

Abstract
Emerging high-throughput screening technologies are rapidly providing opportunities to identify new diagnostic and prognostic markers and new therapeutic targets in human cancer. Currently, cDNA arrays allow the quantitative measurement of thousands of mRNA expression levels simultaneously. Validation of this tool in hospital settings can be done on large series of archival paraffin-embedded tumor samples using the new technique of tissue microarray. On a series of 55 clinically and pathologically homogeneous breast tumors, we compared for 15 molecules with a proven or suspected role in breast cancer, the mRNA expression levels measured by cDNA array analysis with protein expression levels obtained using tumor tissue microarrays. The validity of cDNA array and tissue microarray data were first verified by comparison with quantitative reverse transcriptase-polymerase chain reaction measurements and immunohistochemistry on full tissue sections, respectively. We found a good correlation between cDNA and tissue array analyses in one-third of the 15 molecules, and no correlation in the remaining two-thirds. Furthermore, protein but not RNA levels may have prognostic value; this was the case for MUC1 protein, which was studied further using a tissue microarray containing ∼600 tumor samples. For THBS1 the opposite was observed because only RNA levels had prognostic value. Thus, differences extended to clinical prognostic information obtained by the two methods underlining their complementarity and the need for a global molecular analysis of tumors at both the RNA and protein levels.
The development of genomic, technological, and bioinformatic tools have allowed progress in cancer research. DNA arrays are currently the most used of the new high-throughput methods to analyze the molecular complexity of tumors. Several studies have showed their potential in many types of human cancers.1-4 Even if the clinical benefits for patients remain to be demonstrated, the first results are very encouraging. DNA arrays-based gene expression profiles are improving our understanding of the disease as well as tumor taxonomy by identifying new diagnostic or prognostic subclasses unrecognized by usual parameters. They are expected to lead to the discovery of new potential therapeutic targets, to accurate predictions of survival and response to a given treatment, and eventually to the delivery of a therapy appropriate to each individual patient.
Once a potential marker is identified by this technique, an important next step is its validation and introduction in routine tests in hospital settings.5,6 There, cDNA arrays are not the method of choice because they are still expensive, time-consuming, complex, and require frozen material not always available. Validation studies have been done traditionally by immunohistochemistry (IHC) on paraffin-embedded tissues allowing analysis of many archived samples with a long follow-up. Until recently, pathologists examined sections of tumor slide by slide. Today, the recently developed tissue microarray (TMA) technology7-9 allows the simultaneous analysis of thousands of tumor samples arrayed onto glass slides. This may facilitate the search for correlations between molecular alterations and the histoclinical features of the tumors.
In a recent cDNA array-based, prognosis-oriented study of 55 localized breast carcinoma samples,10 we identified two clusters of discriminator genes (named I and II) the differential expression of which allowed to distinguish subclasses of tumors with significantly different clinical outcome after adjuvant chemotherapy. The aim of the present study was to validate some of these data using TMAs and to evaluate the interest and limitations of this technology as a validation tool. Cylinders from the same 55 tumors were arrayed in a specific tissue-microarray and studied by IHC using antibodies directed against proteins encoded by some of our discriminator genes.
Materials and Methods
Mammary Carcinoma Cell Lines
Nine established mammary carcinoma cell lines were used as positive controls for expression of various genes or proteins. They included: BT-474, MCF-7, MCF-10F, MDA-MB-157, MDA-MB-175, MDA-MB-231, MDA-MB-453, BrCa-MZ-02,11 and HBL-100. All cell lines are derived from carcinomas except HBL-100 and MCF-10F. They were obtained from the American Type Culture Collection, Rockville, MD (http://www.atcc.org/) and grown using the recommended culture conditions.
Breast Tumor Samples and Characteristics of Patients
Tumor samples were obtained from 55 women treated at the Institut Paoli-Calmettes. Inclusion criteria were: 1) localized breast cancer treated with adjuvant anthracyclin-based chemotherapy in addition to loco-regional treatment; 2) tumor material quickly macrodissected and frozen in liquid nitrogen and stored at −160°C; and 3) patient follow-up of 48 months or more after diagnosis. In addition to the axillary lymph node status, four poor prognosis criteria were used to determine whether adjuvant chemotherapy should be administered: patient age less than 40 years, pathological tumor size greater than 20 mm, Scarff-Bloom-Richardson grade equal to 3, and negative estrogen receptor (ER) status as evaluated by IHC with a positivity cutoff value of 1%. Women who received chemotherapy were those with either node-positive tumors or node-negative tumors and one of the poor prognosis criteria if nonmenopausal or two criteria if menopausal. All tumor sections were de novo reviewed by a pathologist (JJ) before analysis; all samples contained more than 50% tumor cells. Tumors were infiltrating adenocarcinomas including, according to the World Health Organization histological typing, 42 ductal, 5 lobular, 5 mixed, and 3 medullary carcinomas.
A second series of breast tumors was analyzed. It was constituted by 592 localized forms of breast cancer collected between 1987 and 1999 (median follow-up, 48 months) for which a sample had been frozen in liquid nitrogen (the 55 tumors previously described were included in this array). There were 401 ductal, 77 lobular, 40 mixed, 4 medullary carcinomas, and 70 other histological types. A total of 297 tumors were node positive and 450 were positive for ER.
Extraction of RNA from Frozen Tissue
Total RNA was extracted from tumor samples by standard methods, as previously described.12 RNA integrity was controlled on denaturing formaldehyde-agarose gel electrophoresis and Northern blots using a 28S-specific oligonucleotide.
DNA Arrays
DNA arrays were made in our facility (Technologies Avanceés pour le Génome et la Clinique)). Nylon filter preparation with spotted polymerase chain reaction (PCR) products derived from ∼1000 selected candidate cancer genes, 33P radioactive hybridization, and data acquisition, normalization, and analysis have been described elsewhere13,14 and can also be consulted on our web site (http:/tagc.univ-mrs.fr/pub/Cancer/).
Reverse Transcription
RNA extracted from frozen tissue was reverse-transcribed in a final volume of 20 μl containing 1× reverse transcriptase (RT)-PCR buffer (Invitrogen Corp., Carlsbad, CA), 5 mmol/L MgCl2 (Invitrogen), 1 mmol/L dXTP (Roche Diagnostics, Meylan, France), 10 mmol/L dithiothreitol (Invitrogen), 5 μmol/L random hexamers (Roche), 20 U of RNase inhibitor (Promega Biosciences, Madison, WI), 200 U of superscript reverse transcriptase (Invitrogen), and 1 μg of total RNA (calibration curve points and patient samples). Samples were incubated at 20°C for 10 minutes and 42°C for 45 minutes; reverse transcriptase was inactivated by heating at 99°C for 3 minutes and cooling at 4°C for 5 minutes.
