

Abstract
Treatment of an unsymmetrical bis(imino)pyridyl-thiolate zinc(II) complex [ZnII(LN3S)(OTf)] (1) with LiAlH4 results in the double reduction of the two imino groups in the ligand backbone, and at the same time causes a rare transmetalation reaction to occur. The products formed in this reaction are two novel aluminium(III) bis(amido)pyridyl-thiolate complexes [(R,S/S,R-[AlIII(LH2N3S)(THF)] (2a) and [(R,R/S,S-[AlIII(LH2N3S)(THF)] (2b), which are diastereomers of each other. These complexes have been characterized by single-crystal X-ray diffraction and 1H NMR spectroscopy. Single crystal X-ray structure analysis shows that the AlIII ion is bound in an almost idealized square pyramidal geometry in 2a, while being held in a more distorted square pyramidal geometry in 2b. The major difference between 2a and 2b arises in the orientation of the terminal methyl groups of the ligand backbone in relation to the AlIIIN3S plane. These two complexes are crystallized at different temperatures (room temperature vs −35 °C), allowing for their separate isolation. Structural analysis shows that these complexes are reduced by the formal addition of one hydride ion to each imino group, resulting in a deprotonated bis(amido)pyridyl-thiolate ligand. A detailed analysis of metrical parameters rules out the possibility of pure one- or two-electron reduction of the π-conjugated bis(imino)pyridine framework. 1H NMR spectra reveal a rich pattern in solution indicating that the solution state structures for 2a and 2b match those observed in the solid-state crystal structures, and reveal that both complexes are severely conformationally restricted. Direct organic synthetic methods failed to produce the reduced bis(amino)pyridyl-thiol ligand in pure form, but during the course of these efforts an unusual unsymmetrical aminopyridyl ketone, 1-(6-(1-(2,6-diisopropylphenylamino)ethyl)pyridin-2-yl)ethanone was synthesized in good yield and can be used as a possible precursor for further ligand development.
Keywords: aluminum, bis(imino)pyridine, transmetalation, zinc, ligand reduction, ligand design
1. Introduction
The tridentate 2,6-bis(imino)pyridine (2,6-BIP) platform has played an important role in a broad range of reactions, including olefin polymerizations, Fe- and Co-centered dinitrogen reduction, and alkane oxidation catalysis [1, 2]. It also exhibits rich redox behavior, providing a site for storing reducing equivalents in conjunction with redox-active metals (e.g. Fe, Ni) [3–10]. Given the usefulness and versatility of this ligand, significant synthetic efforts have gone into modifying the steric and electronic properties of the BIP-type framework, either by substitution of the typical aryl imino substituents, or other methods [11—17]. A particular challenge arises when confronted with preparing desymmetrized analogs of the parent BIP ligand 2,6-(ArN=CMe)2(C5H3N) in which the Ar substituents are different. These unsymmetrical derivatives are desirable for applications from catalysis [18–20] to synthetic bioinorganic analogs of enzyme active sites [21]. With regard to the latter goal, we are interested in developing mixed N,S donor ligands for preparing models of non-heme iron enzymes such as superoxide reductase and cysteine dioxygenase (CDO) [22–27]. Ligand design to target these centers is non-trivial because of the difficulties posed in making unsymmetrical N,S-systems. We recently employed the BIP platform to successfully prepare the unsymmetrical complex [FeII(LN3S)(OTf)] via a metal-assisted template reaction, which served as a structural and functional model of the His3Cys-FeII center found in substrate-bound CDO [26].
To modify, and possibly enhance, the reactivity of our nonheme iron models with O2, we sought a more electron-rich version of the LN3S ligand, and therefore targeted the synthesis of the reduced, bis(amino)pyridine analog. Although bis(amino)pyridine systems have been sought for their use in catalysis, relatively few such compounds have been prepared [14, 28, 29]. For example, the synthesis of 2,6-bis(2,6-diisopropylphenylaminomethyl)pyridine was accomplished by addition of LiNHAr (Ar = 2,6-di-isopropylphenyl) to 2,6-bis(bromomethyl)pyridine [28]. However, this double alkylation strategy is not practical for the synthesis of unsymmetrical derivatives.
Herein we report the successful reduction of the bis(imino) functionalities in the unsymmetrical LN3S ligand, starting from the zinc(II) complex [Zn(LN3S)(OTf)] (1). During this work a rare transmetalation reaction was discovered, leading to the isolation and structural characterization of the neutral AlIII complexes [Al(LH2N3S)(THF)] (2a–b). This facile transmetalation process has potential implications for the role of Al cocatalysts in olefin polymerizations with BIP ligands.
2. Experimental
2.1 General Procedures
All reactions were carried out under an atmosphere of N2 or Ar using a drybox or standard Schlenk techniques unless noted otherwise. All reagents were purchased from commercial vendors and used without further purification unless noted otherwise. Diethyl ether and dichloromethane were purified via a Pure-Solv Solvent Purification System from Innovative Technology, Inc. All solvents were degassed by repeated cycles of freeze-pump-thaw and then stored in an N2-filled drybox. Methanol was distilled over CaH2. The pyridyliminoketone, 2-(O=CMe)-6-(2,6-iPr2-C6H3N=CMe)-C5H3N was synthesized according to the literature [18]. The zinc complex ZnII(LN3S)(OTf) (1) was synthesized as described previously [26].
2.2 Physical Methods
NMR was performed on a Bruker Avance 400 MHz FT-NMR spectrometer at 25 °C. Elemental analysis was performed by Atlantic Microlab Inc., Norcross, GA. LDI-ToF mass spectra were obtained using a Bruker Autoflex III Maldi ToF/ToF instrument (Billerica, MA). Samples were dissolved in Et2O or CH2Cl2 and deposited on the target plate in the absence of any added matrix. Samples were irradiated with a 355 nm UV laser and mass - analyzed by ToF mass spectrometry in the reflectron mode.
