

Abstract
Objectives:
Core CSF changes in Alzheimer disease (AD) are decreased amyloid β1–42, increased total tau, and increased phospho-tau, probably indicating amyloid plaque accumulation, axonal degeneration, and tangle pathology, respectively. These biomarkers identify AD already at the predementia stage, but their diagnostic performance might be affected by age-dependent increase of AD-type brain pathology in cognitively unaffected elderly.
Methods:
We investigated effects of age on the diagnostic performance of CSF biomarkers in a uniquely large multicenter study population, including a cross-sectional cohort of 529 patients with AD dementia (median age 71, range 43–89 years) and 304 controls (67, 44–91 years), and a longitudinal cohort of 750 subjects without dementia with mild cognitive impairment (69, 43–89 years) followed for at least 2 years, or until dementia diagnosis.
Results:
The specificities for subjects without AD and the areas under the receiver operating characteristics curves decreased with age. However, the positive predictive value for a combination of biomarkers remained stable, while the negative predictive value decreased only slightly in old subjects, as an effect of the high AD prevalence in older ages.
Conclusion:
Although the diagnostic accuracies for AD decreased with age, the predictive values for a combination of biomarkers remained essentially stable. The findings highlight biomarker variability across ages, but support the use of CSF biomarkers for AD even in older populations.
Future disease-modifying drugs for Alzheimer disease (AD) will likely be most effective early in the disease, emphasizing the need for early diagnosis.1 Already before dementia, patients with AD have reduced CSF amyloid β1–42 (Aβ42), and increased total tau (t-tau) and phosphorylated tau (p-tau), probably reflecting plaque pathology, axonal degeneration, and intraneuronal tangles, respectively.2 Novel diagnostic criteria for AD propose inclusion of in vivo markers of AD pathology,3,4 thereby enabling diagnosis of patients with mild cognitive impairment (MCI) and positive biomarkers as prodromal AD3 or MCI due to AD.5 To facilitate this groundbreaking step, the diagnostic performance of biomarkers needs to be carefully assessed in relation to possible confounding factors, such as age of subjects.
AD-type brain alterations increase with age in individuals without dementia,6–9 and AD-like CSF biomarker patterns are reported in about a third of cognitively unaffected elderly,10–19 but it is unclear if this reflects asymptomatic AD or if AD-type brain alterations may be insufficient for clinical disease with other factors modulating symptom development. Since most patients with AD are old, overlap toward healthy subjects might undermine broad-scale use of biomarkers. To elucidate the impact of age on biomarker diagnostic performance, we utilized the largest set of patients with AD with CSF data to date,13 investigating patients with AD dementia vs cognitively healthy controls in a cross-sectional cohort and subjects with MCI in a longitudinal cohort. We primarily hypothesized that diagnostic accuracies would decrease with age, but it was unclear how age would affect overall diagnostic usability, since this is also heavily influenced by disease prevalence.
METHODS
Standard protocol approvals and patient consent.
All subjects provided informed consent. Local ethics committees of the participating centers approved the study.
Subjects.
We designed the study following the Standards for the Reporting of Diagnostic Accuracy (STARD).13,20 The study population, enrolled at 12 centers in Europe and the United States between 1990 and 2007, was described previously.13 The cross-sectional cohort included 529 patients with AD dementia and 304 cognitively healthy controls, and the longitudinal cohort 750 patients with MCI, consecutively recruited at memory clinics to prospectively evaluate clinically relevant predictive values. Physicians specialized in cognitive disorders and blinded to the CSF results assessed all participants. The patients with AD dementia met dementia criteria defined by DSM-IV and National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer's Disease and Related Disorders Association criteria for AD.21,22 As controls we included volunteers without cognitive symptoms (Mini-Mental State Examination > 25) and no active neurologic or psychiatric disease. The patients with MCI met the revised Petersen criteria at inclusion.23 Exclusion criteria included known causes of cognitive impairment, such as brain tumor, subdural hematoma, and ongoing alcohol abuse. All patients with MCI were followed for a minimum of 2 years, or until diagnosed with dementia (median follow-up, 3 years; range, 2–11 years). Some patients with MCI remained cognitively stable (SMCI). Patients with MCI deteriorating to AD dementia during the study were called MCI-AD (a clinical diagnosis of AD dementia defined the reference standard). Patients with MCI diagnosed with non-AD dementias during the study were called MCI-other. The requirements of the National Institute of Neurological Disorders and Stroke–Association Internationale pour la Recherche en l'Enseignement en Neurosciences and the criteria established by Roman et al.,24 Erkinjuntti et al.,25 McKeith et al.,26 and Brun et al.27 were used for vascular dementia, dementia with Lewy bodies, and frontotemporal dementia, respectively. All participants were stratified by age at sampling into 3 groups: up to 64 years, 65–74 years, and 75 years and above (table 1; figure e-1, A and B, on the Neurology® Web site at www.neurology.org). These age limits were chosen pre hoc, before statistical analyses, and resulted in overall comparable group sizes. The choice of precisely 3 groups was a tradeoff between obtaining satisfactorily large groups while still allowing detection of age-dependent effects.
Table 1.