Real-Time Quantitative RT-PCR (RQ-PCR)
RQ-PCR analyses for ERBB2, MUC1, and TBP (TATA box binding protein) mRNA were done using the ABI PRISM 7700 Sequence Detection System instrument and software (Perkin Elmer Applied Biosystems, Foster City, CA). Conditions for the analysis of these markers have been described.15,16 Primers and probes for the TaqMan system were designed to meet specific criteria by using Primer Express software (Perkin Elmer) and were synthesized by Genset (Genset Olijos, La Jolla, CA, USA) for the primers and by Roche for the probes. The 5′- and 3′-end nucleotides of the probe were labeled with a reporter (FAM, 6-carboxy-fluorescein) and a quencher dye (TAMRA, 6-carboxy-tetramethylrhodamine). The sequences of the PCR primer pairs and fluorogenic probes used for each gene are shown in Table 1▶ . The oligonucleotides are designated by the nucleotide position relative to ERBB2 GenBank accession no. M11730, MUC1 GenBank accession no. JO5581, TBP GenBank accession no. X54993. The precise amount of total RNA added to each reaction mix (based on absorbance) and its quality (ie, lack of extensive degradation) are both generally difficult to assess. Therefore, the relative expression level of the gene of interest was computed with respect to the internal standard TBP to normalize for variations in the quality of RNA and the amount of input cDNA. Ct (threshold cycle) was used for quantification of the input target number and all experiments were done with duplicates for each data point. All patient samples with a variation >1 Ct for the duplicate were retested. For each experimental sample, the amount of target and endogenous reference was determined from a standard curve. The standard curve was constructed with fivefold serial dilutions of cDNA (1000 ng to 1 ng) from BT-474 (for ERBB2) and MCF-7 (for MUC1) breast carcinoma cell lines, respectively. The relative target gene expression in a tested sample was normalized using a calibrator sample, ie, the HME1 human primary mammary epithelial cell line (Clontech). The level of expression of the target gene was given by the N-ratio, in which each normalized gene value (ERBB2, MUC1) was divided by a calibrator normalized gene value (TBP).
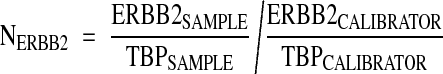 |
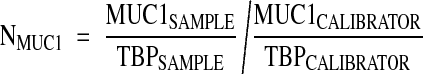 |
PCR was done with 1× TaqMan Universal PCR Master Mix (Perkin Elmer), 300 nmol/L of primers, 200 nmol/L of the probe, and 1 μl of each appropriately diluted reverse transcription sample in a 25-μl final reaction mixture. After a 2-minute incubation at 50°C to allow for uracyl N-glycosylate cleavage, AmpliTaq Gold was activated by an incubation for 10 minutes at 95°C. Each of the 40 PCR cycles consisted of 15 seconds of denaturation at 95°C and hybridization of probe and primers for 1 minute at 60°C.
Table 1.
Sequences of Oligonucleotide Primers and Probes Used in RQ-PCR Experiments
| Gene | Oligonucleotide | Sequence | PCR product size |
|---|---|---|---|
| ERBB2 | Forward primer | 5′-AGCCGCGAGCACCCAAGT-3′ (exon 1) | 147 bp |
| Reverse primer | 5′-TTGGTGGGCAGGTAGGTGAGTT-3′ (exon 2) | ||
| Probe | 5′-CCTGCCAGTCCCGAGACCCACCT-3′ | ||
| MUC1 | Forward primer | 5′-ACCATCCTATGAGCGAGTACC-3′ (exon 6) | 107 bp |
| Reverse primer | 5′-GTTTCTGCAGGTAATGGTGGC-3′ (exon 7) | ||
| Probe | 5′-CCCATGGGCGCTATGTGCCC-3′ | ||
| TBP | Forward primer | 5′-CACGAACCACGGCACTGATT-3′ | 89 bp |
| Reverse primer | 5′-TTTTCTTGCTGCCAGTCTGGAC-3′ | ||
| Probe | 5′-TGTGCACAGGAGCCAAGAGTGAAG-3′ |
TMA Construction
TMAs were prepared as described9 with slight modifications. For each tumor, three representative tumor areas were carefully selected from a hematoxylin- and eosin-stained section of a donor block. Core cylinders with a diameter of 0.6 mm each were punched from each of these areas and deposited into a recipient paraffin block using a specific arraying device (Beecher Instruments, Silver Spring, MD). In addition to tumor tissues, the recipient block also received normal breast tissue and cell line pellets. Five-μm sections of the resulting microarray block were made and used for IHC analysis after transfer to glass slides. Two TMAs were prepared; the first one contained the 55 tumors studied by cDNA arrays (with three cores per sample) and controls, the second one was used for MUC1 study and contained 592 tumor samples (with one core per sample) and controls.
Antibodies and IHC
The characteristics of the antibodies used are listed in Table 2▶ . IHC was performed on 5-μm sections of formalin-embedded tissue specimens. They were deparaffinized in histolemon (Carlo Erba Reagenti, Rodano, Italy) and rehydrated in graded alcohol. Antigen enhancement was done by incubating the sections in target retrieval solution (DAKO, Copenhagen, Denmark) as recommended except for prolactin receptor, in which pretreatment was done with incubation in pepsin (Zymed Laboratories, South San Francisco, CA), for 30 minutes at 37°C, and for MUC1, in which no pretreatment was done. Slides were then transferred to a DAKO autostainer. Staining was done at room temperature as follows: after washes in phosphate buffer, followed by quenching of endogenous peroxidase activity by treatment with 0.1% H2O2, slides were first incubated with blocking serum (DAKO) for 10 minutes and then with the affinity-purified antibody for 1 hour. After washes, slides were incubated with biotinylated antibody against rabbit Ig for 20 minutes followed by streptavidin-conjugated peroxidase (DAKO LSABR2 kit). Diaminobenzidine or 3-amino-9-ethylcarbazole was used as the chromogen, counterstained with hematoxylin, and coverslipped using Aquatex (Merck, Darmstadt, Germany) mounting solution. Slides were evaluated under a light microscope by two pathologists (EC-J, JJ).
Table 2.
List of Proteins Tested by Immunohistochemistry and Characteristics of the Corresponding Antibodies
| Protein | Antibody | Origin | Clone | Dilution |
|---|---|---|---|---|
| Angiogenin (ANG) | Rabbit polyclonal | Santa Cruz Biotechnology | sc-9044 | 1/20 |
| BCL2 | mmab | DAKO | 124 | 1/100 |
| E Cadherin (CDH1) | mmab | Transduction Laboratories | 36 | 1/2000 |
| ERBB2 | mmab | Novocastra Laboratories Ltd. | CB 11 | 1/500 |
| ERBB2 | mmab | Oncogene Research Products | 3B5 | 1/500 |
| ERBB2 | Rabbit polyclonal | DAKO | AO 485 | 1/1000 |
| Estrogen receptor (ESR1/ER) | mmab | Novocastra Laboratories Ltd. | 6F11 | 1/60 |
| FGFR1 | Rabbit polyclonal | Santa Cruz Biotechnology | sc-121 | 1/200 |
| GATA3 | mmab | Santa Cruz Biotechnology | sc-268 | 1/100 |
| Ki67 | mmab | DAKO | KI-67 | 1/100 |
| Melan A/MART1 (MLANA) | mmab | DAKO | A103 | 1/2 |
| MUC1 | mmab | Transgen | H23 | 1/1000 |
| P53 | mmab | Immunotech | DO-1 | 1/4 |
| Progesterone receptor (PR) | mmab | DAKO | PgR 636 | 1/80 |
| Prolactin receptor (PRLR) | mmab | NeoMarkers | B6.2 | 1/200 |
| Transforming acidic coiled-coil 1 TACC1 | Rabbit polyclonal | Upstate Biotechnology | 07-229 | 1/200 |
| Transforming acidic coiled-coil 2 TACC2 | Rabbit polyclonal | Upstate Biotechnology | 07-228 | 1/40 |
| Thrombospondin 1 (THBS1) | mmab | Oncogene Research Products | 46.4 | 1/10 |
mmab, mouse monoclonal.