2.3 Synthesis of R,S/S,R-[AlIII(LH2N3S)(THF)]•1/2Et2O (2a•1/2Et2O)
To a chilled slurry of Zn(LN3S)OTf (430 mg, 0.670 mmol) in 15 mL THF at −35 °C (the solution was pre-cooled in the drybox freezer (−35 °C) for 10 min) was added 0.60 mL of LiAlH4 solution (1 M) dropwise. Bubbling was observed and the solution turned from red to a green-yellow color with the formation of a yellow precipitate as the reaction warmed to room temperature. The solution was stirred for 2 h at r.t., and then the THF was concentrated in vacuo to ca. 1 mL. Diethyl ether (10 mL) was added, and the solution was filtered on a fine frit. The filtrate was concentrated in vacuo, and slow evaporation over 1 d yielded 2a as yellow crystals (30 %). 1H NMR (CD2Cl2) δ 8.02 (t, 1H, J = 7.7 Hz, Py-Hp), 7.53 (d, 1H, J = 7.7 Hz, Py-Hm), 7.45 (d, 1H, J = 7.7 Hz, Py-Hm), 7.14-7.05 (m, 4H, Ar-H), 6.83 (t, 1H, J = 7.6 Hz, Ar-H), 6.43 (d, 1H, J = 7.3 Hz, Ar-H), 6.30 (t, 1H, J = 7.3 Hz, Ar-H), 4.82 (q, 1H, J = 6.4 Hz, CHPy), 4.30 (q, 1H, J = 6.8 Hz, CHPy), 4.00 (br, 2H, CH2 (THF), 3.77 (m, 3H, CH2(THF), one set of CHMe2 resonances is buried under this THF peak), 2.82 (m, 1H, J = 6.8 Hz, CHMe2), 1.89 (br, 4 H, CH2(THF)), 1.55 (d, 3H, J = 6.4 Hz, Me-CHPy), 1.40 (d, 3H, J = 6.8 Hz, Me-CHPy), 1.26 (d, 3H, J = 6.8 CHMe2), 1.25 (d, 3H, J = 6.8 CHMe2), 1.09 (d, 3H, J = 6.8 CHMe2), 1.02 (d, 3H, J = 6.8 CHMe2). Diethyl ether (0.5 equiv.) is found in the 1H NMR spectrum (δ 3.45, 1.15) and is in agreement with the Et2O found in the crystal lattice by X-ray structural analysis. The crystal structure of 2a also suggests that there should be minor contamination from the other racemic pair (S,S/R,R) of diastereomers. However, NMR analysis of bulk crystalline material does not reveal peaks that correspond to this isomer (2b), indicating it is not present in any significant quantity. 13C NMR (CD2Cl2) δ 163.0, 162.5, 149.5, 148.5, 148.4, 140.4, 130.5, 129.4, 124.4, 123.9, 122.9, 122.8, 119.4, 119.1, 113.4, 107.3, 65.8, 28.1, 27.4, 25.8, 25.5, 25.1, 24.0, 21.1, 20.3, 15.2. LDIMS(+): m/z 458 [2a – THF + H]+. Anal. Calcd. for C33H45AlN3O1.5S (2a•1/2(C4H10O)), C, 69.93; H, 8.00; N, 7.41. Found C, 69.38; H, 7.45; N, 8.79.
2.4 Synthesis of R,R/S,S-[AlIII(LH2N3S)(THF)] (2b)
The mother liquor from crystallizations of 2a was separated and layered with pentane (Et2O to pentane ratio 8:1), and left to stand at −35 °C for 2 d, affording X-ray quality crystals of diastereoisomer 2b. 1H NMR (CD2Cl2) δ 7.51 (t, 1H, J = 7.2 Hz, Py-Hp), 7.39 (d, 1H, J = 7.6 Hz, Py-Hm), 7.16 (d, 1H, J = 7.6 Hz, Py-Hm), 7.09 (t, 1H, J = 6.4 Hz Ar-H), 7.04-6.96 (m, 3H, Ar-H), 6.88 (d, 1H, J = 8.0 Hz, Ar-H), 6.58 (t, 2H, J = 7.6 Hz, Ar-H), 4.72 (br, 1H, CHPy), 4.20 (br, 1H, CHPy), 3.92 (br, 2H, CH2 (THF), 3.68 (br, 2H, CH2 (THF)), 3.23 (m, 2H, J = 6.8 Hz, CHMe2), 1.82 (br, 4 H, CH2(THF)), 1.61 (d, 3H, J = 6.4 Hz, Me-CHPy), 1.46 (d, 3H, J = 6.4 Hz, Me-CHPy), 1.22 (d, 6H, J = 6.8 Hz, CHMe2), 1.02 (d, 6H, J = 6.8 Hz, CHMe2).
2.5 Synthesis of 1-(6-(1-(2,6-diisopropylphenylamino)ethyl)pyridin-2-yl)ethanone (3)
Under ambient conditions, 1-(6-(1-(2,6-diisopropylphenylimino)ethyl)pyridin-2-yl)ethanone (1.0 g, 3.10 mmol) was dissolved in a 1:1 mixture of CHCl3/MeOH before NaBH4 (117 mg, 3.10 mmol) was added in small portions. The reaction was monitored by TLC, and after 2 h the reaction had reached completion. The reaction mixture was concentrated in vacuo, and the crude oil was re-dissolved in CHCl3, washed twice with water, dried and concentrated. Purification by column chromatography on silica (EtOAc/hexanes) gave 3 as pure yellow solid. Yield 561 mg (56%). 1H NMR (CDCl3) δ 8.29 (d, 1H), 7.81 (t, 1H), 7.36 (d, 1H), 7.17 (d, 2H), 7.10 (t, 1H), 4.96 (m, 1H), 4.57 (d, 1H), 2.74 (m, 2H), 2.23 (s, 3H), 1.57 (d, 3H), 1.15 (d, 12H); 13C NMR (CDCl3) δ 166.7, 162.1, 154.9, 146.6, 137.7, 136.0, 123.9, 123.3, 121.2, 120.0, 68.8, 28.6, 24.5, 23.5, 23.2, 17.6.
2.6 X-ray structure determinations
All reflection intensities were measured at 110(2) K using a KM4/Xcalibur (detector: Sapphire3) with enhanced graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) under the program CrysAlisPro (Version 1.171.33.55, Oxford Diffraction Ltd., 2010). The program CrysAlisPro (Version 1.171.33.55, Oxford Diffraction Ltd., 2010) was used to refine the cell dimensions. Data reduction was done using the program CrysAlisPro (Version 1.171.33.55, Oxford Diffraction Ltd., 2010). The structure was solved with the program SHELXS-97 (Sheldrick, 2008) and was refined on F2 with SHELXL-97 (Sheldrick, 2008) [30]. Analytical numeric absorption corrections based on a multifaceted crystal model were applied using CrysAlisPro (Version 1.171.33.55, Oxford Diffraction Ltd., 2010). The temperature of the data collection was controlled using the system Cryojet (manufactured by Oxford Instruments). The H-atoms were placed at calculated positions (except for that located on C14) using the instructions AFIX 13, AFIX 23, AFIX 43 or AFIX 137 with isotropic displacement parameters having values 1.2 or 1.5 times Ueq of the attached C atoms. The position and the isotropic temperature factor of the H atom located on C14 (i.e., H14) were refined freely. The distance C14−H14 was restrained to be 1.00(1) Å. Relevant crystallographic data and structure refinement parameters for these complexes are summarized in Table 1.
