

Abstract
Infection caused by hepatitis C virus (HCV) is a significant world health problem for which novel therapies are in urgent demand. The virus is highly prevalent in the Middle East and Africa particularly Egypt with more than 90% of infections due to genotype 4. Nonstructural (NS5B) viral proteins have emerged as an attractive target for HCV antivirals discovery. A potent class of inhibitors having benzisothiazole dioxide scaffold has been identified on this target, however they were mainly active on genotype 1 while exhibiting much lowered activity on other genotypes due to the high degree of mutation of its binding site. Based on this fact, we employed a novel strategy to optimize this class on genotype 4. This strategy depends on using a refined ligand-steered homological model of this genotype to study the mutation binding energies of the binding site amino acid residues, the essential features for interaction and provide a structure-based pharmacophore model that can aid optimization. This model was applied on a focused library which was generated using a reaction-driven scaffold-hopping strategy. The hits retrieved were subjected to Enovo pipeline pilot optimization workflow that employs R-group enumeration, core-constrained protein docking using modified CDOCKER and finally ranking of poses using an accurate molecular mechanics generalized Born with surface area method.
Background:
Hepatitis C virus genotype 4 (HCV-4) is the most common variant of the hepatitis C virus (HCV) in the Middle East and Africa, particularly Egypt. This region has the highest prevalence of HCV worldwide, with more than 90% of infections due to genotype 4. HCV-4 has recently spread in several Western countries, particularly in Europe, due to variations in population structure, immigration, and routes of transmission. Employing HCV proteins as targets, directly acting antiviral agents have been identified and collectively described as ‘specifically targeted antiviral therapy for HCV’ (STAT-C) [1– 3]. Among the nonstructural proteins, NS3–4A protease, NS5B polymerase, NS3 helicase and NS5A have been the object of intense research efforts both by academia and pharmaceutical companies. NS5B RNA-dependent RNA polymerase is recognized as a key target for therapeutic intervention mainly because it is not present in mammalian cells and offers a wide range of possibilities for the discovery of new molecular entities as anti-HCV agents [4– 7]. Mechanistic and structural studies of this enzyme have revealed the existence of multiple allosteric binding sites, and in particular two thumb sites (thumb I and II) and three palm pockets (palm I, II and III) have been identified to date. According to the target site, the different inhibitors will be referred to as palm site I NNIs (PSI-NNIs), palm site II NNIs (PSII-NNIs), palm site III NNIs (PSIII-NNIs), thumb site I NNIs (TSI NNIs) and thumb site II NNIs (TSII-NNIs) [8, 9]. Out of these different allosteric sites and their corresponding inhibitors, we focused this study on palm I site and particularly Benzoisothiazoles dioxide as one of the main Palm I-NNI. The palm I site in genotype 4 shows high degree of mutation with respect to the other genotypes. This has an impact on the activity of the inhibitors of this site where it decreases drastically. This triggered us to study the impact of these mutations on binding by constructing a validated homological model for this genotype and analyzing the ligand-protein interactions. The main aim of this analysis was to optimize the Benzoisothiazoles dioxide on this specific genotype. On the other hand, from a ligand design perspective we attempted to modify this class of ligands such that it has a high diversification capability and high synthetic feasibility that can enable us to optimize it rapidly within the binding site. Thus, we decided to use a reaction-driven scaffold-hopping procedure to achieve this aim.
Methodology:
The protocol consists of two workflows that intersect at some point where the first depends on developing a ligand-protein complex that can be used to study the essential interactions criteria, calculating mutation binding energies and generating a structure-based pharmacophore to filter ligands while the second is ligand dependent where it is used to generate a focused library of synthetically feasible ligands against this target. They intersect at the point where the pharmacophore (first workflow) is used to filter the focused library (second workflow) to handle the hits to an optimization protocol (Enovo) [10]. This is illustrated in (Figure 1).
Figure 1.

The workflow used to optimize the benzisothiazole dioxide activity of NS5b polymerase of genotype 4
First workflow:
The aim of this workflow is to provide a refined homological model of genotype 4 to be used for structure-based pharmacophore screening and docking of the virtual library generated for optimization aim.
Ligand-steered Homological modeling:
Homological model was constructed using Modeler [11] in Discovery Studio. Uniprot was searched for HCV polymerase NS5b sequence for genotype 4a.It was found under accession code O39929 [12]. According to the sequence annotation, the RNA-directed RNA polymerase is represented by the sequence from 2418 to 3008 [13]. Uniprot sequence was blasted using Discovery Studio against PDB_nr95 database. This was done in order to obtain template structure for homology modeling where 3D5M was chosen as template with 77% identity. The sequence alignment was done using align sequence protocol as shown in (Figure 2). After that, the sequence alignment with the template was further used to build a homology model using Modeler while adjusting the settings to high optimization and copying Water molecules and the ligand from the template to the model. 5 models were created. From which, we have chosen the first model that was further minimized using ligandX algorithm, simulated using molecular dynamics protocol in MOE to investigate essential features.
Figure 2.