Subject counts, sex, MMSE, and prevalence numbers in different age groups
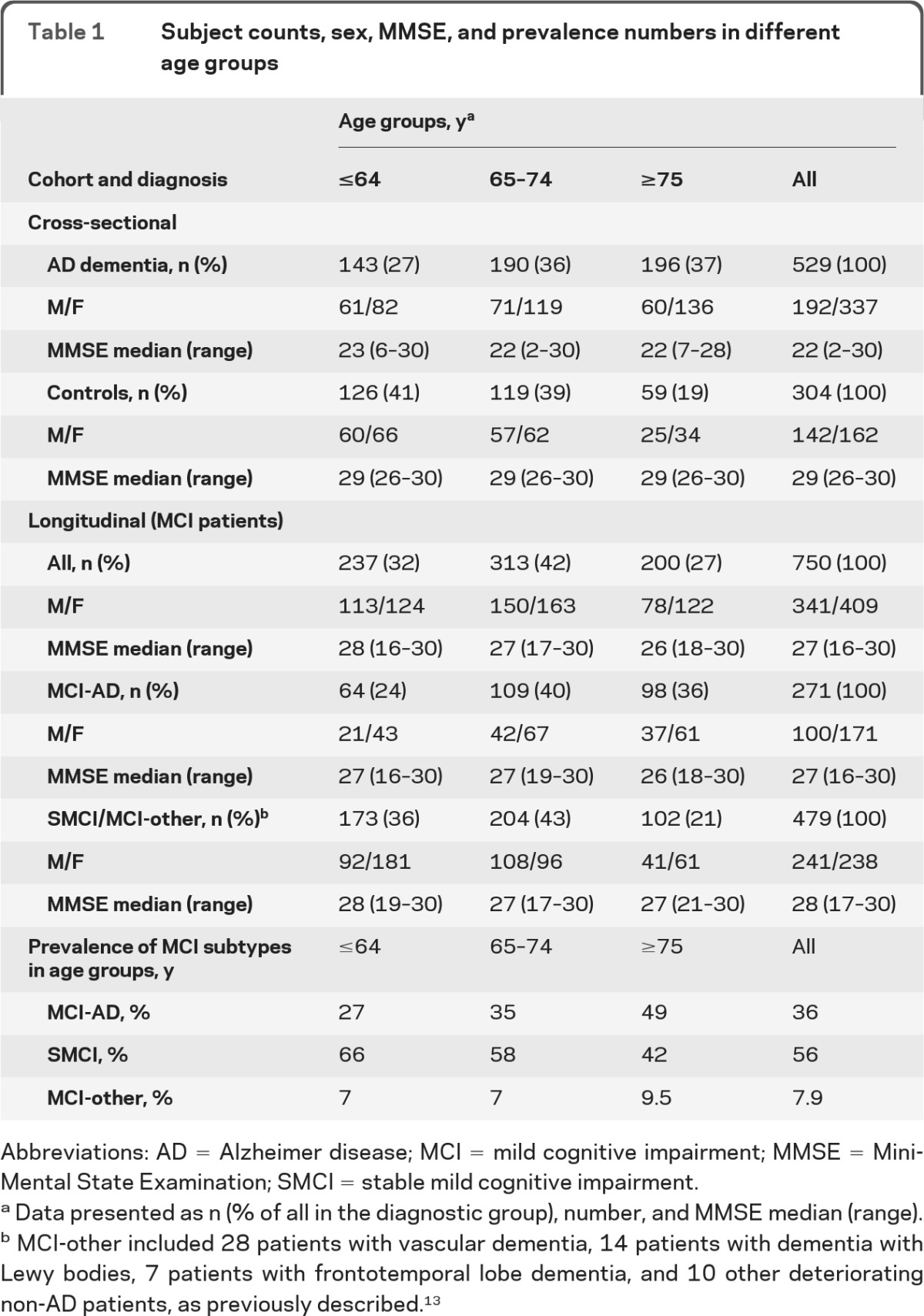
Abbreviations: AD = Alzheimer disease; MCI = mild cognitive impairment; MMSE = Mini-Mental State Examination; SMCI = stable mild cognitive impairment.
Data presented as n (% of all in the diagnostic group), number, and MMSE median (range).
MCI-other included 28 patients with vascular dementia, 14 patients with dementia with Lewy bodies, 7 patients with frontotemporal lobe dementia, and 10 other deteriorating non-AD patients, as previously described.13
Variables.
The main endpoints were differences in diagnostic performance for biomarkers between groups. The main predictor was age. Within each age group, we examined the difference in diagnostic performance between biomarkers.
CSF sampling and biochemical analyses.
All CSF procedures have been described previously.13 All participants underwent lumbar puncture in the L3–4 or L4–5 space. No serious adverse events were reported. The samples were stored in polypropylene tubes at −80°C or −70°C until analysis. Samples were analyzed at the Clinical Neurochemistry Laboratory in Mölndal, Sweden, except for samples from Amsterdam, Kuopio, and Munich, where analyses were done locally. Subsets of samples from these centers were reanalyzed in Mölndal to adjust for interlaboratory variations (necessary for Aβ42 and t-tau).13 CSF biomarkers were determined by ELISA (Innotest®, Innogenetics, Ghent, Belgium).10,13,28,29 For 2 centers, concentrations were determined by Luminex xMAP® (Inno-Bia AlzBio3®, Innogenetics), and converted to ELISA values based on previously published conversion factors.30 Experienced and certified laboratory technicians blinded to all clinical information performed the analyses.
Statistics.
Biomarker distributions.
Differences in biomarker distributions among groups were examined with the nonparametric Kruskal-Wallis test with Dunn post hoc test, correcting for multiple comparisons. The Spearman correlation coefficient was used for analyses of correlations between age and biomarkers. Statistical significance was set to p < 0.05.
Diagnostic accuracy and usefulness.
The performance of a diagnostic test includes its accuracy and usefulness.31 The accuracy is the inherent capacity of a test to discriminate disease from health, affected by the biological variability of the measured factor between compared groups, and by the technical variability in the applied procedures. Our principal estimate of diagnostic accuracy was the area under the receiver operating characteristic curve (AUROC). Receiver operating characteristic (ROC) is a statistic for the goodness of a predictive test in a binary classification task. The ROC curve is a graphic representation of the sensitivity and specificity of the test across the entire range of possible classification cutoffs. AUROC 0.50 indicates random test performance and AUROC 1.00 perfect performance. Sensitivities and specificities were also evaluated at defined cutoffs. The test usefulness refers to the practical value of the information generated by the test, which is affected by factors beyond the test itself, in particular the disease prevalence. Our measurements of diagnostic usefulness were positive (PPV) and negative (NPV) predictive values (appendix e-1, “Diagnostic usefulness”), and positive (LR+) and negative (LR−) likelihood ratios: LR+ = sensitivity/(1−specificity) and LR− = (1 − sensitivity)/specificity. LRs give useful information if the pretest odds is known, since a high LR+ or a small LR− changes the post-test odds significantly from the pretest odds. Most strong tests are characterized by a LR+ >5 or a LR− <0.2.