Immunoreactivities were classified by estimating the percentage (P) of tumor cells showing characteristic staining (from undetectable level or 0%, to homogeneous staining or 100%) and by estimating the intensity (I) of staining (1, weak staining; 2, moderate staining; or 3, strong staining. The cutoff values were the same for all markers tested. Results were scored by multiplying the percentage of positive cells by the intensity, ie, by the so-called quick score (Q) (Q = P × I; maximum = 300). For Ki67, only the percentage (P) of tumor cells was estimated, because intensity does not vary. Expression levels allowed to group tumors into four categories: negative expression (Q = 0 or P = 0 for Ki67), weak expression (0 < Q ≤ 120 or 0 < P < 25 for Ki67), moderate expression (120 < Q ≤ 210 or 25 ≤ P < 60 for Ki67) and strong expression (210 < Q ≤ 300 or 60 ≤ P ≤ 100 for Ki67). Because of its prognostic impact the topographical localization of MUC1 was taken into account and expressed in four categories: absence, apical, circumferential membrane, and cytoplasmic
IHC on Full Tissue Sections
To validate the use of TMAs for immunophenotyping, we compared the protein expression levels of ER, progesterone receptor, P53, and BCL2, on full tissue sections and on TMAs for the group of 55 tumors. The data on full sections were compared to the mean of intensities of the three 0.6-mm core biopsies for 47 cases, or of only two core biopsies for 8 cases.
Statistical Analysis
The concordance between RNA expression levels measured by real-time quantitative RT-PCR and cDNA arrays was examined using Spearman’s rank correlation. Comparison between IHC data from full sections and TMAs analyses was measured using κ statistics (a κ value >0.7 indicated a strong association). Contingency table analysis was used to analyze the relationship between protein expression obtained by IHC on TMAs and RNA expression obtained with cDNA arrays (total chi-square test). Survival analysis used the Kaplan-Meier method and survival curves were compared using the log-rank test (a P value <0.5 was considered as significant). All P values were two-sided. To assess the relationship between two variables assumed to be related (ie, the co-regulated molecules), simple linear regression analyses were performed using Excel Software (Microsoft). For these tests, (O,O) points were removed; the relationship tested was thus for cases with at least one positive value. Each result is given with: N the sample size, a the slope of the regression line, the P value and r2, the coefficient of determination. Thus, for each positive comparison a linear relationship can be determined (eg, y = 0.8x + 20 means BCL2 = 0.8ER + 20).
Results
Selection of Molecules
We previously analyzed the mRNA expression profiles of ∼1000 selected genes in 55 breast carcinoma samples using home-made cDNA arrays. Tumors were homogeneous with respect to histological and clinical parameters, and all patients had received adjuvant anthracyclin-based chemotherapy. Detailed results are described elsewhere.10 Briefly, molecular profiling combined with hierarchical clustering allowed the identification, among this set of poor-prognosis localized breast cancers, of new subclasses distinct with respect to overall and metastasis-free survivals. Such a classification resulted from the differential expression of two discriminator gene clusters (named I and II) and was not possible using classical prognostic factors of disease. Cluster I included the ESR1 gene encoding ER-α. For the present study, we selected 10 of these genes. Interestingly, six of them (BCL2, ERBB2, ESR1, GATA3, MUC1, PRLR) have also been frequently identified as discriminator genes in expression-profiling studies of breast cancer that have addressed the prognosis issue.4,17-19 These genes were thus interesting candidates for further investigation. In addition, other molecules, such as CDH1, Ki67, TP53, progesterone receptor, TACC1,20 and TACC2, were retained because of a known or suspected role in breast cancer. The selection criteria for all molecules also included availability of a commercial antibody. The complete list of the corresponding proteins tested in the following experiments is given in Table 2▶ .
Validation of cDNA Array Data with RQ-PCR
Our cDNA array analyses regularly included extensive experiments and controls designed to ensure reproducibility and reliability of expression measurements.1,13,14,21 Nevertheless, we sought to further validate our data by comparing RNA expression levels of two genes, ERBB2 and MUC1, as measured by cDNA array, to those obtained by RQ-PCR.
RNA from 50 of 55 samples (RNA was no longer available for five cases) was reverse-transcribed and PCR amplification of ERBB2 and MUC1 cDNA was done using a TaqMan device. For ERBB2, 41 tumors displayed mRNA expression levels comparable to normal breast and HME1 control cell line, whereas nine samples (18%) showed overexpression. For MUC1, 17 tumors displayed mRNA expression levels comparable to normal breast and HME1 control cell line, whereas 33 samples (66%) showed overexpression. As shown in Figure 1▶ , mRNA expression levels obtained with both methods were highly similar (Spearman test: ERBB2, ρs = 0.78, P < 0.0001; MUC1, ρs = 0.88, P < 0.0001), further suggesting reliability of our cDNA array data.
Figure 1.

Expression levels of ERBB2 and MUC1 mRNA levels measured by cDNA array analysis and real-time quantitative PCR amplification. ERBB2 and MUC1 mRNA expression levels measured using cDNA arrays (artificially ×30 for visual effect) (left) and real-time quantitative PCR amplification (artificially ×30 for visual effect) (right). Results for each tumor (from top to bottom) are represented as opposite bars. For ERBB2: ρs = 0.78, P < 0.0001; for MUC1: ρs = 0.88, P < 0.0001.
TMA Analysis and Validation of Data
To validate our TMA analyses, we compared the expression of four selected proteins (BCL2, ER, P53, progesterone receptor) measured by IHC using either standard full tissue sections or TMAs in the panel of 55 breast tumors. For BCL2 expression, 38 cases (69%) showed positive cytoplasm staining, whereas 17 cases (31%) were negative on analysis of full sections. In comparison, 37 cases (67%) were positive and 18 cases were negative (33%) on TMA. Overall, the concordance was 91% and the nonconcordance was 9% (five cases), resulting in a strong statistical association between the two methods (κ value, 0.78). An even better correlation was found for nuclear expressions of ER, P53, and progesterone receptor, with only 3 discordant cases of 55 for each of them (concordance, 95%; Kappa values, 0.86 to 0.88). This high degree of concordance between IHC on full sections and on TMAs justified further use of TMAs.
Analysis of Breast Tumors Using TMAs
Fifteen proteins, including the four previously cited, were tested by IHC on TMAs. Most of them corresponded to genes we had identified in our two discriminator gene clusters I and II.10 Other tested molecules corresponded to proteins of interest in breast cancer. Immunostainings were evaluated by the quick score (except for Ki67). Results are shown in Table 3▶ and Figure 2▶ .
Table 3.
