

Abstract
A series of five PIM2 analogues were synthesized and tested for their ability to activate primary macrophages and modulate LPS signaling. Structural changes included replacement of the fatty acid esters of the phosphatidyl moiety of PIM2 with the corresponding ether or amide. An AcPIM2 analogue possessing an ether linkage was also prepared. The synthetic methodology utilized an orthogonally protected chiral myo-inositol starting material that was conveniently prepared from myo-inositol in just two steps. Important steps in the synthetic protocols included the regio- and α-selective glycosylation of inositol O-6 and introduction of the phosphodiester utilizing phosphoramidite chemistry. Replacement of the inositol core with a glycerol moiety gave compounds described as phosphatidylglycerol dimannosides (PGM2). Biological testing of these PIM compounds indicated that the agonist activity was TLR4 dependent. An ether linkage increased agonist activity, removal of the inositol ring enhanced antagonist activity and the presence of an additional lipid chain enhanced LPS-induced cytokine production in primary macrophages. Furthermore, the interruption of the LPS-induced TLR4/MD-2 2:2 signaling complex formation by PIM2 represents a previously unidentified mechanism involved in the bioactivity of PIM molecules.
Introduction
The cell wall of mycobacteria is a rich source of immuno-modulatory molecules that includes lipids, glycoplipids, phosphoglycolipids, lipoproteins and mycolyl-arabinogalactan-peptiglycan motifs.1 Included among these is the family of mannosylated phosphatidylinositols, commonly known as phosphatidylinositol mannosides or PIMs,2 that are of great interest due to their biological activity and synthetic accessibility. Biological activity includes the ability to affect T-cell proliferation,3,4 recruit natural killer T (NKT) cells,5,6,7 activate innate receptors6,8 and function as immune adjuvants.9,10,11 In contrast PIMs have also been shown to suppress allergic airway disease,12,13 negatively regulate lipopolysaccharide (LPS) signaling in macrophages14 and suppress human T-cell proliferation.15
In a previous study16 we synthesized a PIM2 mono-ether analogue, PIM2ME (1), where the glyceryl sn-2 acyl group (COC15H31) had been replaced by an alkyl group (C16H33). Our motivation for this change was to overcome the inherent lability of this acyl group towards hydrolysis giving rise to lyso-PIM species. This analogue, 1, enhanced cytokine production by bone marrow-derived dendritic cells compared to the natural analogous PIM2 compound (2). Here, we report the syntheses and activity of five novel PIM analogues; 3, 4, 5, 6, and 7 where the functionality on the glyceryl moiety and the inositol ring has been varied to probe SAR for this class of compound (Figure 1). The targets were specifically designed to gain as much structural information as possible: target 3 probes amide versus ether or ester functionality at the sn-2 position; 4 is a positional isomer of 1 (sn-1 vs sn-2); analogue 5 investigates the effect of introducing a third lipid functionality; and targets 6 and 7 probe the importance of the inositol ring. The previously reported di-ether 816 was also tested to investigate the impact of two ether linkages. These compounds were then assayed and compared for their ability to induce cytokine production and suppress LPS-induced cytokine production in primary macrophages. We also investigated the interactions of PIMs with the TLR4 signaling pathway.
Figure 1.

Structure of PIM analogues
Results and Discussion
Previous syntheses of PIM compounds have utilized 1-O-allyl-3,4,5-tri-O-benzyl-D-myo-inositol as a convenient starting material.9,17,18,19 This compound is generally obtained from α-methyl D-glucopyranoside by a series of transformations that, although often produce high yields, suffers from the use of stoichiometric amount of a mercuric trifluoroacetate in a carbocyclisation reaction.20 Also, this reaction and the subsequent reduction steps both suffer from the unwanted formation of isomers that are difficult to separate resulting in a relatively un-scalable process. For these reasons, we decided to utilize a different approach starting from the known diol 921 that can be prepared from myo-inositol via the camphanilydene acetal in a scalable two-step process.22
Synthesis of 6-mannosylated-myo-inositol 15
Glycosylation of diol 9 is known to be favored at the C-6 hydroxyl on inositol. Moreover, Martin-Lomas has reported21 the selective glycosylation of 9 with an 2-azido donor in the synthesis of inositol phosphoglycans (IPG’s) and Watanabe demonstrated the synthesis of distearoyl PIM2 using the analogous racemic cylcohexylidene acetal.23,24 In the current work, glycosylation of 9 with mannosyl donor 1025 gave the desired glycoside 11 as the major product along with smaller amounts of the di-glycosylated product. Lowering the temperature of the glycosylation inhibited the formation of the over glycosylated product resulting in the clean formation of 11. Routine functional group manipulation via 12 through 14 afforded diol 1526 that was regioselectively allylated using a minor modification of the literature transformation to 16.26
Synthesis of phosphoramidites 17 and 18
The phosphoramidites 17 and 18 were prepared for the synthesis of targets 3 and 4 respectively. With this in mind, the known mono-ether 1927 was esterified and the para-methoxylbenzyl group removed by hydrogenoloysis to afford alcohol 2128 in good yield. The phosphoramidite 17 was prepared by reaction with benzyloxy-bis-(diisopropylamino)phosphine and 1H-tetrazole.
The commercially available (R)-(+)-2-amino-3-benzyloxy-1-propanol (22) was used as a starting material for the synthesis of the 2-amido containing phosphoramidite 18. Diacylation with palmitoyl chloride and DMAP in chloroform29 afforded 23 (Scheme 2). Hydrogenolysis of 23 over Pearlman’s catalyst proceeded smoothly to give the desired alcohol 24. 1H-Tetrazole activated reaction of alcohol with benzyloxy-bis-(diisopropylamino)phosphine gave the required phosphoramidite 18 in high yield over the three steps.
Scheme 2.

Synthesis of phosphoramidites 17 and 18
Synthesis of 3 and 4
Glycosylation with of 16 with 10 afforded an inseparable mixture of the anomeric glycosides. This result again demonstrates the propensity of the O-2 inositol hydroxyl to form unexpected β-anomers.30 Removal of the benzoate directing group allowed separation of the anomers and access to the desired pseudo-trisaccharide 26. Subsequent benzylation followed by deallylation provided the known inositol dimannoside head group 28.31 The spectroscopic data collected for compound 28 prepared in this way were identical to those reported previously. Inositol dimannoside 28 was then coupled with phosphoramidite 18 to afford the benzyl protected target molecule 30 in 62% yield (Scheme 3). Hydrogenolysis over Pearlman’s catalyst gave the target PIM2-mono-amide analogue that was redissolved in MeOH/H2O with the aid of triethylamine, and then passed through a short column of silica gel. Evaporation and lyophilisation of the residue gave 3, as the partial triethylammonium salt in 75% yield. Coupling of 28 with phosphoramidite 17 followed by oxidation afforded the fully protected 29 that was subsequently deprotected to afford the PIM sn-1 ether analogue 4 in good purity.
Scheme 3.

Synthesis of 3 and 4
For the synthesis of AcPIM2 mono-ether (5), the thioglycoside donor 31 was employed (Scheme 4). This compound was prepared from diol 3232 by regioselective acylation of O-6 with palmitoyl chloride followed by acylation of O-2 with 2-azidomethylbenzoyl chloride (AZMBCl).33 The AZMB protecting group was chosen to provide α-selectivity in the glycosylation, together with an orthogonal mode of deprotection.
Scheme 4.

Synthesis of donor 31
Coupling of 16 and 31 using standard thioglycosylation conditions afforded the α,α-product 34 in 34% yield (Scheme 5). The two anomeric 13C NMR signals of 34 were coincident; however, 1 JCH values for these signals (175 Hz, 175 Hz) could be extracted from a HSQC experiment run without 13C decoupling during acquisition. As observed previously (vide infra), some β-coupled product was also formed which could be removed by column chromatography.30 Deallylation followed by installation of the phosphodiester linkage using the known phosphoramidite 3616 and deprotection afforded the target compound 5 in high yield and purity.
Scheme 5.

Synthesis of 5
To probe the effect of the inositol ring in combination with the above changes to the phosphatidyl group, phosphatidylglycerol dimannoside mono-ether analogue 6 (PGM2ME), where the inositol ring is replaced by a glycerol unit, was prepared. Preparation of the dimannosylated glycerol derivative was effected by DDQ deprotection of the known PMB-ether 3912 to give the desired head group 40 in 72% yield. Reaction of the dimannosylated glycerol alcohol 40 and phosphoramidite 36 provided the protected lipid 41 (Scheme 6). Hydrogenolytic debenzylation to 41 and selective hydrolysis of the acetates gave the sn-2 ether linked PGM2 analogue 6. In a similar manner the bis-acylated congener 7 was prepared from dimannosylated glycerol 40 and phosphoramidite 43.
Scheme 6.

Synthesis of targets 6 and 7
PIM Analogues induce interleukin-6 (IL-6) production and negatively regulate LPS signaling in primary peritoneal macrophages
TLRs play a key role in the innate immune response of macrophages to exogenous pathogens via recognition of pathogen-associated molecular patterns.34,35,36 Whether any specific interaction(s) occur between PIMs and TLRs is currently not clear, but PIM molecules have been reported to act both as agonists of TLR2 and TLR4,6,8,37,38 and antagonists of LPS14 signaling in macrophages. For these reasons, we assessed the ability of the synthetic PIM analogues to both directly activate primary macrophages and modulate LPS-induced macrophage activation.
Primary peritoneal macrophages were isolated from mice and the cells treated with synthetic PIM analogues in vitro. Consistent with previous reports of agonist activity of PIM molecules8,38, all of the PIM analogues tested induced macrophage IL-6 production (Figure 2B, Table 1). In all cases, peak IL-6 production was observed between 0.6-3.1 μM PIM and decreased at higher concentrations without the appearance of cytoxicity39 (Figure 2B and supplementary information). Self regulation of TLR agonist activity has been previously reported for both TLR4 and TLR2.40 Therefore it is likely that the observed pattern of cytokine production for the PIM analogues also results from the engagement of negative feedback mechanisms designed to regulate PIM agonist activity via TLRs.
Figure 2.

Agonist and antagonist activity of PIM compounds. Thioglycollate-induced peritoneal macrophages were treated with (A) LPS (10 ng/ml) or (B) 1 alone for 18 hours. Peritoneal macrophages were treated with LPS (10 ng/ml) in the presence of (C) 1 or (D) 5 for 18 hours. Supernatants were collected and the levels of IL-6 determined by ELISA. Data is representative of at least two replicate experiments. Statistical significance was calculated was by 1-way ANOVA. ** p< 0.01, ***p<0.001 compared to PBS control.
Table 1.
PIM-induced IL-6 production by thioglycollate-elicited macrophages.
| Compound | IL-6max(pg/ml) | SE | Structural variation |
|---|---|---|---|
| 1 | 1456 | 161 | PIM2, sn-2 ether, sn-1 ester |
| 2 | 817 | 210 | PIM2, sn-2 and sn-1 both ester |
| 3 | 664 | 332 | PIM2, sn-2 amide, sn-1 ester |
| 4 | 1130 | 33 | PIM2, sn-2 ester, sn-1 ether |
| 5 | 275 | 62 | AcPIM2, sn-2 ether, sn-1 ester |
| 6 | 923 | 197 | PGM2, sn-2 ether, sn-1 ester |
| 7 | 143 | 31 | PGM2, sn-2 and sn-1 both ester |
| 8 | 494 | 161 | PIM2, sn-2 and sn-1 both ether |
Consistent with our earlier study of dendritic cell responses16, the inclusion of an ether rather than an ester group on the sn-2 position of glycerol in PIMs increased the induction of IL-6 (Table 1, 1 vs 2 or 6 vs 7). Moving the ether to the sn-1 position resulted in a decrease in IL-6 production (Table 1, 1 vs 4); however, compound 4 still enhanced IL-6 secretion compared to 2. In contrast, inclusion of two ether groups, as with compound 8, or exchanging the ether for an amide group, as with compound 3, resulted in decreased cytokine production compared to synthetic PIM2 (2). Interestingly, removal of the inositol ring, but retention of the natural glycerol di-ester (2 vs 7), resulted in a significant loss of agonist activity that was recovered by introducing the sn-2 mono-ether (6 vs 7). Agonist activity was also lost with the addition of a third lipid group, as exemplified by AcPIM2ME (5). Taken together, these results indicate the inositol ring contributes to the agonist activity and that agonist activity is enhanced with a combination of ester and ether linkages on the phosphatidyl group.