Table 1.
Crystallographic data for complexes 2a–b.
| (2a) | (2b) | |
|---|---|---|
| Chemical formula | 2(C31H40AlN3OS)·C4H10O | C31H40AlN3OS |
| Formula weight | 1133.52 | 529.70 |
| Crystal system | Triclinic | Triclinic |
| Space group | P-1 (#2) | P-1 (#2) |
| a (Å) | 8.8009 (2) | 12.54209 (18) |
| b (Å) | 13.7366 (4) | 15.8805 (2) |
| c (Å) | 13.9119 (5) | 17.5832 (3) |
| α (°) | 111.716 (3) | 68.5229 (14) |
| β (°) | 93.621 (2) | 89.6375 (12) |
| γ (°) | 91.141 (2) | 73.0413 (13) |
| V (Å3) | 1557.69 (8) | 3097.32 (8) |
| Z | 1 | 4 |
| μ (mm−1) | 0.16 | 0.16 |
| Crystal size (mm) | 0.40 × 0.25 × 0.14 | 0.42 × 0.38 × 0.21 |
| Tmin, Tmax | 0.965, 0.985 | 0.948, 0.974 |
| No. of measured, independent and observed [I > 2σ (I)] reflections | 15662, 5478, 3770 | 54514, 12182, 9655 |
| Rint | 0.049 | 0.056 |
| R[F2 > 2σ(F2)], wR(F2), S | 0.049, 0.136, 1.02 | 0.042, 0.124, 1.08 |
| No. of reflections | 5478 | 12182 |
| No. of parameters | 402 | 679 |
| No. of restraints | 43 | 0 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.32, −0.36 | 0.41, −0.47 |
2.6.1 Complex 2a
The structure of the compound C31H40AlN3OS·½(C4H10O) 2a was solved and refined in the centrosymmetric space group P 1̄ with Z’ = 1. The asymmetric unit contains one molecule of the Al complex and one lattice diethyl ether solvent molecule (with an occupancy factor of 0.5). The lattice solvent molecule is found disordered over a center of inversion (½, ½, 0). The atom C8/C8’ (methyl group located on C7) is disordered over two positions such that the chirality on C7 is either R or S. The major component of the disorder (C8) refines to an occupancy of 0.819(6). There is no evidence that suggests disorder for atom C15 (methyl group located on C14). Disorder on C15 may not occur because of steric effects from the isopropyl group C22/C23/C24. The C7 and C14 atoms have opposite chirality (i.e., C7RC14S or C7SC14R) approximately 82% of the time, and have the same chirality (i.e., C7RC14R or C7SC14S) approximately the rest of the time.
2.6.2 Complex 2b
The structure of the compound C31H40AlN3OS was solved and refined in the centrosymmetric space group P 1̄ with Z’ = 2. The asymmetric unit contains two crystallographically independent molecules of the Al complex. The diastereoisomers that are found in the crystal lattice are S,S and R,R. Some residual electron density was found in one large void volume located approximately at (½, 0, 1), which includes 65 electrons in a volume of 346 Å3. The void likely contains unresolved solvent molecules. The contribution of the unresolved residual electron density was then taken out for the subsequent stages of the refinement using the program SQUEEZE [31].
3. Results and discussion
3.1 Synthesis
The key desymmetrization for the BIP platform occurs with the synthesis of the pyridyliminoketone, 2-(O=CMe)-6-(2,6-iPr2-C6H3N=CMe)-C5H3N (Scheme 1), previously reported by Bianchini and coworkers [18]. This precursor bears a single 2,6-diisopropylphenyl substituent, which provides steric protection of the subsequently derived thiolate-ligated complexes. An obvious strategy to obtain the bis(amino) derivative involves reductive amination of the imino-ketone with 2-aminothiophenol, but this reaction failed to give the desired product under a variety of conditions. Simple condensation to give the mixed imine was also problematic; the standard methods for Schiff base formation proved unsuccessful, giving only mixtures of unidentified products or recovered starting materials (Scheme 1). A third route involved initial reduction of the pyridyliminoketone to the pyridylaminoketone 2-(O=CMe)-6-(2,6-iPr2-C6H3NHCHMe)-C5H3N 3, which proceeded smoothly with NaBH4 (Scheme 2). However, attempts to form the imine with 2-aminothiophenol and 3 also either failed to react, or gave only unwieldy mixtures of products. Compound 3 also proved to be an ineffective substrate for the metal-assisted template reaction used in the synthesis of 1. It is likely that the difficulties encountered in condensation of 3 with 2-aminothiophenol is a result of the presence of the nucleophilic thiol group. The 2-aminothiophenol is known to be problematic when condensing with ketones because both the N and S groups may add to the carbonyl carbon atom to give a thiazoline [32]. Thus compound 3 should be useful as a key precursor to forming other types of unsymmetrical, polydentate pyridine-based ligands.
Scheme 1.

Scheme 2.

Synthesis of 3.
We next investigated the reduction of the zinc complex (1) as an alternative strategy for synthesizing a reduced bis(amino) derivative (Scheme 3). Complex 1 was prepared via a zinc template reaction, as previously described [26]. Treatment of 1 with LiAlH4 at −35 °C in THF for 1 h led to a color change from red to yellow-green with the formation of a yellow precipitate. The solution was warmed and stirred for an additional 2 h at room temperature. After work-up and recrystallization from Et2O/THF, yellow crystals (blocks) suitable for crystal structure determination were obtained. The X-ray structure revealed that a surprising transmetalation reaction had occurred, and the ZnII ion was replaced by an AlIII ion bound in the tetradentate N3S(thiolate) site, giving complex 2a. The molecular structure for 2a is shown in Figure 1. A THF molecule occupies the fifth coordination site at the AlIII center, while a noncoordinating molecule of Et2O is found in the lattice in the ratio 2 (2a) : 1 Et2O. The absence of any counterions in the crystal lattice indicates that the ligand must carry a 3− charge to balance the trivalent Al ion.