Results of IHC Stainings on Tissue Microarrays
| Protein | Location of staining | Normal | Negative | Weak | Moderate | Strong | |
|---|---|---|---|---|---|---|---|
| cDNA array, cluster I gene-encoded | ANG | Cytoplasm + Stroma | (+) | 17 | 19 | 5 | 14 |
| BCL2 | Cytoplasm | (+) | 23 | 10 | 17 | 5 | |
| ESR1/ER | Nucleus | (+) | 22 | 14 | 5 | 14 | |
| GATA3 | Nucleus | (+) | 26 | 12 | 7 | 10 | |
| MUC1 | Cytoplasm | (+) | 3 | 19 | 8 | 25 | |
| THBS1 | Cytoplasm + Stroma | (+) | 16 | 30 | 9 | 0 | |
| cDNA array, cluster II gene-encoded | MLANA | Cytoplasm | (+) | 22 | 20 | 6 | 7 |
| PRLR | Membrane | (+) | 21 | 12 | 7 | 15 | |
| Others | CDH1 | Membrane | (+) | 6 | 10 | 14 | 25 |
| ERBB2 (CB 11) | Membrane | (−) | 34 | 14 | 4 | 3 | |
| ERBB2 (AO485) | Membrane | (−) | 30 | 11 | 9 | 5 | |
| ERBB2 (3B5) | Membrane | (−) | 37 | 8 | 2 | 8 | |
| FGFR1 | Membrane + Cytoplasm | (+) | 20 | 20 | 12 | 3 | |
| Ki67 | Nucleus | (+) | 4 | 27 | 13 | 11 | |
| P53 | Nucleus | (−) | 33 | 12 | 0 | 10 | |
| TACC1 | Cytoplasm | (+) | 21 | 22 | 9 | 3 | |
| TACC2 | Cytoplasm | (+) | 5 | 18 | 13 | 19 |
(+) and (−) mean expressed or not in normal breast tissue, respectively.
Figure 2.
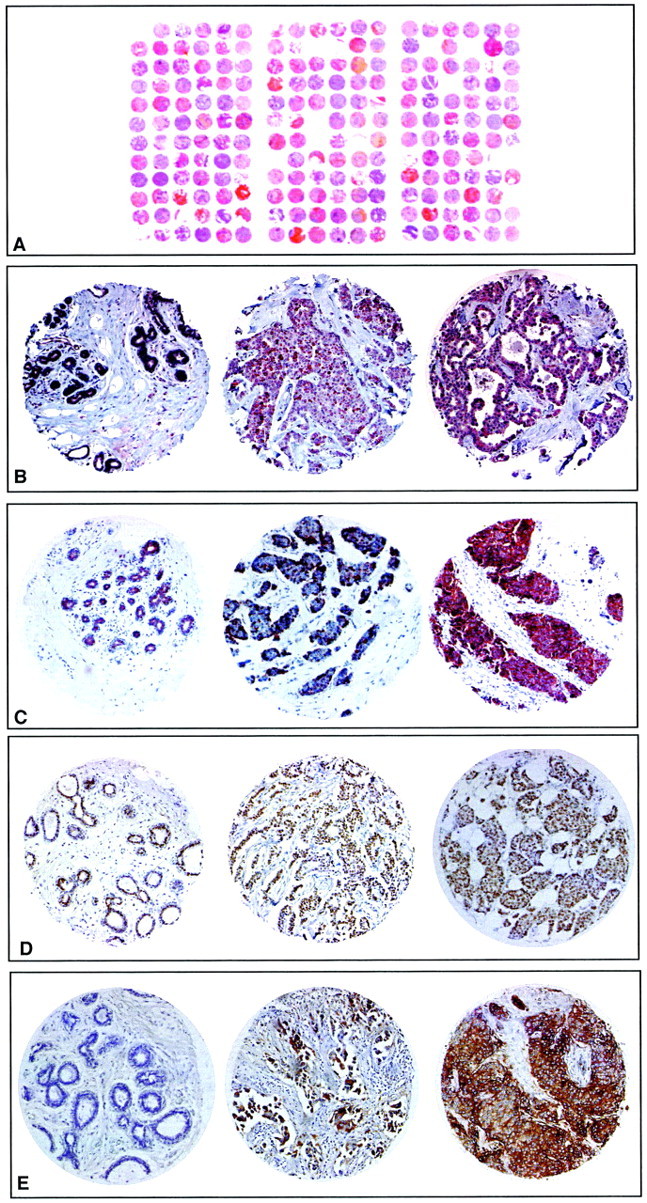
Expression of proteins studied by IHC on TMAs. A: H&E staining of a paraffin block section (25 × 30 mm) from the TMA containing 216 arrayed tumor (3 × 55) and control samples. B: Anti-angiogenin staining. C: Anti-FGFR1 staining. D: Anti-GATA3 staining. E: Anti-PRLR staining. From B to E, the first section is from normal breast tissue, the second and third from tumor tissue (the second illustrates a moderate staining whereas the third illustrates a strong staining). Original magnifications, ×50.
Comparison of the Results Obtained by cDNA Arrays and TMAs
Expression levels obtained by IHC on TMA and by cDNA array hybridizations were compared for the 15 molecules. Data from TMA analyses are discontinuous, whereas those obtained by cDNA array analyses are continuous. To facilitate comparisons, we transformed the cDNA array values into discontinuous data. Tumors were then grouped into two or three classes for each method (Table 4)▶ . Homogeneous classes were defined for TMA, by grouping tumors with an equivalent staining level (see Table 3▶ ). For cDNA arrays, classes were visually defined on examination of the distribution graphs (Figure 3)▶ .
Table 4.
Comparison of Expression Levels Measured Using Analyses of Tissue Microarrays and cDNA Arrays
| Gene | Tissue microarray classes | cDNA array classes | Concordance | ||||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 1 | 2 | 3 | P values | |
| ESR1/ER | 22 (N) | 19 (W + M) | 14 (S) | 15 | 22 | 18 | <0.001 |
| BCL2 | 23 (N) | 10 (W) | 22 (M + S) | 18 | 37 | 0 | <0.02 |
| P53 | 33 (N) | 22 (W + M + S) | / | 46 | 9 | 0 | NS |
| GATA3 | 26 (N) | 12 (W) | 17 (M + S) | 18 | 25 | 12 | <0.001 |
| PRLR | 21 (N) | 19 (W + M) | 15 (S) | 36 | 12 | 7 | NS |
| ERBB2 (3B5) | 37 (N) | 10 (W + M) | 8 (S) | 46 | 4 | 5 | <0.001 |
| CDH1 | 16 (N + W) | 14 (M) | 25 (S) | 42 | 13 | 0 | NS |
| TACC2 | 23 (N + W) | 13 (M) | 19 (S) | 19 | 18 | 18 | NS |
| TACC1 | 21 (N) | 22 (W) | 12 (M + S) | 19 | 18 | 18 | <0.05 |
| MLANA | 22 (N) | 20 (W) | 13 (M + S) | 34 | 21 | 0 | NS |
| FGFR1 | 20 (N) | 20 (W) | 15 (M + S) | 45 | 10 | 0 | NS |
| ANG | 17 (N) | 19 (W) | 19 (M + S) | 17 | 30 | 8 | NS |
| THBS1 | 16 (N) | 30 (W) | 9 (M + S) | 46 | 6 | 8 | NS |
| Ki67 | 31 (N + W) | 24 (M + S) | / | 11 | 33 | 11 | NS |
| MUC1 | 22 (N + W) | 33 (M + S) | / | 39 | 8 | 8 | NS |
Figure 3.