Next, we investigated whether PIM treatment enhanced or suppressed LPS induced IL-6 production by primary macrophages. Previous work on bone marrow-derived macrophages showed that PIM molecules are able to inhibit LPS-induced cytokine production.14 Consistent with this finding, the majority of the PIM analogues inhibited LPS-induced macrophage IL-6 production in a dose-dependent manner (Figure 2C and Table 2). In general, suppressive activity fell in the same concentration range as that observed for shutdown of agonist activity. Whether the same regulatory mechanisms are involved in both the inhibition of PIM agonist activity and PIM-dependent inhibition of LPS-induced IL-6 production has yet to be determined.
Table 2.
Inhibition of IL-6 production by LPS-stimulated thioglycollate-elicited macrophages.
| CN | IC50 (μM) | SE | Structural variation |
|---|---|---|---|
| 1 | 30.3 | 4.9 | PIM2, sn-2 ether, sn-1 ester |
| 2 | 40.2 | 6.1 | PIM2, sn-2 and sn-1 both ester |
| 3 | 22.7 | 1.8 | PIM2, sn-2 amide, sn-1 ester |
| 4 | 28.8 | 3.8 | PIM2, sn-2 ester, sn-1 ether |
| 6 | 42.8 | 5.1 | PGM2, sn-2 ether, sn-1 ester |
| 7 | 12.9 | 3.0 | PGM2, sn-2 and sn-1 both ester |
| 8 | 23.3 | 5.0 | PIM2, sn-2 and sn-1 both ether |
Removal of the inositol moiety in 7 (IC50 12.9 μM) resulted in increased inhibitory activity and decreased agonist activity compared to 2 (IC50 40.2 μM). Therefore, it appeared that the presence of an inositol ring abrogated inhibitory activity.
Interestingly, only 5 enhanced LPS-induced IL-6 production (Figure 2D). The difference between 1, which exhibited inhibitory activity, and 5 is the addition of C-16 (palmitic acid) fatty acyl chain to one of the mannosyl moieties. As such, the observed switch from inhibition to enhancement of LPS-induced IL-6 production may be associated with the greater lipophilicity of 5.
PIM analogue activities act via TLR4
To determine whether PIMs and their analogues may induce cytokine production via TLRs, macrophages from wild type C57 and TLR4-deficient mice were treated with 2. IL-6 production was abrogated in TLR4-deficient macrophages indicating that 2 activates macrophages via the TLR4 signaling pathway (Figure 3).
Figure 3.
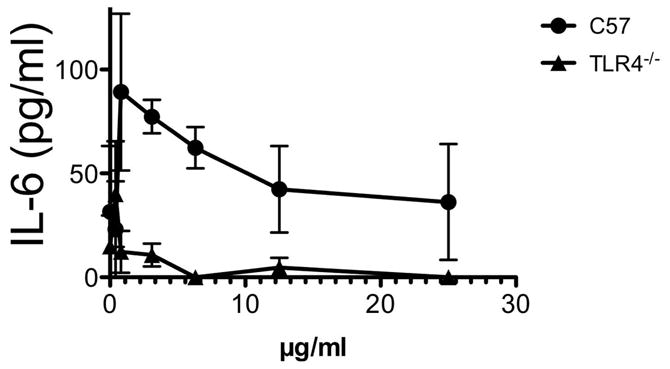
Agonist activity of 2 in thioglycollate-induced macrophages is TLR4 dependent. Data is representative of at least two replicate experiments. Data is representative of at least two replicate experiments. Statistical significance between the curves was calculated by 2-way ANOVA. p<0.0007.
A key event required for LPS signaling via the TLR4 pathway is LPS binding to a heterodimeric 1:1 complex between TLR4 and its co-receptor, myeloid differentiation factor 2 (MD-2), which then induces homodimerization of the 1:1 complex to form a 2:2 complex that leads to activation of the intracellular signaling cascade.41,42,43 To reveal the molecular basis whereby 2 negatively regulates LPS signaling, we examined the effect of 2 on LPS-mediated formation of the active 2:2 TLR4/MD-2 homodimer. As shown by native PAGE, LPS induces the homodimerization of a purified protein complex between the TLR4 ectodomain (sTLR4) and MD-2 (Figure 4A). This LPS-mediated sTLR4/MD-2 homodimerization was partially inhibited by 2 (Figure 4A).
Figure 4.

Compound 2 interferes with LPS binding to TLR4/MD-2 (A) and MD-1 (B) as shown by native PAGE. 2 binding to MD-1 results in a band shift (B, left), whereas 2-induced sTLR4/MD-2 band shift was not observed (A, left) potentially due to a large size of sTLR4/MD-2 (87 kDa), compared to MD-1 (17 kDa).
Given that, for TLR4 activation, LPS is deeply inserted into the hydrophobic cavity of MD-2, we speculated that 2 would inhibit 2:2 TLR4/MD-2 complex formation by competing with LPS for the MD-2 cavity. However, the direct interaction of MD-2 with LPS or 2 could not be addressed due to technical difficulty in recombinant expression of MD-2 alone. Instead, an MD-2 homolog, MD-1, was used as an MD-2 model system. MD-1 has been implicated in the regulation of LPS-induced immune responses and, like MD-2, houses a large hydrophobic cavity that is able to accommodate LPS and presumably other lipogylcan structures such as PIM.44,45 In native gels, 2 was shown to associate with MD-1 in a dose-dependent manner and to compete with LPS for MD-1 binding (Figure 4B), supporting that 2 directly interacts with MD-2. Taken together, these data suggest that PIM molecules, like MPL,43 act as partial agonists of TLR4 and likely inhibit LPS signaling by interfering with LPS binding to the TLR4/MD-2 complex.
In summary, we present concise syntheses of PIM analogues 3, 4 and 5 utilizing the readily available mannosylated chiral inositol 16, and 6 and 7 from a dimannosylated glycerol intermediate. In general, the PIMs and their analogues exhibit partial agonist activity, which is associated with the inositol ring. Furthermore, this activity is enhanced by replacement of a fatty acid ester moiety with that of the corresponding ether on the phosphatidyl group. SAR for the inhibition of LPS signaling resulted in two notable observations. Firstly, loss of the inositol ring enhanced the antagonist activity, and somewhat surprisingly, addition of a third lipid moiety may synergise with LPS to enhance pro-inflammatory activation of macrophages. For the first time, we also show that the activities of PIMs involve regulation of TLR4 signaling, potentially via interactions with TLR4/MD-2. Together these findings identify key structural features that could be used to direct the synthesis of lipoglycans with optimal agonist, antagonist or adjuvant activities and identify an important role for TLR4 in the bioactivity of PIMs.
Experimental Section
General Experimental
NMR spectra are referenced to tetramethylsilane (TMS) (0.0 ppm) or the residual solvent peak (1H CHCl3 δ7.26); 13C CDCl3 δ77.0) or an external reference (31P H3PO4 δ0.0). Anhydrous solvents were sourced commercially and used without further treatment unless stated. Powdered molecular sieves were flame dried under vacuum immediately prior to use. Flash column chromatography was carried out using 40–63 μm silica gel unless otherwise stated. All flash chromatography solvents were AR-grade. Petroleum ether used was bp 60–80° range. All compounds were isolated after silica-gel column chromatography and fractions collected were one spot by TLC. Thin layer chromatography (TLC) plates were visualised under an UV lamp and/or with a spray consisting of 5% w/v dodecamolybdophosphoric acid in ethanol with subsequent heating. See supplementary information for HPLC conditions. HPLC purity of target compounds, 1 (97%), 2 (95%), 3 (94%), 4 (96%), 5 (95%), 6 (96%), 7 (95%) and 8 (97%).
6-(2-O-Benzoyl-3,4,5-tri-O-benzyl-α-D-mannopyranosyl)-3,4-O-(1,1,3,3- tetraisopropyldisiloxanedi-1,3-yl)-1,2-O-((1S,4S)-1,7,7-trimethyl-[2.2.1]bicyclohept-6-ylidene)-D-myo-inositol (11)
TMSOTf (0.20 mL, 1.1 mmol) was added to a mixture of diol 9 (6.00 g, 10.8 mmol), trichloroacetamide 10 (7.53 g, 10.77 mmol) and 4Å molecular sieves in dry Et2O (100 mL) at −70 °C. After stirring for 45 min, the reaction was quenched with saturated NaHCO3 (100 mL) and extracted with Et2O (3×60 mL) and washed with water (100 ml), dried (MgSO4), filtered and the solvent removed. The crude residue was purified on silica gel (EtOAc/petroleum ether = 1:9) to afford compound 11 (8.95 g, 76%) as a white foam. [α]20 D −1.5 (c = 0.4, CH2Cl2); [α]20 365 −26 (c 0.4, CH2Cl2); 1H NMR (300 MHz, CDCl3) δ8.11–8.05 (m, 2H), 7.57–7.50 (m, 1H), 7.41–7.15 (m, 17H), 5.71 (dd, J = 1.8, 2.7 Hz, 1H), 5.40 (d, J = 1.8 Hz, 1H), 4.87–4.74 (m, 3H), 4.60–4.49 (m, 3H), 4.37–4.30 (m, 1H), 4.20 (dd, J = 3.9, 5.4 Hz, 1H), 4.16–4.05 (m, 2H), 4.01–3.85 (m, 4H), 3.84–3.70 (2H, m), 3.36 (ddd, J = 1.8, 8.7, 10.1 Hz, 1H), 2.57 (d, J = 1.8 Hz, 1H), 2.00–1.80 (m, 2H), 1.74–1.57 (m, 2H), 1.46 (d, J = 12.9 Hz, 1H), 1.40–1.13 (m, 3H), 1.12–0.94 (m, 30H), 0.82 (s, 3H), 0.86 (s, 3H); 13C NMR (75 MHz, CDCl3) δ165.7, 138.9, 138.7, 138.3, 132.9, 130.0, 128.3, 128.24, 128.19, 128.1, 128.0, 127.9, 127.5, 127.4, 127.3, 117.7, 96.8, 79.9, 78.6, 77.4, 77.0, 76.9, 76.6, 76.3, 75.7, 75.1, 74.5, 73.3, 72.9, 72.6, 71.6, 71.5, 69.4, 69.2, 51.5, 48.0, 45.2, 45.1, 29.4, 27.1, 20.4, 20.2, 17.6, 17.4, 17.31, 17.26, 17.2, 17.1, 17.0, 13.0, 12.7, 12.5, 12.3, 12.1, 11.7, 9.7. HRMS-ESI [M+Na]+ calculated for C62H84O13Si2Na: 1115.5348. Found 1115.5365.
6-(3,4,5-Tri-O-benzyl-α-D-mannopyranosyl)-3,4-O-(1,1,3,3-tetraisopropyldisiloxanedi-1,3-yl)-1,2-O-((1S,4S)-1,7,7-trimethyl-[2.2.1]bicyclohept-6-ylidene)-D-myo-inositol (12)
Sodium methoxide in MeOH (30% solution, 0.58 mL) was added dropwise to a stirred solution of benzoate 11 (3.91 g, 3.58 mmol) in CH2Cl2/MeOH (1:1, 50 mL). After 22 h, the mixture was partitioned between Et2O (100 mL) and saturated NH4Cl (100 mL), dried (MgSO4), filtered and the solvent removed. The crude residue was purified on silica gel (EtOAc/petroleum ether = 1:9) to afford the title compound 12 (2.75 g, 78%) as a pale yellow oil. [α]20 D +33 (c 0.5, CH2Cl2); 1H NMR (300 MHz, CDCl3) δ7.38–7.15 (m, 15H), 5.38 (d, J = 1.2 Hz, 1H), 4.82 (d, J = 11.0 Hz, 1H), 4.75–4.61 (m, 3H), 4.59–4.45 (m, 2H), 4.31–4.21 (m, 1H), 4.21–4.15 (m, 1H), 4.15–4.08 (m, 1H), 3.97–3.83 (m, 5H), 3.83–3.71 (m, 2H), 3.71– 3.61 (m, 1H), 3.31 (ddd, J = 1.5, 8.3, 9.8 Hz, 1H), 2.61–2.56 (m, 1H), 2.50–2.42 (m, 1H), 2.02–1.87 (m, 2H), 1.78–1.64 (m, 2H), 1.49–1.34 (m, 2H), 1.28–1.14 (m, 1H), 1.13–0.82 (m, 37H); 13C NMR (75 MHz, CDCl3) δ138.7, 138.6, 138.2, 128.4, 128.2, 128.0, 127.9, 127.82, 127.76, 127.4, 127.3, 117.7, 98.2, 80.3, 79.7, 77.4, 77.2, 77.0, 76.9, 76.6, 76.4, 75.8, 74.9, 74.5, 73.2, 73.0, 72.4, 72.0, 70.9, 68.9, 68.8, 51.5, 48.0, 45.4, 45.1, 29.6, 27.1, 20.5, 20.2, 17.6, 17.4, 17.34, 17.28, 17.25, 17.22, 17.1, 17.0, 13.0, 12.7, 12.3, 12.1, 9.7. HRMS-ESI [M+NH4]+ calculated for C55H80O12Si2NH4: 1006.5532. Found 1006.5491.