Comparison of AUROC models.
We compared AUROCs for the same biomarkers across age groups and, since there might be temporal differences in the development of pathologic traits in the brains of patients with AD and subjects without AD, AUROCs for different biomarkers within age groups (appendix e-1, “Comparison of AUROC models”).
Cutoffs.
Sensitivities, specificities, PPVs, NPVs, and LRs were evaluated at defined cutoffs, generated independent of the longitudinal cohort, at 85% sensitivity for AD dementia, which has been suggested as satisfactory for AD biomarkers.32 The cutoffs applied on the cross-sectional cohort were the specific data points that yielded at least 85% sensitivity for AD dementia (appendix e-1, “Cutoffs”), while the cutoffs applied on the longitudinal cohort were linear interpolations at precisely 85% sensitivity. The biomarkers were also combined in a logistic regression model with the 2 covariates Aβ42/p-tau ratio (Y) and t-tau (X), used to construct an analytical expression and cutoff line with 85% sensitivity (Y = 3.694 + 0.0105X, “Combination”).13 To calculate AUROCs for this combination, the 2-dimensional data were transformed into 1 dimension (appendix e-1, “Transformation of scatter plot data into 1 dimension”). Note that for the cross-sectional cohort, those AUROC estimates must be interpreted with caution, since they will be biased toward 1 due to a phenomenon called suboptimization (appendix e-1, “Suboptimization”). From the projected data we also calculated sensitivity, specificity, LRs, PPV, and NPV for the combination.
Outliers.
Outlying biomarkers were defined as values differing >3 standard deviations from the mean (in each age group of controls, AD dementia, and MCI). Few subjects exceeded these limits (appendix e-1, “Outliers”), and excluding them did not affect the main results (similar AUROC models, and similar results when comparing AUROC models between age groups). No restrictions were given to outliers in the final analyses.
Software.
GraphPad Prism 5 (GraphPad Software Inc., La Jolla, CA) was used for general statistics. PASW Statistics 18 (SPSS Inc., Chicago, IL) was used for logistic regression analysis. MedCalc for Windows, version 11.4.4.0 (MedCalc Software, Mariakerke, Belgium), was used for AUROC modeling and other estimates of test performance.
RESULTS
Biomarker distributions are more similar between subjects with AD and subjects without AD in elderly than in young subjects.
The biomarker data have been reported previously,13 but not with detailed studies of accuracy and usefulness in relation to age. As reported, age correlated with t-tau (r = 0.22, p < 0.001) and p-tau (r = 0.23, p < 0.001) in controls, and with Aβ42 in SMCI (r = −0.23, p < 0.001). Since the main aim of the study was to assess diagnostic performance for MCI-AD toward remaining MCI, we merged SMCI and MCI-other patients. This was further justified by these groups' similar biomarker distributions, except for lower Aβ42 in the youngest MCI-other patients (figure 1) (the only bias from this merging is a slight underestimation of the diagnostic performance for nonprogressive MCI in the youngest group, since SMCI/MCI-other shifts toward MCI-AD in Aβ42). All biomarkers differed across ages, but with different patterns in the different diagnostic groups (figure 2, A–F). In sum, older controls and SMCI/MCI-other had more AD-like distributions than younger controls and SMCI/MCI-other, respectively. Likewise, older patients with AD dementia had more control-like Aβ42 and p-tau distributions than younger patients with AD dementia. However, as is evident from figure 2, the main source of variation was diagnosis rather than age.
Figure 1. CSF biomarkers in stable mild cognitive impairment (MCI), MCI-other, and MCI–Alzheimer disease (AD).

Distribution of biomarkers in the longitudinal cohort (MCI) in different age groups. Comparisons between diagnostic groups were done using the nonparametric Kruskal-Wallis test with Dunn post hoc test, with significant differences indicated as *** (p < 0.001) and ** (p < 0.01).
Figure 2. Comparisons of CSF biomarker distributions in different age groups.

Biomarker distributions, presented as cumulative frequencies, in Alzheimer disease (AD) dementia vs controls and stable mild cognitive impairment (SMCI)/mild cognitive impairment (MCI)-other vs MCI-AD (A–F). An “AD-like” profile is characterized by a left shift of the curves for amyloid β1–42 (Aβ42) and a right shift for total tau (t-tau)/phospho-tau (p-tau). As explained previously,13 data on CSF Aβ42, t-tau, or p-tau was missing in 19 patients with AD dementia, 1 control, and 1 patient with MCI.
Diagnostic accuracy for AD decreases with age.
We investigated if the age-dependent variability in biomarkers affected their diagnostic accuracies for AD dementia vs controls, and for MCI-AD vs SMCI/MCI-other. All AUROCs decreased with age (figure e-1, C–H). AUROCs were also compared within age groups to determine if the optimal biomarker differed with age. Generally, the combination of biomarkers had the highest AUROC and p-tau the smallest AUROC. We noted that the ROC curves for Aβ42 and t-tau intersected in most groups, in a way that although Aβ42 achieved superior sensitivity, there were ranges of possible cutoffs where t-tau had superior specificity.
Specificities for non-AD decrease with age while sensitivities for AD are stable.
For all biomarkers, specificities for controls at the 85% sensitivity level decreased with age (table 2). The combination of biomarkers had extraordinary specificity in the youngest controls, and remained superior to any individual marker in all age groups. In the longitudinal cohort, specificities for SMCI/MCI-other decreased with age while sensitivities for MCI-AD were essentially stable (table 2; table e-1).
Table 2.