Scheme 3.

synthesis of the AlIII complexes 2a and 2b.
Figure 1.

Displacement ellipsoid plot (50% probability level) of diastereomer 2a. H atoms and lattice solvent molecules are omitted for clarity.
The trianionic charge of the tetradentate ligand may arise from the reduction of the two imine bonds by addition of two hydride ions, which would lead to the former imino N donors being converted to two monoanionic amido donors. An alternate configuration would result from the addition of two electrons to the conjugated π system of the bis(imino)pyridine framework. The reduction of the conjugated π framework in aryl-substituted bis(imino)pyridine metal complexes by one or two electrons to give monoanionic or dianionic (monoradical or diradical) ligands has been observed for a number of metal complexes, including AlIII bis(imino)pyridine complexes [8]. In general this ligand “non-innocence” is of interest because of fundamental questions related to electronic structure, as well as for the potential implications regarding redox-based reactivity of the ligand-metal complex. Both 1e−- and 2e−-reductions have occurred when [Fe(iPrPDI)(Cl)2] (iPrPDI = 2,6-(2,6-iPr2C6H3N=CMe)2C5H3N) was treated with the hydride donor NaBEt3H, giving mono- or diradical products [33, 34]. Thus the possibility of electron-transfer to the ligand in 1 following treatment with LiAlH4 was carefully considered (vide infra).
3.2 Structural analysis
Relevant bond lengths and angles for 2a are summarized in Table 2, together with data for related complexes. The angles about C(7) and C(14) in 2a range between 105.9 – 115.8°, suggesting sp3 hybridization, as opposed to the angles between 113.7° – 115.7° found in the zinc(II) starting material 1, which contains sp2-hybridized imino carbon atoms. In addition, the methyl groups in 2a (C(8) and C(15)) do not lie in the same plane as the pyridine and former Cimine-N groups, as would be expected for mono- or di-radical ligand structures [8, 34]. The Cimine-Nimine distances in 1 are 1.278(3) and 1.283(3) Å. These distances are significantly elongated in 2a, C(7)–N(1) = 1.462(3) Å and C(14)–N(3) = 1.465(3) Å, whereas the C(7/14)-Cipso bonds in 2a are nearly identical to those in 1 (1.517(4), 1.520(4) Å for 4compared to 1.501(4), 1.497(4) Å in 1). In contrast, pure 1e−- or 2e−-reduction of the bis(imino)pyridine framework is expected to result in only a modest elongation of the Cimine-Nimine bonds; density functional theory (DFT) calculations for a hypothetical bis(imino)pyridine zinc(II) complex reveal 0.03 – 0.05 Å elongation of the Cimine-Nimine bonds upon 1e− or 2e− reduction [34]. Similarly, modest contractions of the Cimine-Cipso single bonds (0.04 – 0.06 Å) are calculated for the reduced ZnII structures. The same trends are observed for the crystallographically characterized 1e−- and 2e−-reduced Fe complexes [Fe(iPrPDI)(Cl)], [Fe(iPrPDI)(N2)2] and [Fe(iPrPDI)(DMAP)] [34]. Finally, the H atom on C(14) was located in the difference map of the X-ray structure for 2a, supporting the assignment of C(14) as an sp3-hybridized tertiary carbon center. All of the metrical parameters for the bis(imino)pyridine backbone in 2a are consistent with hydride addition to the imino bonds in 1, and show no evidence for 1e−- or 2e−-reduction in the conjugated π system.
Table 2.
Selected bond lengths (Å) and angles (°) for 1, 2a, 2b and [FeII(LN3S)OTf].
| 2a | 2ba | 1b | FeII(LN3S)(OTf) | |
|---|---|---|---|---|
| ref | This work | This work | 26 | 26 |
| Me-N1 | 1.887(2) | 1.8763(13) | 2.202(2) | 2.188(2) |
| M-N2 | 1.967(2) | 1.9590(13) | 2.049(2) | 2.072(2) |
| M-N3 | 1.862(2) | 1.8560(13) | 2.173(2) | 2.184(2) |
| M-S1 | 2.2798(9) | 2.2796(6) | 2.2521(8) | 2.2942(8) |
| M-O1 | 1.9320(18) | 1.9485(11) | 2.0940(18) | 2.0870(18) |
| N1-C7 | 1.462(3) | 1.4559(19) | 1.283(3) | 1.293(3) |
| N3-C14 | 1.465(3) | 1.4714(19) | 1.278(3) | 1.284(3) |
| N1-M-N2 | 79.93(9) | 79.91(5) | 74.54(9) | 73.89(9) |
| N1-M-N3 | 154.19(10) | 146.83(6) | 148.80(8) | 146.47(8) |
| N1-M-S1 | 87.18(7) | 88.84(4) | 85.33(6) | 82.95(6) |
| N1-M-O1 | 97.93(9) | 100.96(5) | 97.56(8) | 98.91(8) |
| N2-M-N3 | 81.33(9) | 80.50(5) | 75.51(9) | 74.32(8) |
| N2-M-S1 | 153.69(8) | 165.36(4) | 143.22(7) | 139.13(6) |
| N2-M-O1 | 100.03(8) | 92.17(5) | 99.75(8) | 102.77(8) |
| N3-M-S1 | 102.34(7) | 104.84(4) | 114.62(6) | 115.57(6) |
| N3-M-O1 | 102.69(8) | 106.28(6) | 95.88(8) | 98.20(8) |
| S1-M-O1 | 104.43(6) | 99.20(4) | 113.38(6) | 113.92(6) |
| N1-C7-C9 | 105.9(2) | 106.30(12) | 113.7(2) | 113.9(2) |
| N3-C14-C13 | 107.7(2) | 106.79(12) | 115.7(2) | 115.9(2) |
Data taken from only one of two distinct, but similar molecules in the asymmetric unit.
Data taken from only one of three distinct, but similar molecules in the asymmetric unit.
M equals Al1 in 2a and 2b, Zn1 in 1, Fe1 in the [FeII(LN3S)(OTf)].