Alignment of the sequence of genotype 4 with that of template (3D5M).Red boxes show the amino acids of the binding site.
2D-interaction analysis:
Based on the refined complex, 2D interaction analysis was carried out using MOE 2010 ligand interaction generation. This was very useful to study the actual interactions that are responsible for the activity.
Mutation binding energy studies:
In order to show the impact of the natural polymorphism in genotype 4, we calculated the mutation binding energies of the variable amino acid residues in the Palm I binding site. The difference in binding due to mutation was calculated by finding difference between the free energy of binding in case of mutation and no mutation. The free energy of binding was calculated using CHARMm force field according to this equation: ΔG =αΔGFF +βTΔS where ΔS was calculated using Abagyan and tortov amino acid chain entropy scale with correction according to the side chain solvent accessibility. Calculations were carried out by Accelrys Discovery Studio 3.0 using “calculate mutation binding energy protocol”. The reported energy is the sum of weighed terms: electrostatic (0.45), van-der Walls (0.45) and entropy (0.8). Generalized Born implicit solvent model was used with dielectric constant of 80.
Structure-based pharmacophore:
Using the refined complex of the genotype 4 with benzoisothiazole dioxide, a structure-based pharmacophore was create using the technique that was developed by Wolber and Langer for screening of new compounds instead of the computationally expensive docking[14]. The technique was implemented already in ligandscout software [15]. This algorithm extracts information according to certain rules depending on nearby contact residues. It was used here to rapidly filter ligands of the virtual library used for optimization.
Second workflow:
The aim of this workflow is to generate the virtual library that is focused to this target such that it will be screened using the first workflow.
Reaction-driven scaffold-hopping:
Due to the limited SAR expansion capability of the existing scaffold that can hinder rapid probing of the effect of various substituents on activity, we carried out a reaction-driven scaffold hopping. Initially, the ligand co-crystallized with the protein in 3D5M complex was used as a starting point for retrosynthetic-disconnection [16] into two scaffolds: A and B. A query was built according to scaffold A as shown in (Figure 5). This query was used for substructure search in Scifinder such that the retrieved hits synthesis can be done in not more than 2 steps. Besides, we focused while analyzing the results on the high diversification capability, high synthetic feasibility and the availability of a wide panel of the forming starting materials. Regarding scaffold B, a bioisosteric replacement based on field technology [17] in the Fieldstere software was carried out as shown in the (Figure 5).
Figure 5.