Specificities and likelihood ratios at cutoffs for 85% sensitivity for AD dementiaa
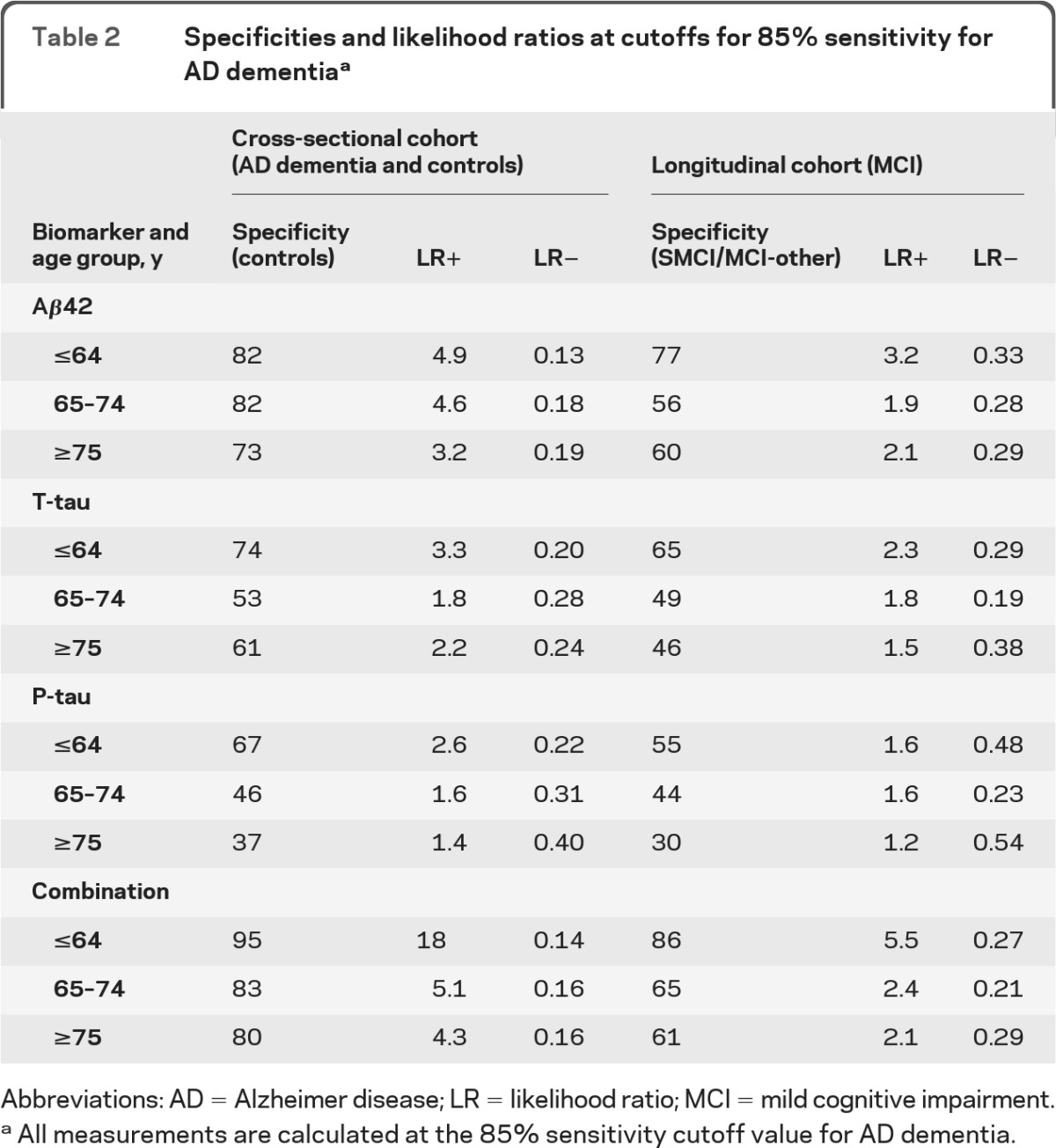
Abbreviations: AD = Alzheimer disease; LR = likelihood ratio; MCI = mild cognitive impairment.
All measurements are calculated at the 85% sensitivity cutoff value for AD dementia.
Likelihood ratios.
The LR+ for the combination of biomarkers was clearly superior to LR+ for individual biomarkers in the youngest subjects, but dropped close to the level of the LR+ for Aβ42 in older subjects (table 2). LR− increased with age for t-tau and p-tau, but remained stable (which is preferable) for Aβ42 and the combination. These results suggest that the relative extra information from combining biomarkers compared to using individual biomarkers is largest to rule in AD in young subjects. LRs in the longitudinal cohort were closer to 1 than in the cross-sectional cohort, indicating lower diagnostic usefulness, but with the same distribution across ages. However, from Bayes' theorem follows that the absolute effect of a diagnostic test depends on disease prevalence (post-test odds = LR × pretest odds, where pretest odds is derived from the prevalence), and as discussed below AD prevalence depended strongly on age.
Predictive values.
The disease probability before a test is carried out equals the prevalence in the surveyed group. The PPV is the revised estimate of the same probability for subjects who are tested positive, and the NPV is the probability that a negative test is correct. From this follows that PPV and NPV are profoundly influenced by disease prevalence. PPV is favored by a high prevalence and NPV is favored by a low prevalence. In the longitudinal cohort, the prevalence of MCI-AD was almost twice as high in the oldest compared to the youngest group (table 1). In line with this, PPVs of the individual biomarkers increased with age and NPVs decreased with age (figure e-1, I–K). At the 85% sensitivity level, the PPV of the combination of biomarkers was higher than the PPV of any individual biomarker in all age groups, while the NPV of the combination of biomarkers was similar to the NPV of Aβ42 alone in all age groups (figure 3). For each biomarker and age group, other cutoffs increased or decreased PPVs with reversed effects on NPVs (figure 3; figure e-1, I–K).
Figure 3. Positive predictive value (PPV) and negative predictive value (NPV).