3,4,5-Tri-O-benzyl-6-(2,3,4,5-tetra-O-benzyl-α-D-mannopyranosyl)-1,2-O-((1S,4S)-1,7,7- trimethyl-[2.2.1]bicyclohept-6-ylidene)-D-myo-inositol (14)
TBAF (1 M, 7.8 mL) was added to a solution of 12 (3.10 g, 3.13 mmol) in THF (50 mL). After stirring for 1 h, the solvent was removed the residue was purified by flash chromatography on silica gel (MeOH/CH2Cl2 = 3:97 to 1:24) to afford partially purified 13 (2.8 g); 1H NMR (300 MHz, CDCl3) inter alia δ7.38–7.16 (m, 15H), 5.18 (d, J = 1.5 Hz, 1H), 4.81 (d, J = 11.0 Hz, 1H), 4.72 (d, J = 11.4 Hz, 1H), 4.67 (d, J = 11.4 Hz, 1H), 4.61–4.45 (m, 3H), 4.28 (dd, J = 4.4, 5.4 Hz, 1H), 3.94–3.84 (m, 2H), 3.82–3.50 (m, 7H), 3.21 (dd, J = 3.0, 9.4 Hz, 1H), 3.06 (br s, 1H), 2.89 (br s, 1H), 2.67 (d, J = 2.3 Hz, 1H), 2.55 (d, J = 7.0 Hz, 1H), 2.06–1.85 (m, 2H), 1.80–1.64 (m, 1H), 1.53–1.33 (m, 2H), 1.28–1.14 (m, 1H), 0.98 (s, 3H), 0.86 (s, 3H), 0.82 (s, 3H). HRMS-ESI [M+NH4]+ calculated for C43H54O11NH4: 764.4010. Found 764.4013. BnBr (3.7 mL, 31 mmol) was added dropwise to a stirred solution of the crude tetraol 13 and NaH (0.94 g, 60% in mineral oil, 23.5 mmol) in dry DMF (75 mL) at 0 °C. The reaction was left to slowly warm to rt over 80 min before being diluted with Et2O (300 mL) and quenched by the slow addition of water (150 mL). The aqueous phase was re-extracted with Et2O (2×100 mL), dried (MgSO4), filtered and the solvent removed. The crude residue was purified on silica gel (EtOAc/petroleum ether = 3:7) to afford the title compound 14 (3.12 g, 90%). [α]20 D +19 (c 1.7, CH2Cl2); 1H NMR (300 MHz, CDCl3) δ7.45–7.00 (m, 35H), 5.53 (d, J = 1.4 Hz, 1H), 4.89 (d, J = 11.0 Hz, 1H), 4.82–4.50 (m, 12H), 4.35 (d, J = 12.1 Hz, 1H), 4.25 (dd, J = 3.4, 6.4 Hz, 1H), 4.09 (dd, J = 8.8, 18.4 Hz, 1H), 4.03–3.95 (m, 2H), 3.90 (dd, J = 3.1, 9.4 Hz, 1H), 3.86–3.71 (m, 4H), 3.63 (dd, J = 3.7, 11.3 Hz, 1H), 3.50 (dd, J = 1.3, 11.3 Hz, 1H), 3.27 (dd, J = 7.1, 9.7 Hz, 1H), 2.00–1.87 (m, 2H), 1.80–1.65 (m, 2H), 1.49–1.33 (m, 2H), 1.29–1.12 (m, 1H), 1.06 (s, 3H), 0.88 (s, 3H), 0.86 (s, 3H); 13C NMR (75 MHz, CDCl3) δ139.1, 138.8, 138.6, 138.5, 138.1, 128.4, 128.3, 128.23, 128.16, 128.1, 127.9, 127.81, 127.77, 127.6, 127.5, 127.4, 127.2, 117.9, 96.1, 81.1, 80.7, 79.6, 77.9, 76.8, 76.4, 75.0, 74.91, 74.85, 74.7, 74.5, 73.8, 73.1, 72.5, 72.1, 71.8, 68.9, 51.6, 47.9, 45.1, 44.9, 29.9, 27.2, 20.6, 20.4, 9.8. HRMS-ESI [M+NH4]+ calculated for C71H78O11NH4: 1124.5888. Found 1124.5896.
3,4,5-Tri-O-benzyl-6-(2,3,4,5-tetra-O-benzyl-α-D-mannopyranosyl)-D-myo-inositol (15)
HCl (37%, 2 mL) was added to a stirred solution of 9 (322 mg, 0.291 mmol) in dioxane/methanol (2:5, 35 mL) at rt. After 48 h, the reaction was diluted with Et2O (100 mL), washed with water (100 mL), the aqueous layer was re-extracted with Et2O, (50 mL) and the combined organic fractions were washed with saturated NaHCO3 (100 mL), brine (100 mL), dried (MgSO4), filtered and the solvent removed. The crude residue was purified on silica gel (EtOAc/petroleum ether = 1:3 to 2:3) to afford the title compound 15 (245 mg, 86%). Analytical data were consistent with those previously reported.26
2-O-Hexadecanoyl-1-O-hexadecyl-3-O-(4-methoxybenzyl)-sn-glycerol (20)
Palmitoyl chloride (0.300 mL, 0.982 mmol) was added dropwise to a stirred solution of alcohol 19 (270 mg, 0.618 mmol) and dry pyridine (0.300 mL, 3.71 mmol) in dry CH2Cl2 (15 mL) at 0 ºC. The reaction mixture was allowed to warm to rt over 17 h before being quenched with water (100 mL). The mixture was extracted with Et2O (2×150 mL) and the ethereal extract washed with a 0.5 M HCl (100 mL), saturated NaHCO3 (100 mL), dried (MgSO4), filtered and the solvent removed. The crude residue was purified by column chromatography on silica gel (EtOAc/petroleum ether = 0:1 to 1:9) to afford the title compound 20 (384 mg, 92%) as a colourless oil. [α]20 D = +1.0 (c 7.7, CHCl3). 1H NMR (300 MHz, CDCl3) δ 7.27–7.21 (m, 2H), 6.90–6.83 (m, 2H), 5.16 (quintet, J = 5.1 Hz, 1H), 4.49–4.43 (m, 2H), 3.79 (s, 3H), 3.60–3.55 (m, 4H), 3.44–3.35 (m, 2H), 2.32 (t, J = 7.5 Hz, 2H), 1.64–1.49 (m, 4H), 1.40–1.20 (m, 50H), 0.92–0.83 (m, 6H); 13C NMR (75 MHz, CDCl3) δ 173.4, 159.3, 130.2, 129.3, 113.8, 73.0, 71.7, 71.4, 69.3, 68.6, 55.3, 34.5, 32.0, 29.8, 29.6, 29.4, 29.2, 26.1, 25.1, 22.8, 14.2. HRMS-ESI [M+Na]+ calculated for C43H78O5Na: 697.5747. Found 697.5734.
2-O-Hexadecanoyl-1-O-hexadecyl-sn-glycerol (21)
Pd(OH)2/C (20%, 88 mg) was added to a mixture of 20 (380 mg, 0.563 mmol) in AcOH/EtOH (1:10, 16.5 mL). The mixture was stirred under an H2 atmosphere for 4 h at rt, then filtered through Celite and the solvent removed. The residue was purified by column chromatography on silica gel (EtOAc/CH2Cl2 = 1:49 to 1:19) to afford the title compound 21 (207 mg, 66%) as an oil. [α]20D = − 2.8 (c 0.72, CHCl3); lit46 = −2.6 (c 2.1, CHCl3); lit28 = −1.2. Analytical data were consistent with those previously reported.28,46
Benzyl (2-O-hexadecanoyl-1-O-hexadecyl-sn-glycero)-diisopropylphosphoramidite (17)
1H-Tetrazole (35 mg, 0.50 mmol) was added to a stirred solution of alcohol 21 (205 mg, 0.369 mmol) and benzyloxy-bis-(diisopropylamino)phosphine (262 mg, 0.775 mmol) in dry CH2Cl2 (20 mL) at rt for 90 min. The solvent was removed and the residue purified by column chromatography on silica gel (Et3N/EtOAc/petroleum ether = 3:10:90) to afford the title compound 17 (267 mg, 91%) as an oil. [α]20 D = + 4.9 (c 0.72, CHCl3). 1H NMR (300MHz, CDCl3) δ 7.37–7.23 (m, 5H), 5.13 (quintet, J = 5.2 Hz, 1H), 4.74–4.62 (m, 2H), 3.83–3.35 (m, 8H), 2.30 (t, J = 7.3 Hz, 2H), 1.65–1.48 (m, 4H), 1.32–1.16 (m, 62H), 0.90–0.83 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 173.3, 139.6, 128.3, 127.3, 127.0, 72.2, 71.7, 69.2, 65.5, 65.3, 62.3, 62.1, 61.9, 43.2, 43.1, 34.6, 32.0, 29.8, 29.7, 29.6, 29.4, 29.2, 26.2, 25.1, 24.8, 24.7, 24.6, 22.8, 14.2. 31P NMR (121.5 MHz, CDCl3) δ 149.5, 149.2. HRMS-ESI [M+H]+ calculated for C48H91NO5P: 729.6635. Found 792.6638.
3-O-Benzyl-2-deoxy-1-O-hexadeconyl-2-hexadeconylamino-sn-glycerol (23)
Palmitoyl chloride (1.54 mL, 5.08 mmol) was added dropwise to a stirred solution of (R)-(+)-2-amino-3-benzyloxy-1-propanol (22) (230 mg, 1.27 mmol) and DMAP (621 mg, 5.08 mmol) in dry CHCl3 (25 mL) at 0 °C. After warming to rt over 6 h, the reaction was diluted with CHCl3 (30 mL) and washed with H2O (30 mL) then 0.5 M HCl (30 mL), dried (MgSO4), filtered and the solvent removed. The crude residue was purified by column chromatography (EtOAc/petroleum ether = 1:4) to afford the title compound 23 (705 mg, 84%) as a gummy solid. 1H NMR (300 MHz, CDCl3) δ 7.38–7.26 (m, 5H), 4.79 (d, J = 8,6 Hz, 1H), 4.51 (br s, 2H), 4.42–4.31 (m, 1H), 4.25 (dd, J = 6.1, 10.9 Hz, 1H), 4.13 (dd, J = 6.0, 10.9 Hz, 1H), 3.59 (dd, J = 3.4, 9.6 Hz, 1H), 3.48 (dd, J = 4.7, 9.6 Hz, 1H), 2.26 (dd, J = 7.4, 7.6 Hz, 2H), 2.14 (dd, J = 7.4, 7.8 Hz, 2H), 1.65–1.53 (m, 4H), 1.30–1.24 (m, 48H), 0.91–0.85 (m, 6H). 13C NMR (75 MHz, CDCl3) δ173.8, 172.9, 137.8, 128.5, 128.0, 127.8, 73.4, 68.8, 63.1, 48.0, 36.9, 34.3, 32.0, 29.9, 29.2, 25.8, 25.0, 22.8, 14.2. HRMS-ESI [M+Na]+ calculated for C42H75NO4Na: 680.5594. Found 680.5588.