The former analysis strongly indicates that the N3S donor in 2a is best described as a fully deprotonated, bis(amido)pyridylthiolate ligand. The short Al–N distances of Al–N(1) = 1.887(2) Å and Al–N(3) = 1.862(2) Å for 2a are in good agreement with other bis(amido) donors. For example, a mixed pyridyl(imino)(amido) aluminum(III) compound derived from treatment of the parent 2,6-bis(imino)pyridine ligand with AlMe3 reported by Bruce and coworkers [35] exhibits a short, deprotonated Namido–Al distance = 1.876(4) Å. These AlIII-N bonds are significantly shorter than the ZnII-N (2.202(2); 2.173(2) Å) and FeII-N (2.188(2); 2.184(2) Å) bond lengths observed in 1 and [FeII(LN3S)(OTf)], respectively [27], or the AlIII-N(H)RR’ bond lengths of 2.090(5) and 2.098(5) Å seen in the complex [((R,R)-R4-salcyan)Al(OH)] [36], where the amine donor is protonated. Furthermore, the AlIII-S distance in 2a (2.2798(9) Å) falls in the reported range for other AlIII-SR bond lengths (2.220(1) to 2.5226(6) Å) [37–39].
The geometry of the AlIII ion is nearly an idealized square pyramid (sp) based on its τ value, which is a quantitative angular parameter that distinguishes between purely square pyramidal and trigonal bipyramidal (tbp) geometries in 5-coordinate complexes. For 2a, τ = 0.01, where (sp) 0 ≤ τ ≤ 1 (tbp)) [40]. The Al atom lies 0.402(3) Å above the basal plane defined by the N3S donor set toward the axially ligated THF molecule. In comparison, the ZnII ion for 1 exhibits a more distorted square pyramidal geometry with τ = 0.09, and sits further away (0.471(4) Å) from the N3S plane. The analogous FeII complex [FeII(LN3S)(OTf)] is also distorted square pyramidal (τ = 0.12) with an even larger Fe-to-plane distance of 0.540(3) Å.
From the former structural data we conclude that complex 1 has been fully reduced by LiAlH4 to give two deprotonated, anionic amido donors, and the ZnII ion has been replaced by AlIII via a novel transmetalation reaction. The fate of the ZnII ion in this process is not known, although we speculate that it is likely released into solution as zinc triflate/hydride salts, that may in part form insoluble byproducts. A white precipitate forms following treatment of 1 with LiAlH4 and warming up of the reaction mixture, and this material may contain some of the displaced ZnII ion as an insoluble byproduct.
3.3 Mechanistic considerations
The facile reduction of imine bonds by strong hydride donors such as LiAlH4 is well known. It is therefore reasonable to conclude that the reaction of 1 with LiAlH4 occurs through hydride addition to the imine bonds in 1 in a first step, which is then followed by transmetallation of ZnII by AlIII. Evidence for this mechanism was obtained from laser desorption ionization mass spectrometry (LDI-MS). Analysis of the white solid that is produced during the reaction was performed by LDI-MS, and revealed a prominent isotopic cluster centered at m/z = 496.14 (Figure S1). The isotope pattern matches that for [ZnIILH4N3S]+ (theor. m/z = 496.18), and can be assigned to the fully reduced ZnII complex, in which both imine bonds have been reduced and the resulting N donors are protonated (from isolation in air). A small peak is also observed at m/z = 494.13, which can be assigned to the ZnII product in which only one of the imine bonds has been reduced ([ZnII(LH2N3S)]+, theor. m/z = 494.16). These data indicate that the reduction of the imine bonds of the zinc(II) starting material most likely occurs prior to the transmetallation process.
To provide further evidence for the proposed mechanistic sequence, we examined the reaction of 1 with a different hydride source, NaBH4, which does not contain the AlIII ion. Addition of 1.5 equiv of NaBH4 to 1 in MeOH/EtOH at r.t. led to the formation of a yellow solid as seen for LiAlH4. This solid was filtered and isolated by aerobic work-up and analyzed by LDI-MS. A prominent isotopic cluster at m/z = 494.00 was observed, corresponding to the singly reduced ZnII complex [ZnII(LH2N3S)]+ (Figure S2). Although this procedure is not useful for obtaining the fully reduced product, it provides clear evidence that imine reduction readily occurs directly for the ZnII complex, and does not require AlIII. This result supports our overall proposed mechanism in which reduction of the imine bonds in 1 is relatively facile, and occurs prior to AlIII transmetalation.
3.4 Stereochemistry
The structural data for the AlIII complex 2a are consistent with one diastereomeric pair (R,S/S,R) being formed in the reduction reaction. However, reduction of 1 with LiAlH4 is not expected to proceed with diastereoselectivity, and we wondered whether the other R,R/S,S diastereomeric pair was formed in this reaction. Recrystallization of the mother liquor that was separated from 2a by layering with pentane at −35 °C afforded a new batch of yellow crystalline blocks (Scheme 2). Structural analysis of these new crystals revealed that they were not a second crop of 2a, but a new complex 2b (Figure 2). Relevant bond lengths and angles are summarized in Table 2 for one of the two independent molecules in the unit cell (both molecules exhibit similar bond distances and angles, and therefore only one set of bond distances and angles are given as representative of the structure). As seen in 2a, the tetradentate LH2N3S ligand forms the basal plane around a transmetalated AlIII ion, with a molecule of THF occupying the axial position. The key difference seen for 2b as compared to 2a lies in the orientation of the methyl groups attached to the former imino carbon atoms (C(7a) and C(14a)). In 2b, these methyl groups can be seen as oriented anti in relation to the AlN3 plane, whereas for 2a these groups are positioned syn across this plane. Reduction of both imine bonds of 2b is confirmed by the former Cimine-Nimine and Cimine-Cipso distances, as well as the non-planarity of C(7a) and C(14a). The Al–N distances of Al–N(1) = 1.8763(13) Å and Al–N(3) = 1.8560(13) Å for 2b are in good agreement with the deprotonated Namido–Al distance. It is worth noting that the geometry of complex 2b is distorted with τ values of 0.31 and 0.24 corresponding to the two independent molecules in the unit cell. In addition, the Al ion is 0.338(2) Å or 0.354(2) Å above the basal plane for the two independent molecules. Thus complex 2b is the second diastereomeric pair (R,R/S,S) produced in the reduction/transmetalation reaction, and shows that there is no strong diastereomeric preference for this reaction. The different orientation of the methyl groups does not cause any significant changes in bond lengths, but subtle changes are induced about the Al III coordination sphere such that the geometry is now significantly distorted from idealized square pyramidal, as seen for 2a.
Figure 2.

Displacement ellipsoid plot (50% probability level) of diastereomer 2b. H atoms and other crystallographically independent molecules are omitted for clarity.