Predictive values across the entire ranges of possible cutoffs in patients with mild cognitive impairment (MCI). The dotted lines indicate specific cutoffs generated at 85% sensitivity for Alzheimer disease (AD) dementia (independent of the MCI cohort). The “Combination” was an analytical expression derived from the combination of biomarkers (using the formula Y[Aβ42/phospho-tau] = 3.694 − 0.0105 × [tau]).13 Data points with 95% confidence intervals ranging more than 50% are excluded from the graphs. SMCI = stable mild cognitive impairment.
Influence of APOE genotype.
APOE genotype was available in 189 controls, 400 AD dementia, 223 MCI-AD, and 393 SMCI/MCI-other patients. APOE ϵ4 had no major effect on correlations between age and biomarkers (figure e-2).
DISCUSSION
Age had distinct effects on the diagnostic performance of CSF AD biomarkers. As hypothesized, the specificities for controls and stable MCI/MCI-other decreased with age. In line with this, the biomarkers were relatively most useful to rule in AD in young subjects, in terms of LRs. The usefulness in absolute terms of a diagnostic test depends on disease prevalence, which increased with age in this study, causing PPVs to increase or remain stable (although at modest levels at the predefined cutoffs), while NPVs dropped slightly in the oldest. A detailed clarification of age distributions and disease prevalence is necessary to determine the true diagnostic usefulness of a biomarker test. For AD, this will likely be important for all diagnostic modalities, including PET amyloid imaging, where measurements correlate tightly with CSF Aβ42.33 Most cutoffs at 85% sensitivity for AD dementia were similar across ages, which is in agreement with earlier reports of CSF biomarker stability during follow-up in AD.34 Age-adjusted cutoffs are therefore not necessary to retain a low number of false-negative AD cases, but unadjusted cutoffs will increase the number of “false-positives” with age, at least with the follow-up time of this study. At carefully selected cutoffs, the diagnostic performance of these biomarkers supports their use in clinical trials for patient selection or stratification (the high NPVs suggest a particular usefulness for AD exclusion). If analyzed at specialized laboratories, minimizing measurement variability and utilizing reliable reference limits, they may also aid in dementia investigations.
Similar to other age-related disorders, including arteriosclerosis, and certain cancer forms (such as prostate cancer), AD forms a continuum with aging6 which lowers the difference in pathology and diagnostic methods between disease and aging with advanced age. In AD, brain changes likely precede clinical symptoms by several years. Therefore, a proportion of cognitively intact elderly have preclinical AD, with an AD-like biomarker profile. This complicates clinical studies on very old patients with AD. Studies with 10–15 years follow-up may be needed to fully clarify how to best use biomarkers in the clinical diagnosis of early-stage AD in the very old. Our results stress the importance for researchers to characterize their control populations carefully with biomarkers, especially in studies of elderly patients. For clinicians today, the biomarkers' high NPVs may be used to exclude AD also in very old patients, while positive biomarkers may be useful to select patients for more detailed follow-up. A more daunting perspective is that AD-type brain alterations are insufficient for clinical disease, and that other factors modulate symptom development.35 Age-dependent reduced diagnostic accuracies would then be an intrinsic problem for biomarkers measuring AD-type alterations, related to larger questions about AD pathogenesis. In relation to this, CSF Aβ42 and p-tau alterations were more pronounced in younger than in older patients with AD, which contributed to the age-dependent overlap toward non-AD. More pronounced differences in younger patients with AD have been shown previously for CSF Aβ4210 and other CSF biomarkers, such as ganglioside GM1 and chromogranin A.36,37 This might reflect a larger heterogeneity in older patients with AD, with several contributing comorbidities.38 Biomarkers for other pathologies, such as vascular pathology or synucleinopathy, might help to clarify this.39–40
A limitation of this study is that clinical rather than autopsy examination served as the diagnostic standard, although the reliability of neuropathologic examination as AD gold standard has been debated.e1 Also, the study included few very old participants (23 subjects were ≥85 years), hindering examination of effects of very high age. In a recent autopsy study, the associations between dementia and plaque and tangle pathology were still strong at 75 years, but attenuated at 95 years, so major effects on the diagnostic performance might appear first in the oldest old.9 The relatively short follow-up time in the longitudinal cohort might have hindered detection of some MCI-AD cases among the patients with SMCI, underestimating the biomarkers' true performance if cognitively stable subjects deteriorated after the end of the study. The results may also have been affected by the inclusion of 4 laboratories (although we tried to normalize for interlaboratory variability) and by the impact of cognitive reserve (since years of education for study subjects was not available). Major strengths of the study include the uniquely large material, enabling age stratifications with large subgroups; the design with independent cutoffs evaluated in a longitudinal cohort; and the multicenter design with several participating clinics and laboratories, favoring international generalization of the results.
In the near future, in vivo measurements of AD pathology in early-stage patients may be included on a broad scale in clinical and research settings.3,5 To enable this, biomarkers need standardization to decrease interlaboratory variability,e2,e3 and their diagnostic performance needs to be carefully determined. Age of study subjects is an important confounding factor in AD biomarker studies, and could explain some of the variability in published diagnostic accuracies and cutoffs. Great care should be taken to control for age in future studies. Studies with longer follow-up time may clarify if effects of age on diagnostic performance are caused by erroneous classifications of study subjects, or related to yet undeciphered variations in the biology of neurodegeneration and normal aging.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Åsa Källén, Monica Christiansson, Sara Hullberg, and Dzemila Secic for technical assistance and volunteers and patients for their participation.