Transformation of continuous cDNA array data into discontinuous data. mRNA expression levels measured by cDNA array are plotted for each sample in an increasing order. For each gene, classes are determined on visual inspection and are separated by vertical bars on the graphs. Results for ER-α (ESR1), prolactin receptor (PRLR), mucin 1 (MUC1), and ERBB2 are shown.
Each tumor sample was then placed into one of the three TMA classes and attributed 1, 2, or 3, and into one of the three cDNA array classes and attributed 1, 2, or 3. Table 4▶ shows the number of samples in each class. Concordance between the two scores was evaluated by a contingency table analysis. A strong concordance was seen for 5 of the 15 comparisons with similar expression levels measured by the two methods: ER, ERBB2, and GATA3 (P < 0.001), BCL2 (P < 0.02), and TACC1 (P < 0.05). No concordance was seen for ANG, CDH1, FGFR1, Ki67, MLANA, MUC1, P53, PRLR, TACC2, and THBS1. Figure 4▶ shows example of comparative graphs.
Figure 4.

Comparison of data obtained by cDNA array and IHC on TMA. Results for each tumor (from top to bottom) are represented as opposite bars, with the value of IHC (quick score) on the left, and the value of the cDNA array analyses (artificially ×30 for visual effect) on the right. Values for ER, GATA3, and ERBB2 show good correlation between the two methods, whereas values for P53, THBS1, and MUC1 do not show such correlation.
Groups of Co-Regulated Molecules
Using cDNA arrays and hierarchical clustering, we had evidenced a co-expression of ESR1 (encoding ER-α), BCL2, and GATA3 at the mRNA level in breast tumors,1,10 with a statistically significant correlation between ESR1 and GATA3 (r = 0.73, R2 = 0.53, P < 0.0001). As shown in Figure 5A▶ , the correlation between the three molecules was statistically confirmed at the protein level as measured by IHC on TMA. FGFR1, TACC1, and TACC2 protein levels also varied together but the correlation was weaker (Figure 5B)▶ . For each pairwise comparison, with the same number of samples (n = 55), we calculated a coefficient of correlation and a P value: BCL2/ER, r = 0.79, R2 = 0.62, P < 0.0001; GATA3/ER, r = 0.74, R2 = 0.54, P < 0.0001; TACC1/FGFR1, r = 0.67, R2 = 0.45, P < 0.001; and TACC2/FGFR1, r = 0.57, R2 = 0.32, P < 0.001.
Figure 5.

Similar variations in expression levels of two groups of proteins. A: The expression levels of ER, BCL2, and GATA3 as measured by IHC on TMAs correlated, as determined by simple linear regression analysis. B: Similarly, the expression levels of FGFR1, TACC1, and TACC2 correlated.
Impact on Survival of RNA and Protein Expression Levels
To further estimate the clinical interest of the cDNA array and TMA combined approach, we examined and compared the prognostic information provided by mRNA and protein expression levels for each of the 15 molecules independently. Only 2 of the 15 tested markers showed individual prognostic value. High THBS1 mRNA levels were associated with a better survival whereas no such correlation was found with protein levels. The opposite was true for MUC1: low levels of MUC1 protein were associated with a better survival, whereas mRNA levels did not correlate with survival (Figure 6)▶ . Thus, depending on the marker, clinically relevant information was differently provided by cDNA or TMA technique, suggesting that both analyses are worth performing simultaneously on the same cases.
Figure 6.

Kaplan-Meier plots of patient overall survival. Left: Survival according to MUC1 mRNA and protein expression levels. Right: Survival according to THBS1 mRNA and protein expression levels (labeled high and low). High and low protein levels correspond to strong plus moderate versus weak plus negative (see Table 3▶ ), respectively, and high and low mRNA levels correspond to classes 2 and 3 versus class 1 (see Figure 4▶ ), respectively.
These results were obtained on a limited number of cases representing a selected population of poor prognosis localized tumors. We sought to confirm the observation on MUC1 on a larger series of cases (Figure 7A)▶ . We studied 592 samples (including the 55) arrayed in a second TMA with anti-MUC1 antibody. MUC1 staining in normal cells is either absent or detected in the apical membrane; tumor cells express MUC1 in two abnormal localizations (cytoplasm or circumferential membrane) and a strong cytoplasmic staining is associated with a poor prognosis.22 For the 55 tumors of the first TMA, the prognostic value of the quantitative quick score was related to a high frequency of abnormal cytoplasmic and circumferential MUC1 localizations (83%) as compared to apical localization and absence (17%). Of the 592 cases of the second TMA, 551 were available for analysis after MUC1 staining: 249 cases (45%) showed apical or no staining, 302 (55%) displayed cytoplasmic or circumferential membrane staining (Figure 7B)▶ . In this larger series the quantitative quick score did not have a significant prognostic value. This was because of the fact that the topographical aspect was significantly different from that of the short series with only 55% versus 83% of cytoplasmic and circumferential localizations. When considered, qualitative assessment of the staining provided prognostic information; the apical localization and the absence of MUC1 strongly correlated with a better evolution (P = 0.0154) (Figure 7C)▶ .
Figure 7.
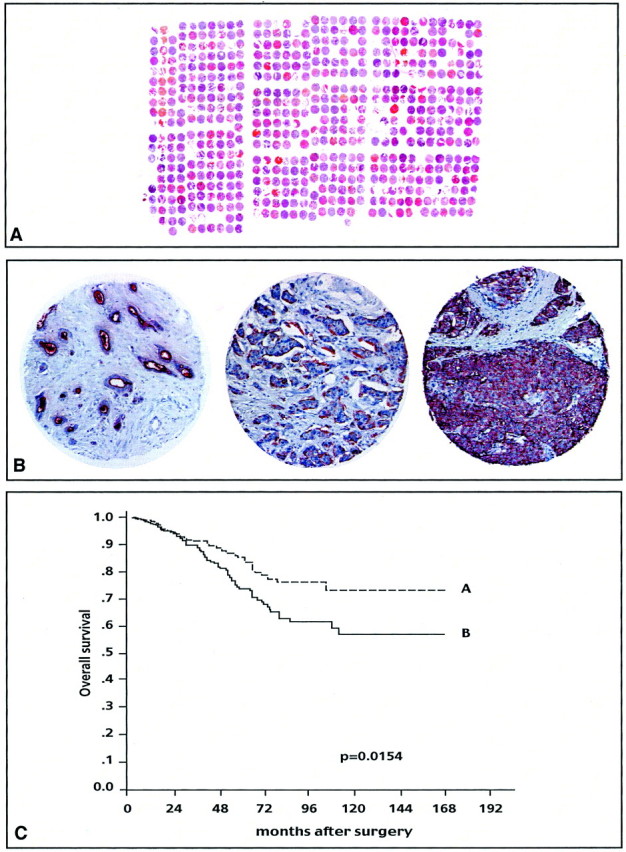
Expression of MUC1 protein studied by IHC on a tissue-microarray. A: H&E staining of a paraffin block section (25 × 30 mm) from the TMA containing 647 arrayed samples, including 592 tumors and 55 controls. B: MUC1 staining: normal breast tissue (left), apical (middle), and cytoplasmic (right) staining in tumors. C: Kaplan-Meier plot of patient overall survival: survival differs significantly according to MUC1 protein localization. A: Absence of staining or apical localization; B: cytoplasmic or circumferential membrane localization.