2-Deoxy-1-O-hexadeconyl-2-N-hexadeconylamino-sn-glycerol (24)29
Pd(OH)2/C (20%, 200 mg) was added to a mixture of 23 (663 mg, 1.01 mmol) in AcOH/EtOH (1:10, 33 mL). The mixture was stirred under an H2 atmosphere for 16 h at rt, then filtered through Celite and the solvent removed. The residue was purified by column chromatography on silica gel (EtOAc/CH2Cl2 = 3:7) to afford the title compound 24 (555 mg, 0.98 mmol, 97%) as a white solid. 1H NMR (300 MHz, CDCl3) δ 6.02 (d, J = 6.6 Hz, 1H), 4.27–4.12 (m, 3H), 3.72–3.56 (m, 2H), 2.96 (br s, 1H), 2.33 (dd, J = 7.6, 7.6 Hz, 2H), 2.19 (dd, J = 7.6, 7.6 Hz, 2H), 1.67–1.55 (m, 4H), 1.35–1.19 (m, 48H), 0.91–0.84 (m, 6H); 13C NMR (75 MHz, CDCl3) δ 177.6, 173.8, 62.5, 62.0, 50.5, 36.8, 34.3, 32.0, 30.0, 29.2, 25.8, 25.0, 22.8. HRMS-ESI [M+Na]+ calculated for C35H69NO4Na: 590.5124. Found 590.5118
2-Deoxy-1-O-hexadeconyl-2-N-hexadeconylamino-sn-glycero-3-O-benzyl-(N,Ndiisopropyl)- phosphoramidite (18)
1H-Tetrazole (28 mg, 0.397 mmol) was added to a stirred solution of alcohol 24 (205 mg, 0.361 mmol) and benzyloxy-bis-(diisopropylamino)phosphine (244 mg, 0.722 mmol) in dry CH2Cl2 (10 mL) at rt for 60 min. The solvent was removed and the residue purified by column chromatography on silica gel (Et3N/EtOAc/petroleum ether = 3:10:90) to afford the title compound 18 (280 mg, 96%) as a clear oil that solidified on standing. 1H NMR (300 MHz, CDCl3) δ 7.37–7.25 (m, 5H), 4.89–4.58 (m, 2H), 4.38–4.25 (m, 1H), 4.22–4.04 (m, 2H), 3.87–3.56 (m, 2H), 2.28 (dd, J = 7.6, 7.6 Hz, 2H), 2.12–2.04 (m, 1H), 1.94–1.87 (m, 1H), 1.64–1.45 (m, 4H), 1.33–1.17 (m, 60H), 0.91–0.85 (m, 6H); 31P NMR (121.5 MHz, CDCl3) δ 150.3, 149.9. HRMS-ESI [M+Na] calculated for C48H89N2O5Na: 827.6407. Found 827.6407.
1-O-Allyl-3,4,5-tri-O-benzyl-6-(2,3,4,5-tetra-O-benzyl-α-D-mannopyranosyl)-2-O-(3,4,5-tri-Obenzyl- α-D-mannopyranosyl)-D-myo-inositol (26)
TMSOTf (12 μL, 0.066 mmol) was added to a mixture of alcohol 1626 (0.339 g, 0.39 mmol) and trichloroacetamide 10 (0.46 g, 0.66 mmol) and 4Å molecular sieves in dry Et2O (50 mL) at −40 °C. After stirring for 1 h, the reaction was quenched with Et3N, filtered through Celite and evaporated to dryness. The crude residue was purified on silica gel (EtOAc/petroleum ether = 1:9 to 1:4) afford the intermediate compound 25 (0.473 g) as an inseparable mixture of anomers. 1H NMR (300 MHz, CDCl3) inter alia δ5.92 (d, J = 2.9 Hz, 0.2H), 5.79–5.60 (m, 2H), 5.58–5.53 (m, 0.8H); 13C NMR (75 MHz, CDCl3) inter alia δ99.2 (1 JCH = 175 Hz), 98.9 (1 JCH = 173 Hz). HRMS-ESI [M+Na]+ calculated for C98H100O17Na: 1571.6858. Found 1571.6818. Sodium methoxide in MeOH (30% solution, 0.1 mL) was added dropwise to a stirred solution of 25 (323 mg) in CH2Cl2/MeOH (1:1, 20 mL). After 24 h, the mixture was quenched by the careful addition of saturated NH4Cl (50 mL) and extracted with Et2O (2×50 mL), dried (MgSO4), filtered and the solvent removed. The crude residue was purified on silica gel (EtOAc/petroleum ether = 1:9 to 3:7) to afford the title compound 26 (208 mg, 63%) as a colourless oil. 1H NMR (300 MHz, CDCl3) δ5.77 (ddd, J = 5.5, 5.5, 10.4, 17.2 Hz, 1H), 4.50 (d, J = 1.4 Hz, 1H), 5.22 (dd, J = 1.4, 17.2 Hz, 1H), 5.21 (d, J = 1.1 Hz, 1H), 5.14 (dd, J = 1.2, 10.4 Hz, 1H), 4.95–4.50 (m, 16H), 4.49–4.41 (m, 2H), 4.38–4.28 (m, 2H), 4.24–3.75 (m, 13H), 3.52–3.20 (m, 6H), 3.16 (dd, J = 1.6, 9.7 Hz, 1H), 2.36 (d,, J = 1.9 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ139.1, 138.8, 138.71, 138.68, 138.6, 138.5, 138.2, 138.1, 138.0, 137.9, 133.9, 128.6, 128.3, 128.23, 128.17, 128.13, 128.05, 128.02, 127.96, 127.91, 127.87, 127.8, 127.6, 127.5, 127.4, 127.32, 127.27, 127.2, 117.7, 100.1, 98.6, 81.6, 81.5, 81.4, 80.1, 79.5, 78.9, 77.4, 77.2, 77.0, 76.6, 76.1, 75.9, 75.6, 75.3, 75.0, 74.9, 74.8, 74.1, 73.4, 73.1, 72.3, 72.2, 72.1, 71.8, 71.0, 70.84, 70.77, 68.7, 68.2. HRMS-ESI [M+Na]+ calculated for C91H96O16Na: 1467.6596. Found 1467.6563.
1-O-Allyl-3,4,5-tri-O-benzyl-2,6-di-O-(2,3,4,6-tetra-O-benzyl-α-D-mannopyranosyl)-Dmyo-inositol (27)
NaH (0.054 g, 60% dispersion in mineral oil, 1.4 mmol) was added to a stirred solution of 26 (0.98 g, 0.68 mmol) and benzyl bromide (0.20 mL, 1.7 mmol) in dry DMF (30 mL) at 0 °C. The mixture was allowed to warm to rt over 18 h before being diluted with Et2O (50 mL) and quenched by the slow addition of water (50 mL). The aqueous phase was re-extracted with Et2O (2×25 mL), dried (MgSO4) filtered and the solvent removed The crude residue was purified on silica gel (EtOAc/petroleum ether = 1:9 to 3:7) to afford the title compound 27 (1.01 g, 97%). Analytical data were consistent with those previously reported.17,31
3,4,5-Tri-O-benzyl-2,6-di-O-(2,3,4,6-tetra-O-benzyl-α-D-mannopyranosyl)-1-O-(2-Ohexadecanoyl- 1-O-hexadecyl-sn-glycero-3-benzylphosphoryl)-D-myo-inositol (29)
1H-Tetrazole (21 mg, 0.30 mmol) was added to a stirred solution of alcohol 28 (90 mg, 0.060 mmol) and phosphoramidite 17 (135 mg, 0.170 mmol) in dry CH2Cl2 (7 mL) at 0 ºC under argon. After 2 h the reaction mixture was cooled to −40 ºC and a dried (MgSO4) solution of m-CPBA (~55%, 90 mg, 0.29 mmol) in CH2Cl2 (10 mL) was added to the reaction. After warming to rt over 1 h, the reaction was quenched by addition of Na2SO3 (10%, 50 mL) and extracted with Et2O (50 mL). The organic phase was washed with saturated NaHCO3 (50 mL), dried (MgSO4), filtered and the solvent removed. The crude residue was purified by column chromatography on silica gel (EtOAc/petroleum ether = 1:9 to 3:7) followed by further purification on silica gel (EtOAc/CH2Cl2 = 1:49 to 1:19) to afford the title compound 29 (68 mg, 52%) as an oil. 1H NMR (300 MHz, CDCl3) mixture of isomers δ 7.38–7.00 (m, 60H), 5.53–5.51 (m, 1H), 5.33–5.32 (m, 1H), 5.05–4.40 (m, 23H), 4.30–3.78 (m, 17H), 3.56–3.20 (m, 9H), 2.23–2.10 (m, 2H), 1.58–1.41 (m, 4H), 1.31–1.15 (m, 50H), 0.89–0.82 (m, 6H). 13C (75 MHz, CDCl3) selected signals δ 173.3, 99.9, 98.9. 31P NMR (121.5 MHz, CDCl3) δ 0.26, 0.00. HRMS-ESI [M+Na]+ calculated for C137H173O22PNa: 2224.2054. Found 2224.2051.
2,6-(Di-O-α-D-mannopyranosyl)-1-O-(2-O-hexadecanoyl-1-O-hexadecyl-sn-glycero-3-phosphoryl)-D-myo-inositol (4)
Pd(OH)2/C (20%, 25 mg) was added to a stirred solution of 29 (68 mg, 0.031 mmol) in THF/MeOH (2:3, 5 mL). The mixture was stirred under an H2 atmosphere for 4 h at rt, then filtered through Celite and the solvent removed. The residue was purified by column chromatography on silica gel (CHCl3/MeOH/H2O = 70:40:0 to 70:60:6) to afford the title compound 4 (26 mg, 74%) as a white powder. [α]D 20 = +39 (c 0.40, CHCl3/MeOH/H2O = 70:60:6). 1H NMR (500 MHz, CDCl3/CD3OD/D2O 70:40:6) δ 5.28–5.20 (m, 1H), 5.15 (br s, 1H), 5.12 (br s, 1H), 4.33 (m, 1H), 4.12–3.96 (m, 7H), 3.87–3.60 (m, 12H), 3.57–3.42m, 3H), 3.30 (t, J = 7.0 Hz, 1H), 2.41 (t, J = 7.2 Hz, 2H), 1.65–1.55 (m, 4H), 1.33–1.22 (m, 50H), 0.92–0.85 (m, 6H); 13C (125 MHz, CDCl3/CD3OD/D2O 70:40:6) δ 176.0, 103.0, 102.9, 79.9, 79.8, 78.2, 78.1, 74.8, 74.4, 74.4, 74.2, 73.5, 73.5, 73.0, 72.1, 72.1, 71.8, 71.6, 70.7, 68.4, 68.4, 65.4, 65.4, 62.8, 62.6, 35.7, 33.2, 31.0, 30.9, 30.7, 30.6, 30.4, 27.4, 26.4, 23.9, 15.0, 10.0; 31P NMR (121.5 MHz, CDCl3/CD3OD/D2O 70:40:6) δ 0.63. HRMS-ESI [M–H]− calculated for C53H100O22P: 1119.6444. Found 1119.6456.
3,4,5-tri-O-benzyl-2,6-di-O-(2,3,4,6-tetra-O-benzyl-α-D-mannopyranosyl)-1-O-(2-deoxy-3-benzylphosphoryl-1-O-hexadeconyl-2-O-hexadeconylamino-sn-glycero)-D-myo-inositol (30)
1H-Tetrazole (10 mg, 0.135 mmol) was added to a stirred solution of alcohol 28 (68 mg, 0.045 mmol) and phosphoramidite 18 (109 mg, 0.135 mmol) in dry CH2Cl2 (10 mL) at 0 ºC under argon. After 1 h, the reaction mixture was cooled to −40 ºC and a dried (MgSO4) solution of m-CPBA (~50%, 47 mg, 0.135 mmol) in CH2Cl2 (5 mL) was added to the reaction. After warming to rt over 2 h the reaction was quenched by addition of Na2SO3 (10%, 50 mL) and extracted with Et2O (50 mL). The organic phase was washed with saturated NaHCO3 (50 mL), dried (MgSO4), filtered and the solvent removed. The crude residue was purified by column chromatography on silica gel (EtOAc/petroleum ether = 1:9 to 1:4) to afford the title compound 30 (61 mg, 62%) as an oil. 1H NMR (300 MHz, CDCl3) mixture of isomers δ 7.48–7.03 (m, 60H), 6.54 (d, J = 7.5 Hz, 3H), 6.40 (d, J = 8.6 Hz, 7H), 5.48 (br s, 3H), 5.44 (br s, 7H), 5.10–5.42 (m, 21H), 4.51–4.27 (m, 5H), 4.26–3.66 (m, 15H), 3.52–3.12 (m, 6H), 2.37–2.27 (m, 1H), 2.17–2.09 (m, 1H), 2.03–1.95 (m, 2H), 1.71–1.37 (m, 4H), 1.35–1.12 (m, 48H), 0.91–0.84 (m, 6H); 13C NMR (125 MHz, CDCl3) selected signals δ 173.8, 173.5, 173.2, 173.1, 98.8, 98.7, 98.4, 98.1; 31P NMR (121.5 MHz, CDCl3) δ 0.94, −0.45. HRMS-ESI [M+Na]+ calculated for C137H172NO5PNa: 2237.2006. Found 2237.2007.