3.5 1H NMR spectra
As seen in the solid-state X-ray structure, reduction of the imine bonds in 1 produces two chiral centers at the ketimine carbon atoms, and the structure of 2a consists of the racemate of the S,R/R,S diastereomer [41]. The 1H NMR spectrum of 2a in CD2Cl2 is shown in Figure 3. The two new C-H resonances resulting from the reduction of the imino carbon atoms are significantly separated and can be found as quartets at δ 4.82 (J = 6.4 Hz) and 4.30 (J = 6.8 Hz) ppm. The adjacent methyl groups are also distinct, and shifted upfield as expected from 2.79 and 2.43 ppm in 1 to 1.55 (J = 6.4 Hz) and 1.40 (J = 6.8 Hz) ppm in 2a. The isopropyl methine resonances are even further separated by ~1 ppm, appearing at 3.77 (overlapped with THF) and 2.82 ppm (septet). This latter ~1 ppm separation clearly indicates that rotation is highly restricted about the C(ipso)-N bond of the diisopropylphenyl substituent. Interestingly, all four iPr-Me groups also give rise to distinct resonances. One pair of doublets appears at 1.09 and 1.01 ppm, while the other pair appears as overlapping doublets at 1.26 ppm. The former data indicate that the rotational freedom about the iPr groups is hindered, pointing to the highly crowded nature of the complex. The solution state structure for 2a matches the solid state, and reveals that the complex is severely conformationally restricted.
Figure 3.
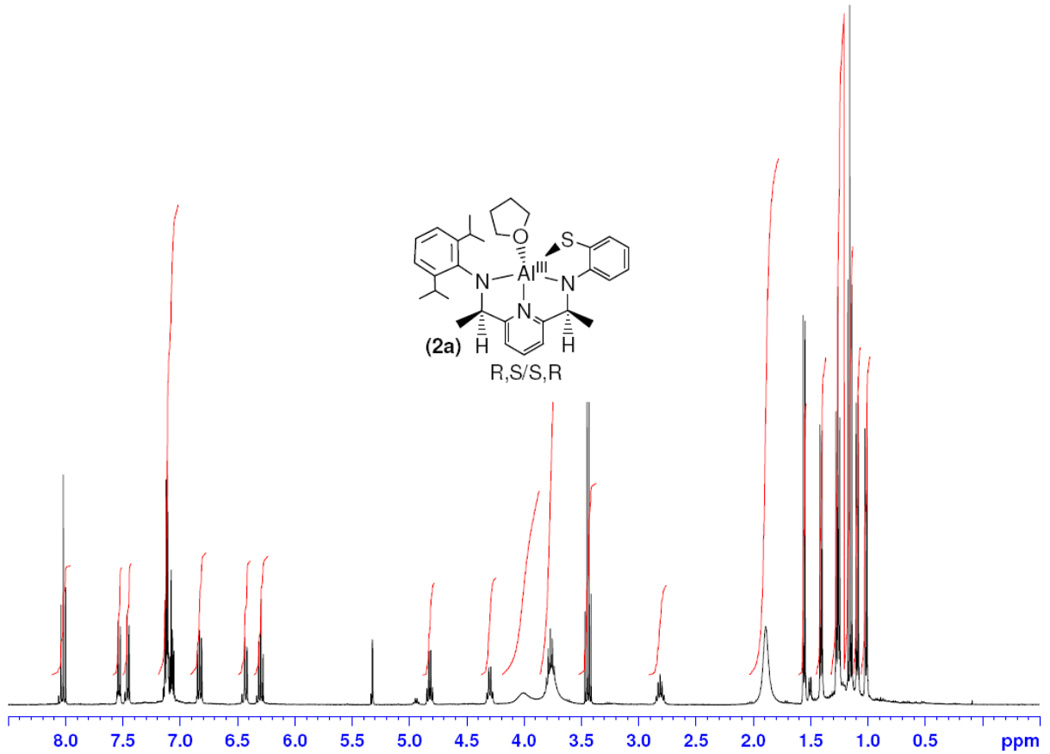
1H NMR spectrum of 2a.
Although the limited solubility of 2b in CD2Cl2 leads to a less well-resolved 1H NMR spectrum (Figure 4), comparisons can still be made between the solution state structures of 2a and 2b, revealing significant differences for the two complexes. The resonances for the new backbone C-H protons in 2b (δ 4.72 and 4.20 ppm) are each shifted upfield by 0.1 ppm in comparison to 2a, while the adjacent methyl groups are shifted slightly downfield for 2b to δ 1.61 and 1.46 ppm. Interestingly, large changes in the resonances for the iPr groups for 2b versus 2a are seen. The iPr-CH methine groups in 2b are no longer separated as in 2a, but appear as a broad, overlapping mutliplet at 3.23 ppm. In addition, all four iPr-CH3 doublets are separated for 2a, but for 2b the iPr-CH3 resonances can be assigned to two doublets centered at 1.22 and 1.02 ppm. The NMR data for 2b are consistent with hindered rotation about the diisopropylphenyl C(ipso)-N bond as seen for 2a, but the fact that the iPr-CH and iPr-CH3 peaks are not well-separated in 2b are consistent with the different solid-state structures of 2b versus 2a. The iPr-C(25) in 2a is likely more crowded than the corresponding C(25A) in 2b because of the positioning of the methyl group C(15), which lies on the same side of the chelate ring as the THF molecule, in proximity to C(25). From the 1H NMR analysis, it is clear that both 2a and 2b maintain their solid state structures in solution.
Figure 4.