GLOSSARY
- Aβ42
amyloid β1–42
- AD
Alzheimer disease
- AUROC
area under the receiver operating characteristic curve
- DSM-IV
Diagnostic and Statistical Manual of Mental Disorders, 4th edition
- LR
likelihood ratio
- MCI
mild cognitive impairment
- NPV
negative predictive value
- p-tau
phospho-tau
- PPV
positive predictive value
- ROC
receiver operating characteristic
- SMCI
stable mild cognitive impairment
- STARD
Standards for the Reporting of Diagnostic Accuracy
- t-tau
total tau
AUTHOR CONTRIBUTIONS
N.M. had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. N.M., K.B., and H.Z. designed the study. O.H., N.A., L.P., S.H., M.W., K.R., E.K., M.C., M.T., E.M., D.A., P.V., J.S., J.M., H.H., P.S., A.W., M.J., B.W., and K.B. performed data acquisition. N.M. and E.R. analyzed the data and performed the statistical analysis. N.M., E.R., K.B., and H.Z. interpreted the data. N.M. wrote the manuscript. All authors revised the manuscript critically for important intellectual content.
STUDY FUNDING
This study was funded with grants from Swedish Brain Power, the Swedish Research Council (projects 14002, 2006-6227, KP2010-63P-21562-01-4, and K2011-61X-20401-05-6), the Alzheimer's Association (NIRG-08-90356), cNEUPRO, the Lundbeck Foundation, the Royal Swedish Academy of Sciences, Sahlgrenska University Hospital, Sahlgrenska Academy, Stiftelsen Psykiatriska Forskningsfonden, Stiftelsen Gamla Tjänarinnor, Uppsala Universitets Medicinska Fakultet stiftelse för psykiatrisk och neurologisk forskning, the Swedish Brain Fund, the Söderberg Foundation, the Alzheimer Foundation, Sweden, and the Dementia Association, Sweden.
DISCLOSURE
Dr. Mattsson has served on a scientific advisory board for Actelion Pharmaceuticals Ltd; serves as an Associate Editor for the Journal of Alzheimer's Disease; and receives research support from Sahlgrenska University Hospital, Sahlgrenska Academy, Stiftelsen Psykiatriska Forskningsfonden, Stiftelsen Gamla Tjänarinnor, Uppsala Universitets Medicinska Fakultet stiftelse för psykiatrisk och neurologisk forskning, the Swedish Brain Fund, the Söderberg Foundation, the Alzheimer Foundation, Sweden, and the Dementia Association, Sweden. Dr. Rosén, Dr. Hansson, and Dr. Andreasen report no disclosures. Dr. Parnetti has served on a scientific board for Innogenetics. Dr. Jonsson has received speaker honoraria from Novartis, Pfizer Inc, and Lundbeck, Inc. and honoraria from Eli Lilly & Company and Bristol-Myers Squibb. Dr. Herukka serves as an Associate Editor for the Journal of Alzheimer's Disease and on the editorial board of the Journal of Alzheimer's Disease and Parkinsonism. Dr. van der Flier reports no disclosures. Dr. Blankenstein has received speaker honoraria from Abbott and Ferring. Dr. Ewers has received funding for travel from the Alzheimer's Association and International College of Geriatric Psychoneuropharmacology and serves as an Associate Editor for the Journal of Alzheimer's Disease. Dr. Rich and Dr. Kaiser report no disclosures. Dr. Verbeek serves as an Associate Editor for the Journal of Alzheimer's Disease; has served as a consultant for Schering-Plough Corp.; and receives/has received research support from Schering-Plough Corp., the AADC Research Trust, Internationale Stichting Alzheimer Onderzoek, Center for Translational Molecular Medicine, American Alzheimer Association, Alzheimer Drug Discovery Foundation, Stichting Internationaal Parkinson Fonds, and Hersenstichting Nederland. Dr. Olde Rikkert has served as a consultant for Janssen, Novartis, Lundbeck, Inc., and Numico. Dr. Tsolaki has served on a scientific advisory board for Novartis and UCB and as a consultant for Pfizer Inc. Dr. Mulugeta reports no disclosures. Dr. Aarsland serves on scientific advisory boards for Lundbeck, Inc., Merck Serono, and DiaGenic ASA; has received funding for travel and speaker honoraria from Lundbeck, Inc, Novartis, GE Healthcare, GlaxoSmithKline, and DiaGenic ASA; serves on the editorial boards of International Psychiogeriatrics and Movement Disorders; and has received research support from Lundbeck, Inc. and Merck Serono. Dr. Visser has served on scientific advisory boards for Bristol-Myers Squibb, Ipsen, Elan Corporation, and Myriad Genetics, Inc.; has received speaker honoraria from Lundbeck, Inc.; serves on the editorial boards of GeroPsych, BMC Geriatrics, Mind and Brain, and the Journal of Psychiatry and as an Associate Editor for the Journal of Alzheimer's Disease; and receives research support from DiaGenic ASA and the European Commission. Dr. Schröder and Dr. Marcusson report no disclosures. Dr. de Leon serves on the scientific advisory board of the French Alzheimer Foundation: has received funding for travel from Nature Medicine; is listed as an author on image analysis patents managed by NYU; and receives research support from the NIH. Dr. Hampel serves on scientific advisory boards or as a consultant for B.R.A.H.M.S. Biotech GmbH, Pfizer Inc/Eisai Inc., GlaxoSmithKline, Novartis and Bristol-Myers Squibb, Novartis, Janssen, and Merz Pharmaceuticals, LLC; has received speaker honoraria from GlaxoSmithKline, Lundbeck, Inc., sanofi-aventis, Eisai Inc., Pfizer Inc, Janssen, Novartis, Merz Pharmaceuticals, LLC, and Schwabe Pharmaceuticals; serves on the editorial boards of Brain Aging, Neuroscience Imaging, the Journal of Alzheimer's Disease, World Journal of Biological Psychiatry, Journal of Medical Psychology, Pharmacopsychiatry, World Journal of Neurology, World Journal of Pharmacology, and Journal of Diagnostics; has