patterned PDMS fused to cover glass, ensuring that the cells were in the focal plane and received fresh media throughout the experiment (Aldridge, B., Fernandez-Suarez, M., Heller, D., Irimia, D., Toner, M., and Fortune, S., 2012). Standard lithography methods were used to manufacture the device. Fluorescent and bright-field images were acquired every ten minutes using a DeltaVision PersonalDV microscope with a heated chamber and a 100× objective (Applied Precision Inc.). Three reviewers independently assessed the cells for the appearance of foci at an old pole, a new pole (associated with recent septation), or ambiguous (could not be properly assigned due to cell movement or clustering).
Creation of targeting substrates for recombineering
Plasmid constructs to be used as recombination substrates were based on pYUB854 and were introduced into the recombineering strain expressing Che9c recombination genes as described (van Kessel and Hatfull, 2008). Oligonucleotides and recombination plasmid substrates used for deletion of saeA-C and eccCb-esxA are described in Tables S2 and S1 respectively. The HygR gene used to replace the target gene was excised by ectopic expression of resolvase. Recombinant strains were verified by PCR and DNA sequence analysis of the resolved res site junction.
Filter mating assays
Quantitative DNA transfer experiments were carried out in triplicate and in parallel with wild-type controls as described previously (Parsons et al., 1998). Two recipient-defective mutant strains were used for matings with a wild-type donor MKD6 (KmR). A transposon insertion mutant of gene eccCbms was described previously (Coros et al., 2008, van Kessel and Hatfull, 2008). A deletion of saeC was also created in this study by a two-step process. First, a hygR allelic replacement of saeC was made in the donor strain by recombineering, as described above. The hygR marker was then transferred into the recipient strain MKD8 (SmR). The precise nature of the deletion was confirmed by PCR and DNA sequence analysis (data not shown). MKD8ΔsaeC was defective for transfer as predicted, and could be complemented in trans with the wild-type gene.
Secretion Assay
Secretion assays were performed as described previously (Coros et al, 2008). Briefly, M. smegmatis strains were grown in 10 ml of Sauton’s media containing 0.05% Tween-80 and grown with shaking at 37°C to 0.6–0.7 OD600. Culture supernatants were concentrated using Amicon Ultra-15 centrifugal filters (3 kDa, MW cut-off). Equivalent cells volumes of supernatant and pellet were heat denatured at 95°C for 15 min in SDS loading buffer. Samples were separated on a 4–12% gradient SDS-PAGE gel, transferred to a PVDF membrane (Amersham Hybond-P), and then probed with appropriate antibodies. Secondary antibodies and detection reagents were from Amersham ECL Plus detection kit (GE Healthcare) and were used according to manufacturer’s instructions. Anti-EsxB polyclonal antibodies were generated in this laboratory. Anti-GroEL and anti-rabbit antibodies were obtained from Enzo Life Sciences and Pierce/Thermo Scientific, respectively.
Supplementary Material
Acknowledgments
The authors wish to thank the Wadsworth Center Applied Genomic Technologies Core for DNA sequencing; the Light Microscopy Core for assistance with fluorescent microscopy; and the Wadsworth Center Digital Photography, Illustration and Video Unit for assistance with image analysis. Brian Callahan made the initial observations with the EccCbmt-YFP expression plasmid. SEW was initially supported by an Emerging Infectious Disease Fellow funded by the CDC, and administered by the Association of Public Health Laboratories. Funding was provided by grants AI042308, R56AI080694 and R21AI088792 to KMD and a New Innovator’s Award, DP2 0D001378, to SF.