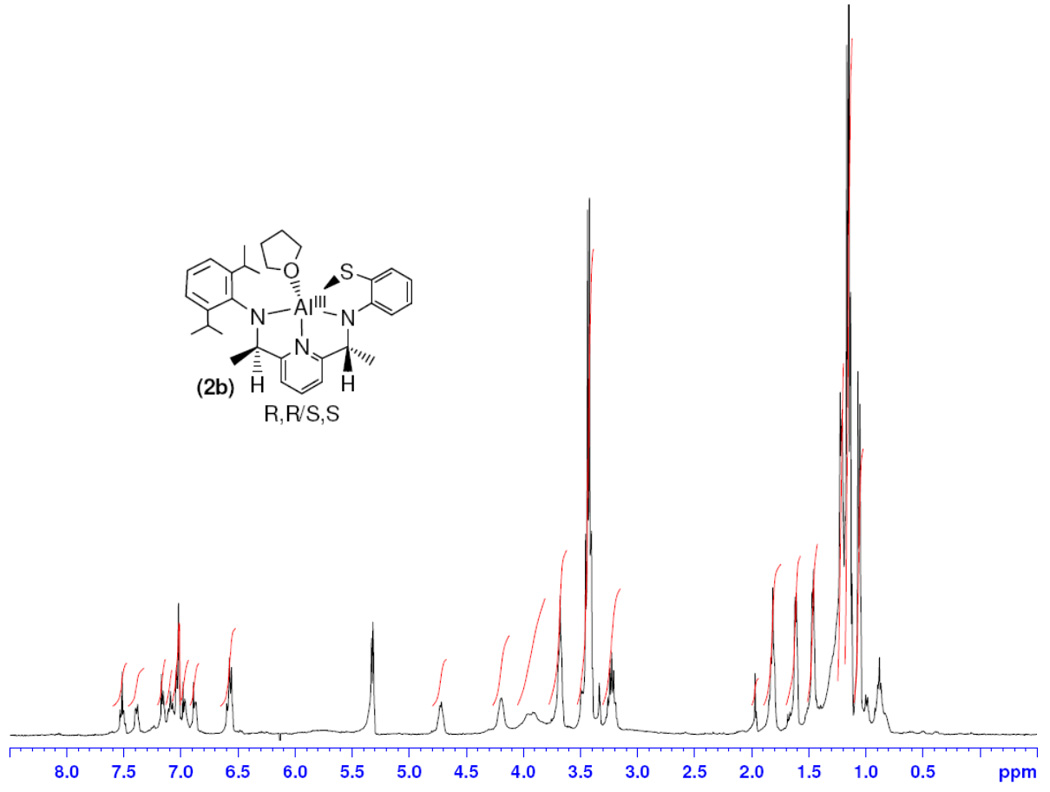
1H NMR spectrum of 2b.
4. Concluding remarks
The synthesis and characterization of two new thiolate-containing Al(III) bis(amido)pyridylthiolate complexes was accomplished via reduction of a ZnII bis(imino)pyridylthiolate precursor complex with LiAlH4. The synthesis of the reduced bis(amino)pyridylthiol ligand could not be accomplished by conventional organic synthetic strategies, and thus the success with the zinc(II) complex 1 as starting material highlights the usefulness of metal template chemistry in the preparation of unsymmetrical frameworks such as 1. Interestingly, during the reduction process a rare transmetallation reaction occurs, producing the AlIII products.
The BIP systems have been extensively studied as supporting ligands for Fe- and Co-catalyzed olefin oligomerizations and polymerizations, and aluminum-containing cocatalysts (methylaluminoxane (MAO), AlR3) are essential for these reactions. Significant efforts have been made to discern the role of the aluminum co-catalysts, but this role remains poorly understood. Evidence has been presented by other workers to indicate that transmetalation with Al may be an important factor in the activity of these catalytic systems [8, 35, 42]. Of particular relevance here is the work from Gambarotta and co-workers, which showed that BIP-FeII complexes react with AlR3 in an unexpected transmetalation process to give Al III products [8]. In this reaction the BIP ligand was reduced by electrons delocalized on to the π-system, resulting in paramagnetic Al complexes. The ability of BIP ligands to exist in such “non-innocent” reduced states [5, 10] suggests the possibility of a non-innocent configuration for 2a–b, and thus such a configuration was carefully considered. However, the structural parameters and diamagnetic 1H NMR spectra for 2a–b clearly rule out this possibility. The transformation of 1 to 2a–b is only the second example of an Al-mediated transmetalation reaction with the BIP system besides that reported by Gambarotta [8], and is the only case in which the BIP framework remains “innocent”. Taken together, these results suggest that aluminum transmetalation may be a more general phenomenon than was apparent from the original report by Gambarotta, and could be of potential importance in the mechanism of olefin polymerizations catalyzed by the widely used Al -dependent (BIP)MII systems.
Supplementary Material
Acknowledgement
The financial support from the National Institutes of Health (GM62309) to D.P.G. is gratefully acknowledged. Y. J. is grateful for an Ernest M. Marks Fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Appendix A. Supplementary material
Supplementary data associated with this article can be found, in the online version, at doi:xxx.
References
- 1.Gibson VC, Redshaw C, Solan GA. Chem. Rev. 2007;107:1745–1776. doi: 10.1021/cr068437y. [DOI] [PubMed] [Google Scholar]
- 2.Knijnenburg Q, Gambarotta S, Budzelaar PHM. Dalton Trans. 2006:5442–5448. doi: 10.1039/b612251e. [DOI] [PubMed] [Google Scholar]
- 3.Bowman AC, Bart SC, Heinemann FW, Meyer K, Chirik PJ. Inorg. Chem. 2009;48:5587–5589. doi: 10.1021/ic9003017. [DOI] [PubMed] [Google Scholar]
- 4.Manuel TD, Rohde JU. J. Am. Chem. Soc. 2009;131:15582–15583. doi: 10.1021/ja9065943. [DOI] [PubMed] [Google Scholar]
- 5.Wieder NL, Gallagher M, Carroll PJ, Berry DH. J. Am. Chem. Soc. 2010;132:4107–4109. doi: 10.1021/ja100894h. [DOI] [PubMed] [Google Scholar]
- 6.Russell SK, Darmon JM, Lobkovsky E, Chirik PJ. Inorg. Chem. 2010;49:2782–2792. doi: 10.1021/ic902162z. [DOI] [PubMed] [Google Scholar]
- 7.Scott J, Vidyaratne I, Korobkov I, Gambarotta S, Budzelaar PHM. Inorg. Chem. 2008;47:896–911. doi: 10.1021/ic701643d. [DOI] [PubMed] [Google Scholar]
- 8.Scott J, Gambarotta S, Korobkov I, Knijnenburg Q, de Bruin B, Budzelaar PHM. J. Am. Chem. Soc. 2005;127:17204–17206. doi: 10.1021/ja056135s. [DOI] [PubMed] [Google Scholar]
- 9.Enright D, Gambarotta S, Yap GPA, Budzelaar PHM. Angew. Chem., Int. Ed. 2002;41:3873–3876. doi: 10.1002/1521-3773(20021018)41:20<3873::AID-ANIE3873>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 10.Gallagher M, Wieder NL, Dioumaev VK, Carroll PJ, Berry DH. Organometallics. 2010;29:591–603. [Google Scholar]
- 11.Griffith GA, Al-Khatib MJ, Patel K, Singh K, Solan GA. Dalton Trans. 2009:185–196. doi: 10.1039/b811309b. [DOI] [PubMed] [Google Scholar]
- 12.Steves JE, Kennedy MD, Chiang KP, Kassel WS, Dougherty WG, Dudley TJ, Zubris DL. Dalton Trans. 2009:1214–1222. doi: 10.1039/b815987d. [DOI] [PubMed] [Google Scholar]