several patents pending; and receives research support from B.R.A.H.M.S. Biotech GmbH, Novartis, Janssen, GlaxoSmithKline, BMBF (German Ministry for Education and Research), Trinity College Dublin, Ireland, State of Hess, Germany, Katharina-Hardt-Foundation, and Thea-Goering-Foundation. Dr. Scheltens has served on scientific advisory boards for Danone Research, Wyeth/Elan Corporation, Bristol-Myers Squibb, Genentech, Inc., Pfizer Inc, GE Healthcare, and Janssen AI; has received speaker honoraria from Lundbeck, Inc., Danone Research, and Nutricia; serves as Book Review Editor for Alzheimer's Disease and Associated Disorders and on the editorial board of Dementia Geriatric Cognitive Disorders; serves as a consultant for Pfizer Inc, Roche, GE Healthcare, Avid Radiopharmaceuticals, Inc./Eli Lilly & Company, Genentech, Inc., and Janssen AI; and receives research support from Alzheimer Nederland, Stichting VUmc fonds, and Innovation Fund. Dr. Wallin serves on a scientific advisory board for Lundbeck, Inc.; has received speaker honoraria from Lundbeck, Inc., Novartis, Phillips Pharmaceuticals Ltd, and Pfizer Inc; serves on the editorial board of Alzheimer's Disease and Associated Disorders; receives publishing royalties for Alzheimers sjukdom och andra kognitiva sjukdomar (Libers förlag, 1995); and receives research support from the Swedish Research Council, Swedish Brain Power, and Stiftelsen Gamla Tjänarinnor; Sahlgrenska University Hospital. Dr. Eriksdotter-Jönhagen receives research support from the Swedish municipalities (SKL), the Swedish Brain Power network, and the Gustav V and Queen Victoria's Freemason Foundation. Dr. Minthon serves on a scientific advisory boards for Pfizer Inc. Dr. Winblad has served on scientific advisory boards for and received funding for travel and honoraria from Pfizer Inc and Eisai Inc. and receives research support from the Swedish Research Council and the Knut & Alice Wallenberg Foundation. Dr. Blennow has served on scientific advisory boards for Pfizer Inc., Innogenetics, Octapharma AG, and Baxter International Inc.; has received speaker honoraria from Janssen AI; serves on the editorial board of Neurodegenerative Diseases; serves as a consultant for AstraZeneca and Bristol-Myers Squibb; and receives research support from Bristol-Myers Squibb, The Research Council, Sweden, Västra Götalandsregionen, Sweden, The Swedish Brain Power Project and The Swedish Council for Working Life and Social Research, The Swedish Alzheimer Foundation, Stiftelsen för Gamla Tjänarinnor, and The King Gustaf V and Queen Victoria's Foundation. Dr. Zetterberg has served on a scientific advisory board for GlaxoSmithKline; serves as an Associate Editor of the Journal of Alzheimer's Disease; and receives research support from The Swedish Research Council and Swedish State Support for Clinical Research.
REFERENCES
- 1. Citron M. Alzheimer's disease: strategies for disease modification. Nat Rev Drug Discov 2010; 9: 387– 398 [DOI] [PubMed] [Google Scholar]
- 2. Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol 2010; 6: 131– 144 [DOI] [PubMed] [Google Scholar]
- 3. Dubois B, Feldman HH, Jacova C, et al. Revising the definition of Alzheimer's disease: a new lexicon. Lancet Neurol 2010; 9: 1118– 1127 [DOI] [PubMed] [Google Scholar]
- 4. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging–Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011; 7: 263– 269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging–Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011; 7: 270– 279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mann DM, Yates PO, Marcyniuk B. Alzheimer's presenile dementia, senile dementia of Alzheimer type and Down's syndrome in middle age form an age related continuum of pathological changes. Neuropathol Appl Neurobiol 1984; 10: 185– 207 [DOI] [PubMed] [Google Scholar]
- 7. Bennett DA, Schneider JA, Arvanitakis Z, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology 2006; 66: 1837– 1844 [DOI] [PubMed] [Google Scholar]
- 8. Green MS, Kaye JA, Ball MJ. The Oregon Brain Aging Study: neuropathology accompanying healthy aging in the oldest old. Neurology 2000; 54: 105– 113 [DOI] [PubMed] [Google Scholar]
- 9. Savva GM, Wharton SB, Ince PG, Forster G, Matthews FE, Brayne C. Age, neuropathology, and dementia. N Engl J Med 2009; 360: 2302– 2309 [DOI] [PubMed] [Google Scholar]
- 10. Andreasen N, Hesse C, Davidsson P, et al. Cerebrospinal fluid beta-amyloid(1–42) in Alzheimer disease: differences between early- and late-onset Alzheimer disease and stability during the course of disease. Arch Neurol 1999; 56: 673– 680 [DOI] [PubMed] [Google Scholar]
- 11. Visser PJ, Verhey F, Knol DL, et al. Prevalence and prognostic value of CSF markers of Alzheimer's disease pathology in patients with subjective cognitive impairment or mild cognitive impairment in the DESCRIPA study: a prospective cohort study. Lancet Neurol 2009; 8: 619– 627 [DOI] [PubMed] [Google Scholar]
- 12. Shaw LM, Vanderstichele H, Knapik-Czajka M, et al. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol 2009; 65: 403– 413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mattsson N, Zetterberg H, Hansson O, et al. CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA 2009; 302: 385– 393 [DOI] [PubMed] [Google Scholar]
- 14. Ewers M, Buerger K, Teipel SJ, et al. Multicenter assessment of CSF-phosphorylated tau for the prediction of conversion of MCI. Neurology 2007; 69: 2205– 2212 [DOI] [PubMed] [Google Scholar]