Discussion
The recent availability of new high-throughput molecular analyses offers the opportunity to tackle the complexity and the combinatorial nature of breast cancer at the molecular level. Expected applications are a better understanding of the disease and the identification of new diagnostic and prognostic markers and therapeutic targets, both needed to improve the management of patients. At the same time it introduces a new challenge for pathologists who, in charge of the first assessment of the tumors, need to know how to optimally use these new methods. The present study directly followed a cDNA array-based analysis of a breast tumor series. The tumor samples were obtained from 55 women with poor prognosis breast cancer treated with adjuvant chemotherapy. Currently such patients have a long-term survival of ∼70% and there is a crucial need to identify parameters that might accurately predict the clinical outcome in individual patients. Our study was designed to evaluate the interest and limitations of IHC on TMA as a natural extension of the cDNA array approach in a hospital setting.
We first confronted cDNA array and TMA analyses to other methods, ie, RQ-PCR and conventional IHC, respectively. The good concordance between mRNA expression levels observed by cDNA arrays and RQ-PCR further confirmed the validity of our cDNA array measurements. TMAs allow to screen large series of tumor samples using several archival materials, but their representation of the entire tumor has been questioned. Our degrees of concordance between stainings on full sections and on TMA were in the same range as published studies. Several authors have reported that TMA constructed with three cores per sample (as in our study) are representative of whole tumor specimens.23-30
As a large-scale validation tool of DNA or RNA data, IHC on TMAs should be interpreted with caution. Indeed, comparison of our cDNA array and TMA data, obtained on the same breast tumor samples, gave different results according to the gene product examined.
For a category of molecules we found important differences between RNA and protein expression levels. This was the case of P53. This discrepancy was rather expected because P53 protein detection is not dependent on mRNA overexpression, but is because of the increased half-life of a mutated protein. In normal cells, P53 protein half-life is short and expression levels are low and undetectable by IHC. In cancer cells, most P53 mutations lead to products that are not ubiquitinated and accumulate in the nuclei where they can then be detected. Other noteworthy cases were MUC1 and THBS1. These differences certainly stem from the fact that different levels of biological information are examined. For many genes, there is little correlation between the abundance of the mRNA transcript with steady-state levels of the encoded protein. Posttranscriptional and posttranslational mechanisms are likely to influence protein expression, thus blurring the correlation between mRNA and protein levels. Proteins encoded by very low levels of RNA, ie, below the detection level of cDNA arrays, can be detected by IHC because of increased protein stability (eg, the case of P53) or high sensitivity of the antibody, and reciprocally, elevated levels of RNA may produce only little amounts of detectable proteins. Special calibration of the antibody aimed to detect only a certain level of protein is another limitation. The chosen antibody may also detect only certain forms of a protein that do not correspond to the cDNA spotted on the DNA array, because of alternative splicings of mRNA for example. This particularly can explain the difference observed between THBS1 mRNA and protein levels, and consequently their different prognostic impact.31 Finally, distinct areas of a heterogeneous tumor may be submitted to RNA and protein analyses.
Conversely, we observed an excellent correlation between RNA and protein levels in one-third of the tested molecules. This was the case for ERBB2, despite the fact that its corresponding antibody is calibrated to detect only overexpression. Among the other molecules with correlated mRNA and protein expression levels were ER, GATA3, and BCL2. We and others had shown that the mRNA levels of the three genes covaried in cDNA array analyses.1,10,32 Here we were able to confirm this co-expression at the protein level. This group of co-regulated genes and proteins may be linked to the hormonal control of the mammary gland. Such identification is important for a better understanding of gene and protein networks that operate in cancer cells; it may lead to the discovery of new molecules to be targeted to block or stimulate a metabolic pathway or function; it may also provide a prognostic information clinically more relevant than that of isolated markers because it better reflects the functional status of a pathway such as the estrogen pathway of breast tumors.
Several studies have shown the interest of TMA studies in cancer research to extend cDNA array data.33 A pioneering analysis was conducted by Moch and colleagues;8 after the identification of vimentin as overexpressed in a renal cancer cell line using cDNA arrays, the authors extended this result to the protein level on a series of 532 tumor specimens arrayed onto a renal cancer TMA. Using TMA of bladder tumors containing 2317 specimens from 1842 patients, Richter and colleagues9 found a positive correlation between CCNE gene amplification measured by fluorescence in situ hybridization and cyclin-E protein overexpression measured by IHC. The combination of cDNA array and TMA allowed the identification of IGFBP2 and HSP27,34 hepsin,2 and AMACR30 as significantly overexpressed in prostate cancer, suggesting their putative diagnostic interest. IGFBP2 was also found as a marker of poor prognosis in a series of 418 brain tumors arrayed onto a TMA.35 A similar study showed the overexpression of the WT protein in ovarian cancer.36 The expression level of PKCβ was measured by IHC on a B-cell lymphoma TMA to validate cDNA array data.3 In breast carcinomas, Hedenfalk and colleagues37 showed that, like mRNA levels, cyclin D1 protein levels were differentially distributed among BRCA1 and BRCA2 hereditary tumors. All these studies showed a good correlation between the two techniques of investigation, but were limited to the analysis of a single highly selected marker and were not, with few exceptions, conducted on the same samples. Our present study is the first deliberate comparative analysis of cDNA and TMAs. It shows a correlation between the two techniques for one-third of the selected markers and the absence of correlation for the other two-thirds.
These discrepancies deserve two commentaries. First, given the flurry of encouraging data associated with the rapidly emerging cDNA array technology, it is paramount to determine to what extent changes in mRNA expression are accompanied or not by similar changes at the protein level. In some cases, the differences may be eliminated by a number of experimental precautions, such as selection of biopsy cores and antibodies, but in other cases, they will remain. If protein levels of a target molecule, or a group of molecules, correlate with its selection by cDNA array, IHC on TMA offers a powerful tool to quickly evaluate the clinical relevance of differentially expressed genes. But if they do not correlate, the cDNA array and TMA results must be considered independently because each can provide distinct information.
Second, even if the intrinsic prognostic power of cDNA array data and clustering analyses derives from the combined expression of several genes, and not from an individual gene, it may be interesting for routine clinical application to test each of these genes as a candidate marker and to determine how its expression may alone distinguish the tumor classes. The main interest of TMA lies in the possibility to test large series of tumor samples with individual markers. In our series of samples, we observed that mRNA, but not protein expression levels of THBS1 had prognostic value, suggesting that they play an important role in the discriminator power of the cDNA array gene cluster. In contrast, for MUC1, as seen earlier,38 low levels of protein were associated with a better prognosis, which was not the case for mRNA; IHC further allowed a qualitative appreciation of the protein localization, which happened to be crucial information for prognosis when an unselected population was studied.