References
- Abdallah AM, Gey van Pittius NC, Champion PA, Cox J, Luirink J, Vandenbroucke-Grauls CM, Appelmelk BJ, Bitter W. Type VII secretion-mycobacteria show the way. Nat Rev Microbiol. 2007;5:883–891. doi: 10.1038/nrmicro1773. [DOI] [PubMed] [Google Scholar]
- Aldridge BB, Fernandez-Suarez M, Heller D, Ambravaneswaran V, Irimia D, Toner M, Fortune SM. Asymmetry and aging of mycobacterial cells lead to variable growth and antibiotic susceptibility. Science. 2012;335:100–104. doi: 10.1126/science.1216166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behr MA, Wilson MA, Gill WP, Salamon H, Schoolnik GK, Rane S, Small PM. Comparative genomics of BCG vaccines by whole-genome DNA microarray. Science. 1999;284:1520–1523. doi: 10.1126/science.284.5419.1520. [DOI] [PubMed] [Google Scholar]
- Berthet FX, Rauzier J, Lim EM, Philipp W, Gicquel B, Portnoi D. Characterization of the Mycobacterium tuberculosis erp gene encoding a potential cell surface protein with repetitive structures. Microbiology. 1995;141:2123–2130. doi: 10.1099/13500872-141-9-2123. [DOI] [PubMed] [Google Scholar]
- Bitter W, Houben EN, Bottai D, Brodin P, Brown EJ, Cox J, Derbyshire KM, Fortune SM, Gao LY, Liu J, Gey van Pittius NC, Pym AS, Rubin EJ, Sherman DR, Cole ST, Brosch R. Systematic genetic nomenclature for Type VII secretion systems. PLoS Pathog. 2009;5:e1000507. doi: 10.1371/journal.ppat.1000507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton B, Dubnau D. Membrane-associated DNA transport machines. Cold Spring Harb Perspect Biol. 2010;2:a000406. doi: 10.1101/cshperspect.a000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson F, Joshi SA, Rangell L, Brown EJ. Polar localization of virulence-related Esx-1 secretion in mycobacteria. PLoS Pathog. 2009;5:e1000285. doi: 10.1371/journal.ppat.1000285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie PJ, Atmakuri K, Krishnamoorthy V, Jakubowski S, Cascales E. Biogenesis, architecture and function of bacterial Type IV secretion sytems. Annual Review of Microbiology. 2005;59:451–485. doi: 10.1146/annurev.micro.58.030603.123630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consaul SA, Pavelka MS., Jr Use of a novel allele of the Escherichia coli aacC4 aminoglycoside resistance gene as a genetic marker in mycobacteria. FEMS Microbiol Lett. 2004;234:297–301. doi: 10.1016/j.femsle.2004.03.041. [DOI] [PubMed] [Google Scholar]
- Converse SE, Cox JS. A protein secretion pathway critical for Mycobacterium tuberculosis virulence is conserved and functional in Mycobacterium smegmatis. J Bacteriol. 2005;187:1238–1245. doi: 10.1128/JB.187.4.1238-1245.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coros A, Callahan B, Battaglioli E, Derbyshire KM. The specialized secretory apparatus ESX-1 is essential for DNA transfer in Mycobacterium smegmatis. Mol Microbiol. 2008;69:794–808. doi: 10.1111/j.1365-2958.2008.06299.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGiuseppe Champion PA, Cox JS. Protein secretion systems in Mycobacteria. Cell Microbiol. 2007;9:1376–1384. doi: 10.1111/j.1462-5822.2007.00943.x. [DOI] [PubMed] [Google Scholar]
- Edgar R, Rokney A, Feeney M, Semsey S, Kessel M, Goldberg MB, Adhya S, Oppenheim AB. Bacteriophage infection is targeted to cellular poles. Mol Microbiol. 2008;68:1107–1116. doi: 10.1111/j.1365-2958.2008.06205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrt S, Guo XV, Hickey CM, Ryou M, Monteleone M, Riley LW, Schnappinger D. Controlling gene expression in mycobacteria with anhydrotetracycline and Tet repressor. Nucleic Acids Res. 2005;33:e21. doi: 10.1093/nar/gni013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flardh K. Cell polarity and the control of apical growth in Streptomyces. Curr Opin Microbiol. 2010;13:758–765. doi: 10.1016/j.mib.2010.10.002. [DOI] [PubMed] [Google Scholar]