2,6-(Di-O-α-D-mannopyranosyl)-1-O-(2-deoxy-1-O-hexadecanoyl-2-N-hexadecanoylaminosn-glycero-3-phosphoryl)-D-myo-inositol (3)
Pd(OH)2/C (20%, 40 mg) was added to a stirred solution of 28 (28 mg, 24.8 mmol) in THF/MeOH (2:3, 5 mL). The mixture was stirred under an H2 atmosphere for 16 h at rt, then filtered through Celite and the solvent removed. The residue was purified by column chromatography on silica gel (CHCl3/MeOH/H2O = 70:40:4) to afford the title compound 3 (21 mg, 75%) as a white powder. [α]D 20 = +35 (c 0.15, CHCl3/MeOH/H2O = 40:40:10). 1H NMR (500 MHz, CDCl3/CD3OD/D2O 40:40:10) δ 5.15 (d, J = 1.6 Hz, 1H), 5.10 (d, J = 1.5 Hz, 1H), 4.32–4.28 (m, 2H), 4.15 (dd, J = 8.0, 11.4 Hz, 1H), 4.08–4.03 (m, 3H), 4.01–3.93 (m, 4H), 3.85–3.78 (m, 5H), 3.77–3.59 (m, 5H), 3.48 (dd, J = 7.6, 10.1 Hz, 1H), 3.30 (dd, J = 9.2, 9.3 Hz, 1H), 3.16 (q, J = 7.3 Hz, 6H), 2.33 (dd, J = 8.3, 6.9 Hz, 2H), 2.24 (dd, J = 7.5, 7.5 Hz, 2H), 1.65–1.55 (m, 4H), 1.31 (t, J = 7.3 Hz, 9H), 1.29–1.26 (m, 48H), 0.91–0.87 (m, 6H); 13C NMR (125 MHz, CDCl3/CD3OD/D2O 40:40:10) δ176.3, 175.5, 102.4, 102.2, 79.3, 77.55, 77.50, 74.1, 73.8, 73.7, 73.6, 71.5, 71.4, 71.11, 71.07, 70.9, 67.8, 67.7, 65.2, 65.1, 64.2, 62.2, 62.0, 47.4, 37.0, 34.8, 32.6, 30.5, 30.42, 30.37, 30.35, 30.31, 30.30, 30.26, 30.1, 30.01, 30.00, 29.9, 26.7, 25.5, 23.3, 14.5. 31P NMR (121.5 MHz, CDCl3) δ 4.63. HRMS-ESI [M–H]− calculated for C53H99NO22P: 1132.6396. Found 1132.6382.
Phenyl 3,4-di-O-benzyl-6-O-hexadecanoyl-1-thio-α-D-mannopyranoside (33)
Hexadecanoyl chloride (2.6 mL, 8.5 mmol) was added dropwise to a stirred solution of diol 3232 (3.50 g, 7.73 mmol) and pyridine (6.25 mL, 77 mmol) in dry CH2Cl2 (30 mL) at 0 °C and the reaction was allowed to warm to rt overnight. The reaction mixture was diluted with CH2Cl2 (70 mL), washed with 1 M aq HCl (100 mL), saturated aq NaHCO3, dried (MgSO4), filtered and the solvent removed. The residue was purified by column chromatography on silica gel (EtOAc/petroleum ether = 1:9 to 3:7) to afford the title compound 33 (4.82 g, 90%) as a yellow oil. [α]20 D = +148 (c 1.3, CH2Cl2); 1H NMR (300 MHz, CDCl3) δ 7.47–7.43 (m, 2H), 7.39–7.21 (m, 13H), 5.57 (d, J = 1.5 Hz, 1H), 4.87 (d, J = 10.8 Hz, 1H), 4.71 (s, 2H), 4.58 (d, J = 10.8 Hz, 1H), 4.38–4.30 (m, 3H), 4.26–4.23 (m, 1H), 3.90, (dd, J = 3.1, 9.0 Hz, 1H), 3.81 (app t, J = 9.2 Hz, 1H), 2.65 (d, J = 2.5 Hz, 1H), 2.27–2.22 (m, 2H), 1.62–1.52 (m, 2H), 1.33–1.24 (m, 24H), 0.90–0.86 (m, 3H); 13C NMR (75 MHz, CDCl3) δ 173.5, 137.8, 137.4, 133.6, 131.6, 129.0, 128.6, 128.5, 128.2, 128.01, 127.99, 127.9, 127.5, 87.1, 80.3, 75.2, 74.4, 72.2, 70.5, 69.7, 63.1, 34.1, 31.9, 29.65, 29.62, 29.57, 29.4, 29.3, 29.2, 29.1, 24.8, 22.7, 14.1; HRMS-ESI [M+Na]+ Calculated for C42H58O6SNa 713.3852. Found 713.3881.
Phenyl 2-O-(2-azidomethylbenzoyl)-3,4-di-O-benzyl-6-O-hexadecanoyl-1-thio-α-D-mannopyranoside (31)
2-Azidomethylbenzoyl chloride (AZMBCl) was prepared as described47 and used without purification. A solution of 32 (1.35 g, 1.95 mmol) in pyridine (10 mL) was added to ice-cooled AZMBCl (0.49 g, 2.5 mmol) and the stirred mixture was allowed to warm to rt overnight. Excess pyridine was concentrated under reduced pressure, and the residue was taken up in EtOAc (150 mL), washed with 1 M aq HCl (2 x 50 mL), saturated aq NaHCO3 (2 x 50 mL), dried (MgSO4), filtered and the solvent removed. The crude residue was purified by column chromatography on silica gel (EtOAc/petroleum ether = 7.5:92.5) to afford the title compound 31 (1.016 g, 61%) as an oil. [α]20 D = +55 (c 0.9, CH2Cl2); 1H NMR (300 MHz, CDCl3) δ 8.04, (dd, J = 1.2, 7.8 Hz, 1H), 7.58 (dt, J = 1.2, 7.4 Hz, 1H), 7.52–7.48 (m, 3H), 7.42–7.24 (m, 14H), 5.83 (dd, J = 1.6, 3.0 Hz, 1H), 5.60 (d, J = 1.6 Hz, 1H), 4.92 (d, J = 10.9 Hz, 1H), 4.80 (d, J = 11.3 Hz, 1H), 4.75 (s, 2H), 4.64 (d, J = 11.3 Hz, 1H), 4.61 (d, J = 10.9 Hz, 1H), 4.48–4.32 (m, 3H), 4.07, (dd, J = 3.0, 9.2 Hz, 1H), 3.92 (app t, J = 9.3 Hz, 1H), 2.29–2.24 (m, 2H), 1.63–1.53 (m, 2H), 1.32–1.23 (m, 24H), 0.90–0.86 (m, 3H); 13C NMR (75 MHz, CDCl3) δ 173.4, 165.6, 137.8, 137.6, 137.4, 133.2, 133.0, 132.1, 131.4, 129.7, 129.1, 128.4, 128.2, 128.1, 127.95, 127.1, 86.1, 78.5, 75.3, 74.3, 71.9, 70.9, 70.8, 63.1, 52.9, 34.1, 31.9, 29.7, 29.63, 29.58, 29.5, 29.3, 29.23, 29.20, 24.8, 22.7, 14.1; HRMS-ESI [M+Na]+ Calculated for C50H63N3O7SNa: 872.4284. Found 872.4283.
1-O-Allyl-2-O-(2-O-[2-azidomethylbenzoyl]-3,4-di-O-benzyl-6-O-hexadecanoyl-α-D-mannopyranosyl)- 6-O-(2,3,4,6-tetra-O-benzyl-α-D-mannopyranosyl)-3,4,5-tri-O-benzyl-D-myo-inositol (34)
Acceptor 1626 (160 mg, 0.16 mmol) was dissolved in dry CH2Cl2 (5 mL) and cooled to −60 °C. In a separate flask, a mixture of donor 31 (125 mg, 0.147 mmol), diphenyl sulfoxide (83 mg, 0.412 mmol) and 2,6-di-tert-butyl-4-methylpyridine (DTBMP, 91 mg, 0.441 mmol) was co-evaporated from dry CH2Cl2 (10 mL). The reagents were redissolved in CH2Cl2 (5 mL) and cooled to −60 °C with stirring, before the addition of Tf2O (35 μL, 0.21 mmol). After 5 min, the solution of acceptor was transferred to the reaction vessel (cannula), rinsing with CH2Cl2 (2 mL), and the reaction mixture was allowed to warm to 0 °C over 3 h. The reaction was quenched with saturated aq NaHCO3 and extracted with CH2Cl2. The organic extracts were washed with saturated aq NaHCO3, dried (MgSO4), filtered and the solvent removed. The residue was purified by column chromatography on silica gel (EtOAc/petroleum ether = 15:85) followed by a second column (CH2Cl2/petroleum ether/diethyl ether = 30:20:1) to afford the title compound 34 (88 mg, 34%). [α]20 D = +19 (c 0.96, CH2Cl2); 1H NMR (300 MHz, CDCl3) δ 8.03, (dd, J = 1.1, 7.8 Hz, 1H), 7.59–7.50 (m, 2H), 7.41–7.08 (m, 46H), 5.82–5.68 (m, 2H), 5.57 (d, J = 1.6 Hz, 1H), 5.26–5.19 (m, 2H), 5.08–5.03 (m, 1H), 4.94–4.45 (m, 19H), 4.31–3.82 (m, 15H), 3.40 (dd, J = 3.1, 11.3 Hz, 1H), 3.33–3.25 (m, 3H), 3.17 (dd, J = 1.7, 9.7 Hz, 1H), 2.24–2.19 (m, 2H), 1.59–1.51 (m, 2H), 1.33–1.21 (m, 24H), 0.90–0.86 (m, 3H); 13C NMR (75 MHz, CDCl3) δ 173.3, 165.1, 139.1, 138.85, 138.77, 138.6, 138.4, 138.2, 138.1, 137.7, 137.6, 133.8, 132.7, 131.3, 129.4, 128.7, 128.5, 128.4, 128.32, 128.29, 128.2, 128.1, 128.04, 127.95, 127.82, 127.78, 127.7, 127.5, 127.40, 127.35, 127.21, 127.15, 117.4, 98.9, 81.5, 81.4, 80.9, 80.0, 78.5, 75.9, 75.7, 75.3, 75.1, 74.9, 74.8, 73.6, 73.2, 72.55, 72.50, 72.1, 72.0, 71.5, 70.8, 69.8, 69.1, 68.6, 62.8, 52.9, 34.1, 31.9, 29.7, 29.63, 29.59, 29.5, 29.3, 29.23, 29.20, 24.8, 22.7, 14.1; HRMS-ESI [M+Na]+ Calculated for C108H125N3O18Na: 1774.8856. Found 1774.8870.