- 15. De Meyer G, Shapiro F, Vanderstichele H, et al. Diagnosis-independent Alzheimer disease biomarker signature in cognitively normal elderly people. Arch Neurol 2010; 67: 949– 956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Peskind ER, Li G, Shofer J, et al. Age and apolipoprotein E*4 allele effects on cerebrospinal fluid beta-amyloid 42 in adults with normal cognition. Arch Neurol 2006; 63: 936– 939 [DOI] [PubMed] [Google Scholar]
- 17. Glodzik-Sobanska L, Pirraglia E, Brys M, et al. The effects of normal aging and ApoE genotype on the levels of CSF biomarkers for Alzheimer's disease. Neurobiol Aging 2009; 30: 672– 681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bouwman FH, Schoonenboom NS, Verwey NA, et al. CSF biomarker levels in early and late onset Alzheimer's disease. Neurobiol Aging 2009; 30: 1895– 1901 [DOI] [PubMed] [Google Scholar]
- 19. Kester MI, Blankenstein MA, Bouwman FH, van Elk EJ, Scheltens P, van der Flier WM. CSF biomarkers in Alzheimer's disease and controls: associations with APOE genotype are modified by age. J Alzheimers Dis 2009; 16: 601– 607 [DOI] [PubMed] [Google Scholar]
- 20. Bossuyt PM, Reitsma JB, Bruns DE, et al. Towards complete and accurate reporting of studies of diagnostic accuracy: the STARD initiative: Standards for Reporting of Diagnostic Accuracy. Clin Chem 2003; 49: 1– 6 [DOI] [PubMed] [Google Scholar]
- 21. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders: DSM-III-R. Washington, DC: American Psychiatric Association; 1987 [Google Scholar]
- 22. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984; 34: 939– 944 [DOI] [PubMed] [Google Scholar]
- 23. Winblad B, Palmer K, Kivipelto M, et al. Mild cognitive impairment: beyond controversies, towards a consensus: report of the International Working Group on Mild Cognitive Impairment. J Intern Med 2004; 256: 240– 246 [DOI] [PubMed] [Google Scholar]
- 24. Roman GC, Erkinjuntti T, Wallin A, Pantoni L, Chui HC. Subcortical ischaemic vascular dementia. Lancet Neurol 2002; 1: 426– 436 [DOI] [PubMed] [Google Scholar]
- 25. Erkinjuntti T, Inzitari D, Pantoni L, et al. Research criteria for subcortical vascular dementia in clinical trials. J Neural Transm Suppl 2000; 59: 23– 30 [DOI] [PubMed] [Google Scholar]
- 26. McKeith IG, Perry EK, Perry RH. Report of the second dementia with Lewy body international workshop: diagnosis and treatment: Consortium on Dementia with Lewy Bodies. Neurology 1999; 53: 902– 905 [DOI] [PubMed] [Google Scholar]
- 27. Brun A, Englund E, Gustafson L, et al. Clinical and neuropathological criteria for frontotemporal dementia: The Lund and Manchester Groups. J Neurol Neurosurg Psychiatry 1994; 57: 416– 418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Blennow K, Wallin A, Agren H, Spenger C, Siegfried J, Vanmechelen E. Tau protein in cerebrospinal fluid: a biochemical marker for axonal degeneration in Alzheimer disease? Mol Chem Neuropathol 1995; 26: 231– 245 [DOI] [PubMed] [Google Scholar]
- 29. Vanmechelen E, Vanderstichele H, Davidsson P, et al. Quantification of tau phosphorylated at threonine 181 in human cerebrospinal fluid: a sandwich ELISA with a synthetic phosphopeptide for standardization. Neurosci Lett 2000; 285: 49– 52 [DOI] [PubMed] [Google Scholar]
- 30. Olsson A, Vanderstichele H, Andreasen N, et al. Simultaneous measurement of beta-amyloid(1–42), total tau, and phosphorylated tau (Thr181) in cerebrospinal fluid by the xMAP technology. Clin Chem 2005; 51: 336– 345 [DOI] [PubMed] [Google Scholar]
- 31. Zweig MH, Campbell G. Receiver-operating characteristic (ROC) plots: a fundamental evaluation tool in clinical medicine. Clin Chem 1993; 39: 561– 577 [PubMed] [Google Scholar]
- 32. Consensus report of the Working Group on Molecular and Biochemical Markers of Alzheimer's Disease. The Ronald and Nancy Reagan Research Institute of the Alzheimer's Association and the National Institute on Aging Working Group. Neurobiol Aging 1998; 19: 109– 116 [PubMed] [Google Scholar]
- 33. Fagan AM, Mintun MA, Mach RH, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol 2006; 59: 512– 519 [DOI] [PubMed] [Google Scholar]
- 34. Zetterberg H, Pedersen M, Lind K, et al. Intra-individual stability of CSF biomarkers for Alzheimer's disease over two years. J Alzheimers Dis 2007; 12: 255– 260 [DOI] [PubMed] [Google Scholar]
- 35. Rolstad S, Nordlund A, Eckerstrom C, Gustavsson MH, Zetterberg H, Wallin A. Biomarkers in relation to cognitive reserve in patients with mild cognitive impairment–proof of concept. Dement Geriatr Cogn Disord 2009; 27: 194– 200 [DOI] [PubMed] [Google Scholar]
- 36. Blennow K, Davidsson P, Wallin A, et al. Gangliosides in cerebrospinal fluid in ‘probable Alzheimer's disease'. Arch Neurol 1991; 48: 1032– 1035 [DOI] [PubMed] [Google Scholar]
- 37. Blennow K, Davidsson P, Wallin A, Ekman R. Chromogranin A in cerebrospinal fluid: a biochemical marker for synaptic degeneration in Alzheimer's disease? Dementia 1995; 6: 306– 311 [DOI] [PubMed] [Google Scholar]
- 38. Jellinger KA, Attems J. Prevalence of dementia disorders in the oldest-old: an autopsy study. Acta Neuropathol 2010; 119: 421– 433 [DOI] [PubMed] [Google Scholar]
- 39. Ohrfelt A, Grognet P, Andreasen N, et al. Cerebrospinal fluid alpha-synuclein in neurodegenerative disorders-A marker of synapse loss? Neurosci Lett 2009; 450: 332– 335 [DOI] [PubMed] [Google Scholar]
- 40. Sjogren M, Blomberg M, Jonsson M, et al. Neurofilament protein in cerebrospinal fluid: a marker of white matter changes. J Neurosci Res 2001; 66: 510– 516 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.