In the period of validation studies that has now begun, for which retrospective IHC studies on archival paraffin-embedded material are required,6 it is particularly important to bear in mind that differences between mRNA and protein expression levels are possible with respect to intensities and to prognostic relevance. These differences underline the complementarity or synergy between expression measurements from cDNA arrays and IHC on TMA, and also the need for other high-throughput technologies such as cDNA arrays containing alternatively spliced transcripts,39 protein arrays,40 and in situ hybridizations on TMAs.41 The combination of these complementary approaches will accelerate even more the identification of new diagnostic and prognostic markers as well as new therapeutic targets and will improve the management of breast cancer patients.
Acknowledgments
We thank J. Hassoun, G. Houvenaegel, D. Maraninchi, and C. Mawas for encouragement; and I. Bièche (Unité propre de recherche et d’enseignement supérieur JE2195, Paris, France), J. M. Durey (Institut Paoli-Calmettes, Marseille, France), O. P. Kallioniemi (National Human Genome Research Institute, Bethesda, MD), S. Lakhani (The Breakthough Toby Robins Breast Cancer Research Centre, London, UK), R. Lidereau (E0017 INSERM, St-Cloud, France), and B. Puig (Institut Paoli-Calmettes, Marseille, France) for help and advice.
Footnotes
Address reprint requests to Daniel Birnbaum, U119 INSERM, Institut Paoli-Calmettes, Département d’Oncologie Moléculaire, 27 Bd. Leï Roure, 13009 Marseille, France. E-mail: birnbaum@marseille.inserm.fr.
Supported by INSERM, Institut Paoli-Calmettes, and grants from l’Association pour la Recherche sur le Cancer and la Ligue Nationale contre le Cancer.
ECJ and FB contributed equally to this work.
References
- 1.Bertucci F, Houlgatte R, Benziane A, Granjeaud S, Adelaide J, Tagett R, Loriod B, Jacquemier J, Viens P, Jordan B, Birnbaum D, Nguyen C: Expression profiling in primary breast carcinomas using arrays of candidate genes. Hum Mol Genet 2000, 9:2981-2991 [DOI] [PubMed] [Google Scholar]
- 2.Dhanasekaran SM, Barrette TR, Ghosh D, Shah R, Varambally S, Kurachi K, Pientas KJ, Rubin MA, Chinnaiyan AM: Delineation of prognostic biomarkers in prostate cancer. Nature 2001, 412:822-825 [DOI] [PubMed] [Google Scholar]
- 3.Shipp MA, Ross KN, Tamayo P, Weng A, Kutok JL, Aguiar RCT, Gaasenbeek M, Angelo M, Reich M, Pinkus GS, Ray TS, Koval MA, Last KW, Norton A, Lister TA, Mesirov J, Neuberg DS, Lander ES, Aster JC, Golub TR: Diffuse large B-cell lymphoma outcome prediction by gene-expression profiling and supervised machine learning. Nat Med 2002, 8:68-74 [DOI] [PubMed] [Google Scholar]
- 4.Van’t Veer LJ, Dai H, van de Vijver M, He YD, Hart AAM, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, Schreiber GJ, Kerkhoven RM, Roberts C, Linsley PS, Bernards R, Friend SH: Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002, 415:530-535 [DOI] [PubMed] [Google Scholar]
- 5.Bertucci F, Houlgatte R, Nguyen C, Viens P, Jordan B, Birnbaum D: Gene expression profiling of cancer using DNA arrays: how far from the clinic? Lancet Oncol 2001, 2:674-682 [DOI] [PubMed] [Google Scholar]
- 6.Lakhani S, Ashworth A: Microarray and histopathological analysis of tumours: the future and the past? Nat Rev Cancer 2001, 1:151-157 [DOI] [PubMed] [Google Scholar]
- 7.Kononen J, Bubendorf L, Kallioniemi A, Barlund M, Schraml P, Leighton S, Torhorst J, Mihatsch MJ, Sauter G, Kallioniemi OP: Tissue microarrays for high-throughput molecular profiling of tumors specimens. Nat Med 1998, 4:844-847 [DOI] [PubMed] [Google Scholar]
- 8.Moch H, Schraml P, Bubendorf L, Mirlacher M, Kononen J, Gasser T, Mihatsch MJ, Kallioniemi OP, Sauter G: High-throughput tissue microarray analysis to evaluate genes uncovered by cDNA microarray screening in renal cell carcinoma. Am J Pathol 1999, 154:981-986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richter J, Wagner U, Kononen J, Fijan A, Bruderer J, Schmid U, Ackerman D, Maurer R, Alund G, Knönagel H, Rist M, Wilber K, Anabitarte M, Hering F, Hardmeier T, Schönenberger A, Flury R, Jäger P, Fehr JL, Schrami P, Moch H, Mihatsch MJ, Gasser T, Kallioniemi OP, Sauter G: High-throughput tissue microarray analysis of cyclin E gene amplification and overexpression in urinary bladder cancer. Am J Pathol 2000, 157:787-794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bertucci F, Nasser V, Granjeaud S, Eisinger F, Tagett R, Adélaïde J, Loriod B, Benziane A, Giaconia A, Devilard E, Jacquemier J, Viens P, Nguyen C, Birnbaum D, Houlgatte R: Gene expression profiles of poor prognosis primary breast cancer correlate with survival. Hum Mol Genet 2002, 11:863-872 [DOI] [PubMed] [Google Scholar]
- 11.Möbus VJ, Moll R, Gerharz CD, Kieback DG, Merk O, Runnebaum IB, Linner S, Dreher L, Grill HJ, Kreienberg R: Differential characteristics of two new tumorigenic cell lines of human breast carcinoma origin. Int J Cancer 1998, 77:415-423 [DOI] [PubMed] [Google Scholar]
- 12.Theillet C, Adélaïde J, Louason G, Bonnet-Dorion F, Jacquemier J, Adnane J, Longy M, Katsaros D, Sismondi P, Gaudray P, Birnbaum D: FGFR1 and PLAT genes and DNA amplification at 8p12 in breast and ovarian cancers. Genes Chromosom Cancer 1993, 7:219-226 [DOI] [PubMed] [Google Scholar]
- 13.Bertucci F, Van Hulst S, Bernard K, Loriod B, Granjeaud S, Tagett R, Starkey M, Nguyen C, Jordan B, Birnbaum D: Expression scanning of an array of growth control genes in human tumor cell lines. Oncogene 1999, 18:3905-3912 [DOI] [PubMed] [Google Scholar]
- 14.Bertucci F, Bernard K, Loriod B, Chang YC, Granjeaud S, Birnbaum D, Nguyen C, Peck K, Jordan BR: Sensitivity issues in DNA array-based expression measurements: advantages of Nylon microarrays for small samples. Hum Mol Genet 1999, 8:1715-1722 [DOI] [PubMed] [Google Scholar]
- 15.Bièche I, Franc B, Vidaud D, Vidaud M, Lidereau R: Analyses of MYC, ERBB2 and CCND1 genes in benign and malignant thyroid follicular cell tumors by real-time polymerase chain reaction. Thyroid 2001, 11:147-152 [DOI] [PubMed] [Google Scholar]