2-O-(2-O-[2-Azidomethylbenzoyl]-3,4-di-O-benzyl-6-O-hexadecanoyl-α-D-mannopyranosyl)- 6-O-(2,3,4,6-tetra-O-benzyl-α-D-mannopyranosyl)-3,4,5-tri-O-benzyl-D-myo-inositol (35)
(1,5-Cyclooctadiene)bis(methyldiphenylphosphine)iridium(I) hexafluorophosphate (29 mg, 34 mmol) was stirred with a solution of 34 (301 mg, 172 mmol) in THF (10 mL) under Ar at rt for 10 min. The reaction was exposed to an atmosphere of H2 for ~30 s, during which time the colour changed from red to pale yellow, then stirred under Ar for 45 min, at which point TLC (20% EtOAc/petroleum ether) indicated complete consumption of the starting material. The mixture was concentrated under reduced pressure and dissolved in CH2Cl2/MeOH (2:1, 12 mL) containing 0.45 mL AcCl. After 1 h at rt, solid NaHCO3 was added and the mixture stirred for a further 5 min. The reaction mixture was diluted with water, extracted with CH2Cl2, dried (MgSO4), filtered and the solvent removed. The crude residue was purified by column chromatography on silica gel (EtOAc/petroleum ether = 1:4) to afford the title compound 35 (240 mg, 82%). [α]20 D +32 (c 1.1, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ8.07, (d, J = 7.9 Hz, 1H), 7.59–7.50 (m, 2H), 7.39–7.14 (m, 46H), 5.70–5.69 (m, 1H), 5.40 (d, J = 1.6 Hz, 1H), 5.30 (d, J = 2.0 Hz, 1H), 4.91–4.26 (m, 20H), 4.19–3.76 (m, 11H), 3.64–3.56 (m, 2H), 3.47 (dd, J = 6.6, 10.3 Hz, 1H), 3.36–3.27 (m, 3H), 2.26–2.21 (m, 2H), 1.61–1.52 (m, 2H), 1.33–1.21 (m, 24H), 0.90–0.86 (m, 3H); 13C NMR (75 MHz, CDCl3) δ 173.4, 165.3, 138.53, 138.49, 138.4, 138.2, 138.1, 137.80, 137.76, 137.7, 132.8, 131.4, 129.3, 128.6, 128.44, 128.41, 128.38, 128.30, 128.26, 128.2, 128.0, 127.9, 127.8, 127.7, 127.63, 127.59, 127.54, 127.46, 127.4, 99.0, 96.0, 81.2, 80.4, 80.1, 79.4, 78.2, 76.1, 75.6, 75.3, 75.1, 75.02, 74.97, 74.5, 73.7, 73.3, 72.5, 72.2, 72.14, 72.09, 71.6, 71.1, 69.7, 69.3, 69.2, 62.8, 52.9, 34.1, 31.9, 29.7, 29.62, 29.58, 29.5, 29.3, 29.23, 29.20, 24.8, 22.7, 14.1; HRMS-ESI [M+Na]+ Calculated for C105H121N3O18Na: 1734.8543. Found 1734.8586.
2-O-(2-O-[2-Azidomethylbenzoyl]-3,4-di-O-benzyl-6-O-hexadecanoyl-α-D-mannopyranosyl)- 1-O-(1-O-hexadecanoyl-2-O-hexadecyl-sn-glycero-3-benzylphosphoryl)-6-O-(2,3,4,6-tetra-O-benzyl-α-D-mannopyranosyl)-3,4,5-tri-O-benzyl-D-myo-inositol (37)
1H-Tetrazole (13 mg, 0.19 mmol) was added to a stirred solution of alcohol 35 (106 mg, 0.062 mmol) and phosphoramidite 36 (147 mg, 0.186 mmol) in dry CH2Cl2 (8 mL) under Ar at 0 ºC. After stirring at rt for 3 h, the reaction mixture was cooled to −40 ºC and a solution of m-CPBA (50%, 86 mg, 0.25 mmol) in CH2Cl2 (10 mL) was transferred by cannula into the reaction mixture. After being stirred at rt for 1 h, the reaction was quenched by addition of 10% aq Na2SO3 (50 mL) and the combined mixture extracted with Et2O (100 mL). The ethereal extract was washed with saturated aq NaHCO3 (3 × 50 mL), dried (MgSO4), filtered and the solvent removed. The residue was purified by column chromatography on silica gel (EtOAc/petroleum ether = 1:9 to 1:4) followed by a second column (acetone/toluene = 1:50 to 3:97) to afford the title compound 37 (59 mg, 0.024 mmol, 39%) as an oil. 1H NMR (300 MHz, CDCl3) δ 8.04–7.98 (m, 1H), 7.58–7.43 (m, 2H), 7.36–7.03 (m, 51H), 5.66–5.62 (m, 1H), 5.56–5.62 (m, 1H), 5.33–5.29 (m, 1H), 5.11–5.04 (m, 2H), 4.95–4.39 (m, 20H), 4.36–3.82 (m, 17H), 3.60–3.22 (m, 7H), 2.35–2.13 (m, 4H), 1.61–1.48 (m, 4H), 1.42–1.17 (m, 76H), 0.90–0.86 (m, 9H); 13C (75 MHz, CDCl3) selected signals δ 173.2, 99.6, 98.6; 31P NMR (121 MHz, CDCl3) δ 0.1, 0.0; HRMS-ESI [M+Na]+ calcd for C147H196N3O24NaP: 2441.3845. Found 2441.3855.
2-O-(3,4-Di-O-benzyl-6-O-hexadecanoyl-α-D-mannopyranosyl)-1-O-(1-O-hexadecanoyl-2-O-hexadecyl-sn-glycero-3-benzylphosphoryl)-6-O-(2,3,4,6-tetra-O-benzyl-α-D-mannopyranosyl)-3,4,5-tri-O-benzyl-D-myo-inositol (38)
Tri-n-butyl phosphine (34 μL, 0.14 mmol) was added to a degassed solution of AZMB ester 37 (58 mg, 0.024 mmol) in THF/H2O (9:1, 10 mL) under Ar. After stirring at rt for 3 h, toluene (20 mL) was added and the solvent removed in vacuo. The residue was purified by column chromatography on silica gel (EtOAc/petroleum ether = 1:4 to 3:7) to afford the title compound 38 (42 mg, 78%) as an oil. 1H NMR (300 MHz, CDCl3) δ 7.40–6.99 (m, 50H), 5.46–5.42 (m, 1H), 5.19 (m, 1H), 5.11–4.39 (m, 20H), 4.19–3.78 (m, 18H), 3.62–3.20 (m, 7H), 2.54–2.49 (m, 1H), 2.28–2.18 (m, 4H), 1.60–1.40 (m, 6H), 1.33–1.22 (m, 74H), 0.90–0.86 (m, 9H); 13C (75 MHz, CDCl3) selected signals δ 173.5, 101.5, 98.4; 31P NMR (121 MHz, CDCl3) δ 0.0, −0.2; HRMS-ESI [M+Na]+ calcd for C139H191O23PNa: 2282.3412. Found 2282.3401.
1-O-(1-O-Hexadecanoyl-2-O-hexadecyl-sn-glycero-3-phosphoryl)-2-O-(6-O-hexadecanoyl-α-D-mannopyranosyl)-6-O-(α-D-mannopyranosyl)-D-myo-inositol (5)
Pd(OH)2/C (20%, 36 mg) was added to a stirred solution of compound 38 (42 mg, 0.019 mmol) in THF/MeOH (2:3, 5 mL). The mixture was stirred under a H2 atmosphere for 3.5 h at rt, then filtered through Celite and the solvent removed. The residue was purified by column chromatography on silica gel (CHCl3/MeOH/H2O = 70:40:0 to 70:40:8) afforded the title compound 5 (19 mg, 75%) as a white powder. 95% pure by HPLC system 1 [α] 20 D = +30 (c 0.10, CHCl3/CH3OH/H2O, 70:40:6); 1H NMR (500 MHz, CDCl3/CD3OD/D2O 70:40:6) δ 5.15 (br s, 1H), 5.10 (br s, 1H), 4.38–4.08 (m, 8H), 4.05 (dd, J = 1.7, 3.3 Hz, 1H), 4.01–3.96 (m, 2H), 3.94–3.89 (m, 1H), 3.84–3.73 (m, 6H), 3.70–3.65 (m, 3H), 3.60 (app t, J = 9.5 Hz, 1H), 3.55– 3.51 (m, 1H), 3.46 (dd, J = 2.5, 10 Hz, 1H), 3.29 (t, J = 9.3 Hz, 1H), 2.39–2.33 (m, 4H), 1.65–1.54 (m, 6H), 1.37–1.27 (m, 74H), 0.90–0.87 (m, 9H); 13C NMR (126 MHz, CDCl3/CD3OD/D2O 70:40:6) δ 175.7, 175.3, 102.3, 102.2, 79.4, (br d), 79.2, 77.5, 77.4, 74.0, 73.8, 73.5, 71.44, 71.35, 71.3, 71.2, 71.0, 70.9, 70.7, 67.8, 67.6, 64.9, 64.7, (br d), 64.3, 61.9, 34.9, 34.5, 32.51, 30.46, 30.40, 30.37, 30.3, 30.23, 30.18, 30.03, 29.97, 29.9, 29.8, 26.7, 25.6, 25.4, 23.2, 14.5; 31P NMR (202 MHz, CDCl3/CD3OD/D2O 70:40:6) δ −0.3; HRMS-ESI [M-H]− calcd for C69H130O23P: 1357.8741. Found 1357.8729.
1,3-bis-(2-O-Acetyl-3,4,5-tri-O-benzyl-α-D-mannopyranosyl)-2-hydroxypropane (40)
DDQ (35 mg, 0.154 mmol) was added to a stirred solution of 39 (149 mg, 0.128 mmol) in CH2Cl2/H2O (9:1, 4 mL). After 50 min the reaction was diluted with Et2O (80 mL), washed with water (3×20 mL), saturated NaHCO3 (3×20 mL), dried (MgSO4), filtered and the solvent removed. The crude residue was purified by column chromatography on silica gel (EtOAc/petroleum ether = 2:3) to afford the title compound 40 (96 mg, 72%) as waxy solid. [α]20 D +30 (c 0.82, CH2Cl2). 1H NMR (500 MHz, CDCl3) δ7.35–7.22 (m, 26H), 7.18–7.13 (m, 4H), 5.38–5.35 (m, 2H), 4.87–4.82 (m, 4H), 4.71–4.62 (m, 4H), 4.55–4.50 (m, 2H), 4.50–4.45 (m, 4H), 3.97–3.90 (m, 3H), 3.89–3.78 (m, 4H), 3.78–3.72 (m, 2H), 3.72–3.65 (m, 3H), 3.62 (dd, J = 6.2, 10.7 Hz, 1H), 3.56 (dd, J = 4.5, 10.7 Hz, 1H), 3.51 (dd, J = 6.6, 10.6 Hz, 1H), 2.13 (s, 6H); 13C NMR (125 MHz, CDCl3) δ170.3,138.09, 138.07, 137.9, 128.34, 128.28, 128.26, 128.0, 127.8, 127.7, 127.6, 127.5, 98.5 (J1 CH = 170 Hz), 98.3 (J1 CH = 170 Hz), 78.1, 78.0, 75.11, 75.09, 74.2, 73.4, 71.89, 71.85, 71.7, 69.8, 69.6, 69.2, 68.79, 68.77, 68.7, 68.6, 21.0. HRMS-ESI [M+Na]+ calculated for C61H68O15Na: 1063.4456. Found 1063.4445.