- 13.Ionkin AS, Marshall WJ, Adelman DJ, Fones BB, Fish BM, Schiffhauer MF. Organometallics. 2006;25:2978–2992. [Google Scholar]
- 14.Britovsek GJP, England J, Spitzmesser SK, White AJP, Williams DJ. Dalton Trans. 2005:945–955. doi: 10.1039/b414813d. [DOI] [PubMed] [Google Scholar]
- 15.Lappalainen K, Yliheikkila K, Abu-Surrah AS, Polamo M, Leskela M, Repo T, Anorg Z. Allg. Chem. 2005;631:763–768. [Google Scholar]
- 16.Zhang TZ, Sun WH, Li T, Yang XZ. J. Mol. Catal. A-Chem. 2004;218:119–124. [Google Scholar]
- 17.Davis RN, Tanski JM, Adrian JC, Tyler LA. Inorg. Chim. Acta. 2007;360:3061–3068. [Google Scholar]
- 18.Bianchini C, Mantovani G, Meli A, Migliacci F, Zanobini F, Laschi F, Sommazzi A. Eur. J. Inorg. Chem. 2003:1620–1631. [Google Scholar]
- 19.Ionkin AS, Marshall WJ, Adelman DJ, Fones BB, Fish BM, Schiffhauer MF, Soper PD, Waterland RL, Spence RE, Xie TY. J. Polym. Sci., Part A: Polym. Chem. 2008;46:585–611. [Google Scholar]
- 20.Xie GY, Li TC, Zhang AQ. Inorg. Chem. Commun. 2010;13:1199–1202. [Google Scholar]
- 21.Kendall AJ, Zakharov LN, Gilbertson JD. Inorg. Chem. 2010;49:8656–8658. doi: 10.1021/ic101408e. [DOI] [PubMed] [Google Scholar]
- 22.Krishnamurthy D, Kasper GD, Namuswe F, Kerber WD, Sarjeant AAN, Moënne-Loccoz P, Goldberg DP. J. Am. Chem. Soc. 2006;128:14222–14223. doi: 10.1021/ja064525o. [DOI] [PubMed] [Google Scholar]
- 23.Namuswe F, Kasper GD, Sarjeant AAN, Hayashi T, Krest CM, Green MT, Moënne-Loccoz P, Goldberg DP. J. Am. Chem. Soc. 2008;130:14189–14200. doi: 10.1021/ja8031828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Namuswe F, Hayashi T, Jiang YB, Kasper GD, Sarjeant AAN, Moenne-Loccoz P, Goldberg DP. J. Am. Chem. Soc. 2010;132:157–167. doi: 10.1021/ja904818z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stasser J, Namuswe F, Kasper GD, Jiang YB, Krest CM, Green MT, Penner-Hahn J, Goldberg DP. Inorg. Chem. 2010;49:9178–9190. doi: 10.1021/ic100670k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang YB, Widger LR, Kasper GD, Siegler MA, Goldberg DP. J. Am. Chem. Soc. 2010;132:12214–12215. doi: 10.1021/ja105591q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Badiei YM, Siegler MA, Goldberg DP. J. Am. Chem. Soc. 2011;133:1274–1277. doi: 10.1021/ja109923a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Britovsek GJP, Gibson VC, Mastroianni S, Oakes DCH, Redshaw C, Solan GA, White AJP, Williams DJ. Eur. J. Inorg. Chem. 2001:431–437. [Google Scholar]
- 29.Jie S, Diaconescu PL. Organometallics. 2010;29:1222–1230. [Google Scholar]
- 30.Sheldrick GM. Acta Crystallogr. A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 31.Spek AL. Acta Crystallogr. D. 2009;65:148–155. doi: 10.1107/S090744490804362X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fahmi N, Singh RV. Transit. Metal Chem. 1994;19:453–456. [Google Scholar]
- 33.Bouwkamp MW, Bart SC, Hawrelak EJ, Trovitch RJ, Lobkovsky E, Chirik PJ. Chem. Commun. 2005:3406–3408. doi: 10.1039/b504063a. [DOI] [PubMed] [Google Scholar]
- 34.Bart SC, Chlopek K, Bill E, Bouwkamp MW, Lobkovsky E, Neese F, Wieghardt K, Chirik PJ. J. Am. Chem. Soc. 2006;128:13901–13912. doi: 10.1021/ja064557b. [DOI] [PubMed] [Google Scholar]
- 35.Bruce M, Gibson VC, Redshaw C, Solan GA, White AJP, Williams DJ. Chem. Commun. 1998:2523–2524. [Google Scholar]
- 36.Nixon TD, Dalgarno S, Ward CV, Jiang ML, Halcrow MA, Kilner C, Thornton-Pett M, Kee TP, Chimie CR. 2004;7:809–821. [Google Scholar]
- 37.Lien YL, Chang YC, Chuang NT, Datta A, Chen SJ, Hu CH, Huang WY, Lin CH, Huang JH. Inorg. Chem. 2010;49:136–143. doi: 10.1021/ic9016189. [DOI] [PubMed] [Google Scholar]
- 38.Chen IC, Ho SM, Chen YC, Lin CY, Hu CH, Tu CY, Datta A, Huang JH, Lin CH. Dalton Trans. 2009:8631–8643. doi: 10.1039/b908164j. [DOI] [PubMed] [Google Scholar]
- 39.Reddy ND, Kumar SS, Roesky HW, Vidovic D, Magull J, Noltemeyer M, Schmidt HG. Eur. J. Inorg. Chem. 2003:442–448. [Google Scholar]
- 40.Addison AW, Rao TN, Reedjik J, van Rijn J, Verschoor GC. Dalton Trans. 1984:1349–1456. [Google Scholar]
- 41.Some minor positional disorder of C(8) is observed for 2a, and modeling of this disorder leads to an estimate of the population of the two diastereomers of 82% for the S,R/R,S pair, and 18% for the S,S/R,R pair. However, 1H NMR data show no evidence of the S,S/R,R isomer in the bulk solid, suggesting the population of the minor component is much less than 18%.
- 42.Knijnenburg Q, Smits JMM, Budzelaar PHM. Organometallics. 2006;25:1036–1046. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.