- 16.Mitas M, Mikhitarian K, Walters C, Baron PL, Elliott BM, Brothers TE, Robison JG, Metcalf JS, Palesch YY, Zhang Z, Gillanders WE, Cole DJ: Quantitative real-time RT-PCR detection of breast cancer micrometastasis using a multigene marker panel. Int J Cancer 2001, 93:162-171 [DOI] [PubMed] [Google Scholar]
- 17.Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, Van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Lonning PE, Borresen-Dale AL: Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA 2001, 98:10869-10874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahr A, Karn T, Solbach C, Seiter T, Strebhardt K, Holtrich U, Kaufmann M: Identification of high risk breast-cancer patients by gene expression profiling. Lancet 2002, 359:131-132 [DOI] [PubMed] [Google Scholar]
- 19.Bertucci F, Eisinger F, Houlgatte R, Viens P, Birnbaum D: Gene expression profiling of breast cancer and prognosis. Lancet 2002, 360:173-174 [DOI] [PubMed] [Google Scholar]
- 20.Conte N, Charafe-Jauffret E, Delaval B, Adélaïde J, Ginestier C, Geneix J, Isnardon D, Jacquemier J, Birnbaum D: Carcinogenesis and translational controls: TACC1 is down-regulated in human cancers and associates with mRNA regulators. Oncogene 2002, 21:5619-5630 [DOI] [PubMed] [Google Scholar]
- 21.Ugolini F, Adélaïde J, Charafe-Jauffret E, Nguyen C, Jacquemier J, Jordan B, Birnbaum D, Pébusque MJ: Differential expression assay of chromosome arm 8p genes identifies Frizzled-related (FRP1/FRZB) and fibroblast growth factor receptor 1 (FGFR1) as candidate breast cancer genes. Oncogene 1999, 18:1903-1910 [DOI] [PubMed] [Google Scholar]
- 22.Rahn JJ, Dabbagh L, Pasdar M, Hugh JC: The importance of MUC1 cellular localization in patients with breast carcinoma. Cancer 2001, 91:1973-1982 [DOI] [PubMed] [Google Scholar]
- 23.Hoos A, Cordon-Cardo C: Tissue microarray profiling of cancer specimens and cell lines: opportunities and limitations. Lab Invest 2001, 81:1331-1338 [DOI] [PubMed] [Google Scholar]
- 24.Nocito A, Kononen J, Kallioniemi OP, Sauter G: Tissue microarrays (TMAs) for high-throughput molecular pathology research. Int J Cancer 2001, 94:1-5 [DOI] [PubMed] [Google Scholar]
- 25.Rimm DL, Camp RL, Charette LA, Olsen DA, Provost E: Amplification of tissue by construction of tissue microarrays. Exp Mol Pathol 2001, 70:255-264 [DOI] [PubMed] [Google Scholar]
- 26.Camp RL, Charette LA, Rimm DL: Validation of tissue microarray technology in breast carcinoma. Lab Invest 2000, 80:1943-1949 [DOI] [PubMed] [Google Scholar]
- 27.Hoos A, Urist MJ, Stojadinovic A, Mastorides S, Dudas ME, Leung DHY, Kuo D, Brennan MF, Lewis JL, Cordon-Cardo C: Validation of the tissue microarray for immunohistochemical profiling of cancer specimens using the example of human fibroblastic tumors. Am J Pathol 2001, 158:1245-1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Torhorst J, Bucher C, Kononen J, Haas P, Zuber M, Kochli OR, Mross F, Dieterich H, Moch H, Mihatsch M, Kallioniemi OP, Sauter G: Tissue microarray for rapid linking of molecular changes to clinical endpoints. Am J Pathol 2001, 159:2249-2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rubin MA, Dunn R, Strawderman M, Pienta KJ: Tissue microarray sampling strategy for prostate cancer biomarker analysis. Am J Surg Pathol 2002, 26:312-319 [DOI] [PubMed] [Google Scholar]
- 30.Rubin MA, Zhou M, Dhanasekaran SM, Varambally S, Barrette TR, Sanda MG, Pienta KJ, Ghosh D, Chinnaiyan AM: Alpha-methylacyl coenzyme A racemase as a tissue biomarker for prostate cancer. JAMA 2002, 287:1662-1670 [DOI] [PubMed] [Google Scholar]
- 31.Matthias LJ, Gotis-Graham I, Underwood PA, McNeil HP, Hogg PJ: Identification of monoclonal antibodies that recognize different disulfide bonded forms of thrombospondin 1. Biochem Biophys Acta 1996, 1296:138-144 [DOI] [PubMed] [Google Scholar]
- 32.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lonning PE, Borresen-Dale AL, Brown PO, Botstein D: Distinctive gene expression patterns in human mammary epithelial cells and breast cancers. Proc Natl Acad Sci USA 1999, 96:9212-9217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mousses S, Kallioniemi A, Kauraniemi P, Elkahloun A, Kallioniemi OP: Clinical and functional target validation using tissue and cell microarrays. Curr Opin Chem Biol 2001, 6:97-101 [DOI] [PubMed] [Google Scholar]
- 34.Bubendorf L, Kolmer M, Kononen J, Koivisto P, Mousses S, Chen Y, Mahlamaki E, Schraml P, Moch H, Willi N, Elkahloun AG, Pretlow TG, Gasser TC, Mihatsch MJ, Sauter G, Kallioniemi OP: Hormone therapy failure in human prostate cancer: analysis by complementary DNA and tissue microarrays. J Natl Cancer Inst 1999, 91:1758-1764 [DOI] [PubMed] [Google Scholar]
- 35.Sallinen SL, Sallinen PK, Haapasalo HK, Helin HJ, Helen PT, Schraml P, Kallioniemi OP, Kononen J: identification of differentially expressed genes in human gliomas by DNA microarray and tissue chip techniques. Cancer Res 2000, 60:6617-6622 [PubMed] [Google Scholar]
- 36.Su AI, Welsh JB, Sapinoso LM, Kern SG, Dimitrov P, Lapp H, Schultz PG, Powell SM, Moskaluk CA, Frierson HF, Hampton GM: Molecular classification of human carcinomas by use of gene expression signatures. Cancer Res 2001, 61:7388-7393 [PubMed] [Google Scholar]
- 37.Hedenfalk I, Duggan D, Chen Y, Radmacher M, Bittner M, Simon R, Meltzer P, Gusterson B, Esteller M, Kallioniemi OP, Wilfond B, Borg A, Trent J: Gene-expression profiles in hereditary breast cancer. N Engl J Med 2001, 344:539-548 [DOI] [PubMed] [Google Scholar]
- 38.McGuckin MA, Walsh MD, Hohn BG, Ward BG, Wright RG: Prognostic significance of MUC1 epithelial mucin expression in breast cancer. Hum Pathol 1995, 26:432-439 [DOI] [PubMed] [Google Scholar]
- 39.Yeakley JM, Fan JB, Doucet D, Luo L, Wickham E, Ye Z, Chee MS, Fu XD: Profiling alternative splicing on fiber-optic arrays. Nat Biotechnol 2002, 20:353-358 [DOI] [PubMed] [Google Scholar]
- 40.Emili AQ, Cagney G: Large-scale functional analysis using peptide or protein arrays. Nat Biotechnol 2000, 18:393-397 [DOI] [PubMed] [Google Scholar]
- 41.Fejzo MS, Slamon DJ: Frozen tumor tissue microarray technology for analysis of tumor RNA, DNA, and proteins. Am J Pathol 2001, 159:1645-1650 [DOI] [PMC free article] [PubMed] [Google Scholar]