2-O-(Benzyloxy-1-O-hexadecanoyl-2-O-hexadecyl-sn-glycero-3-phosphoryl)-1,3-bis-(2-O-acetyl-3,4,5-tri-O-benzyl-α-D-mannopyranosyl)glycerol (41)
A mixture of alcohol 40 (86 mg, 0.083 mmol) and phosphoramidite 36 (98 mg, 0.124 mmol) was co-evaporated from dry CH2Cl2 (10 mL) then placed under high vacuum for 1 h. The reagents were dissolved in dry CH2Cl2 (2 mL) and stirred for 1 h at rt over 4Å molecular sieves. The solution was cooled to 0 °C before the addition of 4,5-dicyanoimidazole (16 mg, 0.14 mmol) then warmed to rt over 18 h. The solution was then cooled to −15 °C before the addition of dried (MgSO4) mCPBA (~60%, 50 mg) in CH2Cl2. After warming to rt over 1 h, the reaction mixture was diluted with Et2O (30 mL) and washed Na2S2O3 (10%, 30 mL), saturated NaHCO3 (3 × 20 mL), brine (30 mL), dried (MgSO4), filtered and the solvent removed. The crude residue was purified by column chromatography on silica gel (EtOAc/petroleum ether = 1:4 to 2:3) to afford the title compound 41 (93 mg, 64%). 1H NMR (500 MHz, CDCl3) mixture of isomers δ 7.37–7.19 (m, 31H), 7.17–7.10 (m, 4H), 5.11–5.01 (m, 2H), 4.90–4.79 (m, 4H), 4.70–4.57 (m, 5H), 4.52–4.39 (m, 6H), 4.17–4.10 (m, 1H), 4.09–3.73 (m, 10H), 3.71–3.58 (m, 4H), 3.58–3.52 (m, 1H), 3.50–3.37 (m, 2H), 2.26–2.21 (m, 2H), 2.12 (s, 3H), 2.11 (s, 3H), 1.62–1.51 (m, 2H), 1.50–1.41 (m, 2H), 1.35–1.15 (m, 50H), 0.93–0.80 (m, 6H); 13C NMR (125 MHz, CDCl3) δ 173.3, 170.2, 138.4, 138.2, 137.9, 135.8, 135.75, 135.71, 135.69, 128.6, 128.5, 128.4, 128.32, 128.30, 128.25, 128.2, 128.1, 128.0, 127.81, 127.80, 127.78, 127.7, 127.5, 98.32, 98.31, 98.15, 98.11, 78.20, 78.19, 75.79, 75.77, 75.73, 75.71, 75.4, 75.31, 75.26, 75.10, 75.07, 74.04, 74.00, 73.40, 73.38, 71.92, 71.88, 71.85, 71.83, 70.65, 70.61, 69.42, 69.38, 68.71, 68.66, 68.6, 68.4, 68.1, 66.72, 66.68, 66.42, 66.38, 66.3, 62.5, 34.1, 31.9, 29.9, 29.7, 29.64, 29.62, 29.5, 29.32, 29.28, 29.1, 26.0, 24.8, 22.6, 21.0; 31P NMR (202 MHz, CDCl3) δ −1.44, − 1.48; HRMS-ESI [M+Na]+ calculated for C103H143O21PNa: 1769.9757. Found 1769.9751.
2-O-(1-O-Hexadecanoyl-2-O-hexadecyl-sn-glycero-3-phosphoryl)-1,3-bis-(2-O-acetyl-α-D-mannopyranosyl) glycerol (42)
A suspension of 41 (90 mg, 51 mmol) and Pd(OH)2 in 2:3 THF/MeOH (5 mL) were stirred under an atmosphere of H2 ambient temperature for 4 h. The mixture was filtered through Celite, concentrated and purified by purified by column chromatography (neat CHCl3 to 2:1 CHCl3/MeOH) to give 42 (40 mg, 70%) as a white powder. [α]20 D = +27 (c 0.74, 2:1 CHCl3/MeOH); 1H NMR (500 MHz, 2:1 CDCl3/CD3OD) δ 5.08–5.03 (m, 2H), 4.83 (d, J = 0.9 Hz, 1H), 4.81 (d, J = 1.1 Hz), 4.54–4.46 (m, 1H), 4.29 (dd, J = 2.8, 11.6 Hz), 4.17–4.11 (m, 1H), 4.06–3.86 (m, 8H), 3.80–3.51 (m, 11H), 2.35 (t, J = 7.5 Hz, 1H), 2.12 (2 × s, 6H), 1.67–1.52 (m, 4H), 1.38–1.22 (m, 50H), 0.92–0.85 (m, 6H); 13C NMR (125 MHz, 2:1 CDCl3/CD3OD) δ 174.8, 171.61, 171.58, 98.1, 98.0, 76.8, 76.7, 74.4, 74.3, 73.7, 73.6, 72.72, 72.69, 71.2, 69.8, 68.7, 68.5, 67.0, 65.43, 65.41, 64.0, 62.5, 62.4, 34.6, 32.3, 30.3, 30.05, 30.00, 29.90, 29.87, 29.7, 29.5, 26.4, 25.3, 23.0, 20.9, 14.2; 31P NMR (202 MHz, 2:1 CDCl3/CD3OD) δ 2.7; HRMS-ESI [M-H]− Calculated for C54H100O21P: 1115.6495. Found 1115.6488.
2-O-(1-O-Hexadecanoyl-2-O-hexadecyl-sn-glycero-3-phosphoryl)-1,3-bis-O-(α-D-mannopyranosyl) glycerol (6)
Sodium methoxide in MeOH (0.5 M solution, 50 μL) was added dropwise to a stirred solution of 42 (26 mg, 0.023 mmol) in MeOH (5 mL). After 2 h the mixture was quenched with Dowex 50W8-100 (H+) resin, filtered and subsequently made neutral by treatment with Dowex 50W8-100 (Na+) resin. After filtration and concentration, the crude residue was purified on silica gel (CHCl3/MeOH/H2O = 70:35:1.75 to 70:35:6) to afford the title compound 6 (17 mg, 71%). [α]20 D = +39 (c 0.14, CHCl3/MeOH/H2O 70:40:6); 1H NMR (500 MHz, CDCl3/CD3OD/D2O 70:35:6) δ 4.87 (br, 1H), 4.84 (br, 1H), 4.39 (br, 1H), 4.30 (dd, J = 2.7, 11.8 Hz, 1H), 4.11 (dd, J = 7.3, 11.7 Hz, 1H), 3.95–3.83 (m, 8H), 3.77–3.59 (m, 12H), 3.53 (dt, J = 7.0, 9.4 Hz, 1H), 2.35 (t, J = 7.6 Hz, 2H), 1.65–1.60 (m, 2H), 1.59–1.53 (m, 2H), 1.34–1.27 (m, 50H), 0.90–0.87 (m, 6H); 13C NMR (125 MHz, CDCl3/CD3OD/D2O 70:35:6) δ 175.2, 100.8, 100.7, 77.3, 77.2, 73.5, 73.4, 71.5, 71.3, 70.94, 70.90, 67.8, 67.7, 67.3, 67.1, 64.9, 64.7, 64.6, 61.9, 61.8, 34.8, 32.4, 30.4, 30.23, 30.19, 30.14, 30.08, 30.05, 29.8, 29.7, 26.5, 25.5, 23.1, 14.4; 31P NMR (202 MHz, CDCl3/CD3OD/D2O 70:35:6) δ −0.8; HRMS-ESI [M-H]− Calculated for C50H96O19P: 1031.6283. Found 1031.6288.
Cytokine release assay
Thioglycollate-elicited peritoneal murine macrophages were generated as previously described.48 Macrophages were cultured in 96 well plates 2 x 105 cells/well in supplemented RPMI-1640 (100 U/mL penicillin-streptomyocin, 10% fetal calf serum). Compounds and 10 ng/ml LPS were added to macrophage cultures to a final culture volume of 200 μl/well and incubated at 37°C, 5% CO2 for 18h. Supernatants were collected and IL-6 levels measured by ELISA. Cell viability was measured by trypan blue exclusion.
Expression and purification of sTLR4/MD-2
A complex between TLR4 ectodomain (sTLR4) and MD-2 was expressed by a baculovirus expression system using a modified dual expression vector, pAcUW51 (BD Biosciences). sTLR4 and MD-2 are appended to a C-terminal His6-tag and a Strep-tag II, respectively. sTLR4/MD-2 expressing baculovirus was generated by co-transfecting SF9 insect cells with the sTLR4/MD-2 expression vector and linearized baculovirus DNA, BaculoGold (BD Biosciences). For sTLR4/MD-2 expression, Hi5 insect cells were infected with the amplified recombinant virus. The secreted sTLR4/MD-2 protein was initially purified by Ni-NTA and Strep-Tactin affinity chromatographies and its C-terminal tags were removed by thrombin. The resultant protein was further purified by size exclusion chromatography.
Expression and purification of MD-1
MD-1 was expressed by a baculovirus expression system using a modified pAcGP67 expression vector and purified by three steps including Ni-NTA affinity, anion exchange, and gel filtration chromatography, as described previously.44
Native PAGE
Purified sTLR4/MD-2 protein was mixed with 2 first and then immediately with LPS in 20 mM Hepes pH 7.4/150 mM NaCl and the mixture was incubated at room temperature for 1 hour. The mixture was analyzed by native PAGE, which was performed at room temperature at pH 8.8 using a 4–20% gradient polyacrylamide gel (Bio-Rad).
Supplementary Material
Scheme 1.

Synthesis of common intermediate 16
Acknowledgments
The work was supported by the New Zealand Foundation for Research and Technology Contract C08X0808 and NIH grant AI1042266 (IAW) and the Skaggs Institute for Chemical Biology of the Scripps Research Institute. The authors thank Dr Regan Anderson for his very helpful contribution in the preparation of this manuscript.