- Flint JL, Kowalski JC, Karnati PK, Derbyshire KM. The RD1 virulence locus of Mycobacterium tuberculosis regulates DNA transfer in Mycobacterium smegmatis. Proc Natl Acad Sci U S A. 2004;101:12598–12603. doi: 10.1073/pnas.0404892101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortune SM, Jaeger A, Sarracino DA, Chase MR, Sassetti CM, Sherman DR, Bloom BR, Rubin EJ. Mutually dependent secretion of proteins required for mycobacterial virulence. Proc Natl Acad Sci U S A. 2005;102:10676–10681. doi: 10.1073/pnas.0504922102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gey Van Pittius NC, Gamieldien J, Hide W, Brown GD, Siezen RJ, Beyers AD. The ESAT-6 gene cluster of Mycobacterium tuberculosis and other high G+C Gram-positive bacteria. Genome Biol. 2001;2:1–18. doi: 10.1186/gb-2001-2-10-research0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn J, Maier B, Haijema BJ, Sheetz M, Dubnau D. Transformation proteins and DNA uptake localize to the cell poles in Bacillus subtilis. Cell. 2005;122:59–71. doi: 10.1016/j.cell.2005.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes CS, Aoki SK, Low DA. Bacterial contact-dependent delivery systems. Annu Rev Genet. 2010;44:71–90. doi: 10.1146/annurev.genet.42.110807.091449. [DOI] [PubMed] [Google Scholar]
- Hsu T, Hingley-Wilson SM, Chen B, Chen M, Dai AZ, Morin PM, Marks CB, Padiyar J, Goulding C, Gingery M, Eisenberg D, Russell RG, Derrick SC, Collins FM, Morris SL, King CH, Jacobs WR., Jr The primary mechanism of attenuation of bacillus Calmette-Guerin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proc Natl Acad Sci U S A. 2003;100:12420–12425. doi: 10.1073/pnas.1635213100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judd PK, Kumar RB, Das A. Spatial location and requirements for the assembly of the Agrobacterium tumefaciens type IV secretion apparatus. Proc Natl Acad Sci U S A. 2005;102:11498–11503. doi: 10.1073/pnas.0505290102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufenstein M, van der Laan M, Graumann PL. The three-layered DNA uptake machinery at the cell pole in competent Bacillus subtilis cells is a stable complex. J Bacteriol. 2011;193:1633–1642. doi: 10.1128/JB.01128-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutter S, Buhrdorf R, Haas J, Schneider-Brachert W, Haas R, Fischer W. Protein subassemblies of the Helicobacter pylori Cag Type IV secretion system revealed by localization and interaction studies. J Bacteriol. 2008;190:2161–2171. doi: 10.1128/JB.01341-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenarcic R, Halbedel S, Visser L, Shaw M, Wu LJ, Errington J, Marenduzzo D, Hamoen LW. Localisation of DivIVA by targeting to negatively curved membranes. EMBO J. 2009;28:2272–2282. doi: 10.1038/emboj.2009.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis KN, Liao R, Guinn KM, Hickey MJ, Smith S, Behr MA, Sherman DR. Deletion of RD1 from Mycobacterium tuberculosis mimics bacille Calmette-Guerin attenuation. J Infect Dis. 2003;187:117–123. doi: 10.1086/345862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGurn JA, Raghavan S, Stanley SA, Cox JS. A non-RD1 gene cluster is required for Snm secretion in Mycobacterium tuberculosis. Mol Microbiol. 2005;57:1653–1663. doi: 10.1111/j.1365-2958.2005.04800.x. [DOI] [PubMed] [Google Scholar]