Abbreviations used
- TLR
Toll-Like Receptor
- PIM
phosphatidylinositol mannoside
- NKT
natural killer T
- LPS
lipopolysaccharide
- MD-2
myeloid differentiation factor 2
Footnotes
Supporting Information Available: This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kaur D, Guerin ME, Skovierová H, Brennan PJ, Jackson M. Adv Appl Microbiol. 2009;69:23. doi: 10.1016/S0065-2164(09)69002-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cao B, Williams SJ. Chemical approaches for the study of the mycobacterial glycolipids phosphatidylinositol mannosides, lipomannan and lipoarabinomannan. Nat Products Reports. 2010;27:919–947. doi: 10.1039/c000604a. [DOI] [PubMed] [Google Scholar]
- 3.de la Salle H, Mariotti S, Angenieux C, Gilleron M, Garcia-Alles LF, Malm D, Berg T, Paoletti S, Maitre B, Mourey L, Salamero J, Cazenave JP, Hanau D, Mori L, Puzo G, De Libero G. Assistance of microbial glycolipid antigen processing by CD1e. Science. 2005;310:1321–1324. doi: 10.1126/science.1115301. [DOI] [PubMed] [Google Scholar]
- 4.Mahon RN, Rojas RE, Fulton SA, Franko JL, Harding CV, Boom WH. Mycobacterium tuberculosis cell wall glycolipids directly inhibit CD4+ T-cell activation by interfering with proximal Tcell-receptor signaling. Infect Immun. 2009;77:4574–4583. doi: 10.1128/IAI.00222-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fischer K, Scotet E, Niemeyer M, Koebernick H, Zerrahn J, Maillet S, Hurwitz R, Kursar M, Bonneville M, Kaufmann SH, Schaible UE. Mycobacterial phosphatidylinositol mannoside is a natural antigen for CD1d-restricted T cells. Proc Natl Acad Sci U S A. 2004;101:10685–10690. doi: 10.1073/pnas.0403787101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gilleron M, Quesniaux VF, Puzo G. Acylation state of the phosphatidylinositol hexamannosides from Mycobacterium bovis bacillus Calmette Guerin and mycobacterium tuberculosis H37Rv and its implication in Toll-like receptor response. J Biol Chem. 2003;278:29880–29890. doi: 10.1074/jbc.M303446200. [DOI] [PubMed] [Google Scholar]
- 7.Mempel M, Ronet C, Suarez F, Gilleron M, Puzo G, Van Kaer L, Lehuen A, Kourilsky P, Gachelin G. Natural killer T cells restricted by the monomorphic MHC class 1b CD1d1 molecules behave like inflammatory cells. J Immunol. 2002;168:365–371. doi: 10.4049/jimmunol.168.1.365. [DOI] [PubMed] [Google Scholar]
- 8.Jones BW, Means TK, Heldwein KA, Keen MA, Hill PJ, Belisle JT, Fenton MJ. Different Toll-like receptor agonists induce distinct macrophage responses. J Leukoc Biol. 2001;69:1036–1044. [PubMed] [Google Scholar]
- 9.Boonyarattanakalin S, Liu X, Michieletti M, Lepenies B, Seeberger PH. Chemical synthesis of all phosphatidylinositol mannoside (PIM) glycans from Mycobacterium tuberculosis. J Am Chem Soc. 2008;130:16791–16799. doi: 10.1021/ja806283e. [DOI] [PubMed] [Google Scholar]
- 10.Parlane NA, Denis M, Severn WB, Skinner MA, Painter GF, La Flamme AC, Ainge GD, Larsen DS, Buddle BM. Phosphatidylinositol Mannosides are Efficient Mucosal Adjuvants. Immunol Invest. 2008;37:129–142. doi: 10.1080/08820130701690782. [DOI] [PubMed] [Google Scholar]
- 11.Sprott GD, Dicaire CJ, Gurnani K, Sad S, Krishnan L. Activation of Dendritic Cells by Liposomes Prepared from Phosphatidylinositol Mannosides from Mycobacterium bovis Bacillus Calmette-Guérin and Adjuvant Activity In Vivo. Infect Immun. 2004;72:5235–46. doi: 10.1128/IAI.72.9.5235-5246.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harper JL, Hayman CM, Larsen DS, Painter GF, Singh-Gill G. A PIM analogue suppresses allergic airway disease. Bioorg Med Chem. 2011;19:917–925. doi: 10.1016/j.bmc.2010.11.058. [DOI] [PubMed] [Google Scholar]
- 13.Sayers I, Severn W, Scanga CB, Hudson J, Le Gros G, Harper JL. Suppression of allergic airway disease using mycobacterial lipoglycans. J Allergy Clin Immunol. 2004;114:302–309. doi: 10.1016/j.jaci.2004.03.057. [DOI] [PubMed] [Google Scholar]
- 14.Doz E, Rose S, Court N, Front S, Vasseur V, Charron S, Gilleron M, Puzo G, Fremaux I, Delneste Y, Erard F, Ryffel B, Martin OR, Quesniaux VF. Mycobacterial phosphatidylinositol mannosides negatively regulate host Toll-like receptor 4, MyD88-dependent proinflammatory cytokines, and TRIF-dependent co-stimulatory molecule expression. J Biol Chem. 2009;284:23187–23196. doi: 10.1074/jbc.M109.037846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ainge GD, Compton BJ, Hayman CM, Martin WJ, Toms SM, Larsen DS, Harper JL, Painter GF. Chemical Synthesis and Immunosuppressive Activity of Dipalmitoyl Phosphatidylinositol Hexamannoside. J Org Chem. 2011;76:4941–4951. doi: 10.1021/jo200588u. [DOI] [PubMed] [Google Scholar]
- 16.Ainge GD, Parlane NA, Denis M, Dyer BS, Harer A, Hayman CM, Larsen DS, Painter GF. Phosphatidylinositol mannoside ether analogues: syntheses and interleukin-12-inducing properties. J Org Chem. 2007;72:5291–5296. doi: 10.1021/jo070639m. [DOI] [PubMed] [Google Scholar]
- 17.Ainge GD, Hudson J, Larsen DS, Painter GF, Gill GS, Harper JL. Phosphatidylinositol Mannosides: Synthesis and Suppression of Allergic Airway Disease. Bioorg Med Chem. 2006;14:5632–5642. doi: 10.1016/j.bmc.2006.04.037. [DOI] [PubMed] [Google Scholar]
- 18.Liu X, Stocker BL, Seeberger PH. Total Synthesis of Phosphatidylinositol Mannosides of Mycobacterium tuberculosis. J Am Chem Soc. 2006;128:3638–3648. doi: 10.1021/ja0565368. [DOI] [PubMed] [Google Scholar]
- 19.Jayaprakash KN, Lu J, Fraser-Reid B. Synthesis of a key Mycobacterium tuberculosis biosynthetic phosphoinositide intermediate. Bioorg Med Chem Lett. 2004;14:3815–3819. doi: 10.1016/j.bmcl.2004.04.103. [DOI] [PubMed] [Google Scholar]
- 20.Bender SL, Budhu R. J Biomimetic Synthesis of Enantiomerically Pure D-myo-Inositol derivatives. J Am Chem Soc. 1991;113:9883–9885. [Google Scholar]
- 21.Dietrich H, Espinosa JF, Chiara JF, Jimenez-Barbero J, Leon Y, Varela-Nieto I, Mato JM, Cano FH, Foces-Foces C. Martín-Lomas Glycosyl Inositol Derivatives Related to Inositolphosphoglycan Mediators: Synthesis Structure and Biological Activity M. Chem Eur J. 1999;1:320. [Google Scholar]
- 22.Bruzik KS, Tsai MD. Efficient and Systematic Syntheses of Enantiomerically Pure and Regiospecifically Protected myo-Inositols. J Am Chem Soc. 1992;114:6361–6374. [Google Scholar]
- 23.Watanabe Y, Yamamoto T, Okazaki T. Synthesis of 2,6-di-O-α-D-Mannopyranosylphosphatidyl-D-myo-inositol. Utilization of glycosylation and phosphorylation based on phosphite chemistry. Tetrahedron. 1997;53:903–918. [Google Scholar]
- 24.Watanabe Y, Yamamoto T, Ozaki S. Regiospecific Synthesis of 2,6-Di-O-(α-D-mannopyranosyl) phosphatidyl-D-myo-inositol. J Org Chem. 1996;61:14–15. [Google Scholar]
- 25.Hölemann A, Stocker BL, Seeberger PH. Synthesis of a Core Arabinomannan Oligosaccharide of Mycobacterium tuberculosis. J Org Chem. 2006;71:8071–8088. doi: 10.1021/jo061233x. [DOI] [PubMed] [Google Scholar]
- 26.Elie CJJ, Verduyn R, Dreef CE, Brounts DM, van der Marel GA, van Boom JH. Synthesis of 6-0-(α-D-mannopyranosyl)-D-myo-inositol: a fragment from mycobacteria phospholipids. Tetrahedron. 1990;46:8243–8254. [Google Scholar]
- 27.Wang DS, Hsu AL, Chen CS. A phosphatidylinositol 3,4,5-trisphosphate analogue with low serum protein-binding affinity. Bioorg Med Chem. 2001;9:133–139. doi: 10.1016/s0968-0896(00)00227-3. [DOI] [PubMed] [Google Scholar]
- 28.Ogawa T, Horisaki T. Synthesis of 2-O-hexadecanoyl-1-O-hexadecyl-[alpha-Glc-6SO3 Na-(1--6) - alpha-Glc-(1–6)-alpha-Glc-(1–3)]-sn-glycerol: a proposed structure for the glyceroglucolipids of human gastric secretion and of the mucous barrier of rat-stomach antrum. Carbohydr Res. 1983;123:C1–C4. doi: 10.1016/0008-6215(83)88396-7. [DOI] [PubMed] [Google Scholar]
- 29.Zhai X, Bartel M, Brezesinski G, Rattay B, Möhwald H, Li J. Small angle X-ray scattering (SAXS) and differential scanning calorimetry (DSC) studies of amide phospholipids. Chem Phys Lipids. 2005;133:79. doi: 10.1016/j.chemphyslip.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 30.Ainge GD, Parlane NA, Denis M, Hayman CM, Larsen DS, Painter GF. Phosphatidylinositol mannosides: Synthesis and adjuvant properties of phosphatidylinositol di- and tetramannosides. Bioorg Med Chem. 2006;14:7615–7624. doi: 10.1016/j.bmc.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 31.Elie CJJ, Verduyn R, Dreef CE, Van der Marel GA, Van Boom JH. J Carbohydr Chem. 1992;11:715–739. [Google Scholar]
- 32.Dudkin VY, Miller JS, Dudkina AS, Danishefsky SJ, Antczak C, Scheinberg DA. Toward a Prostate Specific Antigen-Based Prostate Cancer Diagnostic Assay: Preparation of Keyhole Limpet Hemocyanin-Conjugated Normal and Transformed Prostate Specific Antigen Fragments. J Am Chem Soc. 2008;130:13598–13607. doi: 10.1021/ja8028137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Doi T, Kinbara A, Inoue H, Takahashi T. Donor-bound glycosylation for various glycosyl acceptors: bidirectional solid-phase semisynthesis of vancomycin and its derivatives. Chem Asian J. 2007;2:188. doi: 10.1002/asia.200600301. [DOI] [PubMed] [Google Scholar]
- 34.Ruiperez V, Astudillo AM, Balboa MA, Balsinde J. Coordinate regulation of TLR-mediated arachidonic acid mobilization in macrophages by group IVA and group V phospholipase A2s. J Immunol. 2009;182:3877–83. doi: 10.4049/jimmunol.0804003. [DOI] [PubMed] [Google Scholar]
- 35.Raoust E, Balloy V, Garcia-Verdugo I, Touqui L, Ramphal R, Chignard M. Pseudomonas aeruginosa LPS or flagellin are sufficient to activate TLR-dependent signaling in murine alveolar macrophages and airway epithelial cells. PLoS One. 2009;4:e7259. doi: 10.1371/journal.pone.0007259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Palma C, Iona E, Ebensen T, Guzman CA, Cassone A. The Toll-like Receptor 2/6 Ligand MALP-2 Reduces the Viability of Mycobacterium tuberculosis in Murine Macrophages. Open Microbiol J. 2009;3:47–52. doi: 10.2174/1874285800903010047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Drage MG, Tsai HC, Pecora ND, Cheng TY, Arida AR, Shukla S, Rojas RE, Seshadri C, Moody DB, Boom WH, Sacchettini JC, Harding CV. Mycobacterium tuberculosis lipoprotein LprG (Rv1411c) binds triacylated glycolipid agonists of Toll-like receptor 2. Nat Struct Mol Biol. 2010;17:1088–1095. doi: 10.1038/nsmb.1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jones BW, Heldwein KA, Means TK, Saukkonen JJ, Fenton MJ. Differential roles of Toll-like receptors in the elicitation of proinflammatory responses by macrophages. Ann Rheum Dis. 2001;60(Suppl 3):iii6–12. doi: 10.1136/ard.60.90003.iii6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.In the concentration ranges reported this class of compound have also previously be shown to be non-toxic in both in vitro in vivo assay systems – form examples see ref [14] Denis M, Ainge GD, Larsen DS, Severn W, Painter GF. A synthetic analogue of phosphatidylinositol mannoside is an efficient adjuvant. Immunopharmacology and Immunotoxicology. 2009;31:577–582. doi: 10.3109/08923970902824862.
- 40.Dobrovolskaia MA, Medvedev AE, Thomas KE, Cuesta N, Toshchakov V, Ren T, Cody MJ, Michalek SM, Rice NR, Vogel SN. Induction of In Vitro Reprogramming by Toll-Like Receptor (TLR)2 and TLR4 Agonists in Murine Macrophages: Effects of TLR “Homotolerance” Versus “Heterotolerance” on NF-kappa B. J Immunol. 2003;170:508–519. doi: 10.4049/jimmunol.170.1.508. [DOI] [PubMed] [Google Scholar]
- 41.Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, Miyake K, Kimoto M. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J Exp Med. 1999;189:1777–1782. doi: 10.1084/jem.189.11.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Re F, Strominger JL. Monomeric recombinant MD-2 binds toll-like receptor 4 tightly and confers lipopolysaccharide responsiveness. J Biol Chem. 2002;277:23427–23432. doi: 10.1074/jbc.M202554200. [DOI] [PubMed] [Google Scholar]
- 43.Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458:1191–1195. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]
- 44.Yoon SI, Hong MG, Han GW, Wilson IA. Crystal structure of soluble MD-1 and its interaction with lipid IVa. Proc Natl Acad Sci U S A. 2010;107:10990–10995. doi: 10.1073/pnas.1004153107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Divanovic S, Trompette A, Atabani SF, Madan R, Golenbock DT, Visintin A, Finberg RW, Tarakhovsky A, Vogel SN, Belkaid Y, Kurt-Jones EA, Karp CL. Negative regulation of Toll-like receptor 4 signaling by the Toll-like receptor homolog RP105. Nat Immunol. 2005;6:571–578. doi: 10.1038/ni1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lindberg J, Svensson SCT, Pahlsson P, Konradsson P. Synthesis of galactoglycerolipids found in the HT29 human colon carcinoma cell line. Tetrahedron. 2002;58:5109–5117. [Google Scholar]
- 47.Wada T, Ohkubo A, Mochizuki A, Sekine M. 2-(Azidomethyl)benzoyl as a new protecting group in nucleosides. Tetrahedron Lett. 2001;42:1069–1072. [Google Scholar]
- 48.Michl J, Pieczonka MM, Unkeless JC, Silverstein SC. Effects of immobilized immune complexes on Fc- and complement-receptor function in resident and thioglycollate-elicited mouse peritoneal macrophages. J Exp Med. 1979;150:607–621. doi: 10.1084/jem.150.3.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.