- Mahairas GG, Sabo PJ, Hickey MJ, Singh DC, Stover CK. Molecular analysis of genetic differences between Mycobacterium bovis BCG and virulent M. bovis. J Bacteriol. 1996;178:1274–1282. doi: 10.1128/jb.178.5.1274-1282.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat Biotechnol. 2002;20:87–90. doi: 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- Pallen MJ. The ESAT-6/WXG100 superfamily -- and a new Gram-positive secretion system? Trends Microbiol. 2002;10:209–212. doi: 10.1016/s0966-842x(02)02345-4. [DOI] [PubMed] [Google Scholar]
- Papanikou E, Karamanou S, Economou A. Bacterial protein secretion through the translocase nanomachine. Nat Rev Microbiol. 2007;5:839–851. doi: 10.1038/nrmicro1771. [DOI] [PubMed] [Google Scholar]
- Parsons LM, Jankowski CS, Derbyshire KM. Conjugal transfer of chromosomal DNA in Mycobacterium smegmatis. Mol Micro. 1998;28:571–582. doi: 10.1046/j.1365-2958.1998.00818.x. [DOI] [PubMed] [Google Scholar]
- Pym AS, Brodin P, Brosch R, Huerre M, Cole ST. Loss of RD1 contributed to the attenuation of the live tuberculosis vaccines Mycobacterium bovis BCG and Mycobacterium microti. Mol Microbiol. 2002;46:709–717. doi: 10.1046/j.1365-2958.2002.03237.x. [DOI] [PubMed] [Google Scholar]
- Ramamurthi KS. Protein localization by recognition of membrane curvature. Curr Opin Microbiol. 2010;13:753–757. doi: 10.1016/j.mib.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramamurthi KS, Lecuyer S, Stone HA, Losick R. Geometric cue for protein localization in a bacterium. Science. 2009;323:1354–1357. doi: 10.1126/science.1169218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renshaw PS, Panagiotidou P, Whelan A, Gordon SV, Hewinson RG, Williamson RA, Carr MD. Conclusive evidence that the major T-cell antigens of the Mycobacterium tuberculosis complex ESAT-6 and CFP-10 form a tight, 1:1 complex and characterization of the structural properties of ESAT-6, CFP-10, and the ESAT-6-CFP-10 complex. Implications for pathogenesis and virulence. J Biol Chem. 2002;277:21598–21603. doi: 10.1074/jbc.M201625200. [DOI] [PubMed] [Google Scholar]
- Reuther J, Gekeler C, Tiffert Y, Wohlleben W, Muth G. Unique conjugation mechanism in mycelial streptomycetes: a DNA-binding ATPase translocates unprocessed plasmid DNA at the hyphal tip. Mol Microbiol. 2006;61:436–446. doi: 10.1111/j.1365-2958.2006.05258.x. [DOI] [PubMed] [Google Scholar]
- Rokney A, Shagan M, Kessel M, Smith Y, Rosenshine I, Oppenheim AB. E. coli transports aggregated proteins to the poles by a specific and energy-dependent process. J Mol Biol. 2009;392:589–601. doi: 10.1016/j.jmb.2009.07.009. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow florescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- Simeone R, Bottai D, Brosch R. ESX/Type VII secretion systems and their role in host-pathogen interaction. Curr Opin Microbiol. 2009;12:4–10. doi: 10.1016/j.mib.2008.11.003. [DOI] [PubMed] [Google Scholar]
- Stover CK, de la Cruz VF, Fuerst TR, Burlein JE, Benson LA, Bennett LT, Bansal GP, Young JF, Lee MH, Hatfull GF, et al. New use of BCG for recombinant vaccines. Nature. 1991;351:456–460. doi: 10.1038/351456a0. [DOI] [PubMed] [Google Scholar]
- Tseng TT, Tyler BM, Setubal JC. Protein secretion systems in bacterial-host associations, and their description in the Gene Ontology. BMC Microbiol. 2009;9(Suppl 1):S2. doi: 10.1186/1471-2180-9-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kessel JC, Hatfull GF. Mycobacterial recombineering. Methods Mol Biol. 2008;435:203–215. doi: 10.1007/978-1-59745-232-8_15. [DOI] [PubMed] [Google Scholar]
- Zhu W, Banga S, Tan Y, Zheng C, Stephenson R, Gately J, Luo ZQ. Comprehensive identification of protein substrates of the Dot/Icm Type IV transporter of Legionella pneumophila. PLoS One. 2011;6:e17638. doi: 10.1371/journal.pone.0017638. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.