Reaction-driven scaffold hopping. The disconnection approach resulted in two scaffolds A and B. A was modified using a simple scifinder substructure query limiting results on 2 steps synthetic pathways while B was modified using a fieldbased approach which was influenced too by the ease of synthesis and overall yield. Regarding synthesis of A: (a) for aromatic aldehydes :i—RCHO, TEA, MgSO4, THF, 25°C, 12 h; ii—NaBH4, MeOH, 25 °C, 1 h; for aliphatic aldehydes: RCHO, NaBH3CN, MeOH, 25°C, 12 h; (b) HO2CCH2CO2Et, EDC_HCl, TEA, DCM, 25 _C,12 h; (c) (i) NaOEt, EtOH, 25°C, 12 h; (ii) 1 M H2SO4 (aq), reflux, 1 h. Regarding synthesis of the new A analogue: (d) malonic acid,SOCl2 ;(e)RNCO,140°C, 10 min.
Library design:
Based on the new scaffolds retrieved (Figure 1), we constructed a reaction-based virtual library where enumeration of ligands was carried out according to the reaction used to synthesize scaffold A. Library design “Enumeration by reaction” module in Accelrys discovery studio was used.
Pharmacophore-based screening:
Due to the fact that many of the ligands enumerated in the virtual library will show steric hindrance with the binding site, a rapid screening was carried out to filter those ligands which show steric clash with the binding site. This was based on the presence of excluded volumes in the structure-based pharmacophore created.
E-novo optimization workflow:
The filtered library was screened using E-novo protocol. This protocol is usually applied for structure-based lead optimization as it is based on using core-constrained docking. A scaffold core is generated from the ligand-bound protein homology model. After that, Ligands are generated from that scaffold using R-group fragmentation/enumeration tool such that the cores are aligned. The ligands side chains are conformationally sampled and are subjected to core-constrained protein docking using modified CDOCKER. Finally, a physics– based binding energy scoring function is applied to rank top ligand CDOCKER poses using more accurate molecular mechanics generalized Born with surface area method.
Results and Discussion:
The sequence alignment and the homological model clearly indicate which amino acids are mutated in the palm I site. This aided us to conduct the mutation binding energy calculations on those varied amino acids. The results are shown in (Table 1, see supplementary material). They show that the mutation of Met414 to valine is a strong effector on binding and that optimization should focus on efficient binding with valine in genotype 4 (it has shorter side chain than that of Met). Applying minimization and molecular dynamics on the complex enabled us to carry out a ligand-protein interaction analysis as depicted in (Figure 3). This analysis shows that importance of methansulfonamide group where it hydrogen bonds with Asp318 and Asn291. It also showed that the hydroxyl group of the tetramic acid is an important feature where it hydrogen bonds with Tyr448. Regarding the important hydrophobic features of the ligand, it is shown that the tertiary butyl group and the substituted phenyl ring attached to the tetramic acid interact with Val414, Pro197 and Leu384. Additionally, the refined complex was used as a starting point to create a structure-based pharmacophore that is totally dependent on the actual interactions between the ligand and the protein as shown in (Figure 4). The pharmacophore clearly takes into consideration the excluded volume (amino acids of the binding site that the ligand should not sterically clash with) besides the important features that are responsible for the aforementioned interactions in the 2D interaction analysis.
Figure 3.
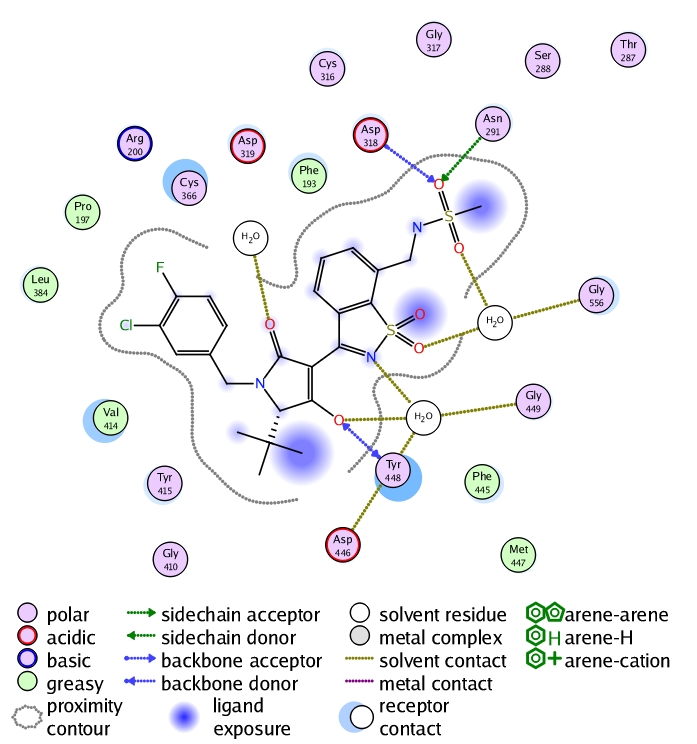
2D-interaction analysis of the reference ligand in the homological model of NS5b polymerase enzyme of genotype 4.
Figure 4.

Structure-based pharmacophore created using the ligand-homological model of NS5b polymerase. The pharmacophore represents HHAAA where H is hydrophobic feature and A is the acceptor feature. Grey spheres represents excluded volumes, blue represents hydrophobic features while the green represent acceptor features.
The optimization of the benzisothiazole dioxide inhibitory activity against genotype 4 was carried out with the aid of the homological model and the pharmacophore. Initially, we carried out a retrosynthetic dissection for the ligand as shown in (Figure 5) into two scaffolds A and B. In our case, we wanted to find a bioisoster for A that is synthetically feasible and with an economic capability of diversification such that it enables the very rapid probing of different substituents in that binding site. One of the best hits in scifinder that is based on two steps synthetic procedure was that of pyrazolidine-3, 5-Dione: A substituted hydrazine condensation with the readily available diethylmalonate yield the desired product which can be further substituted by any isocyante to form a urea using a simple workup. On the contrary, tetramic acid derivatives require a suitable amino acid that should be protected followed by reductive amination with a suitable aldehyde in a reaction that requires tedious purification by chromatographic techniques and higher number of steps. Regarding the scaffold B, a fieldstere hit was used based on the alignment of that hit with the original scaffold. This was carried out in order to minimize number of synthetic steps and avoid protection-deprotection schemes which affect the final yield as depicted in (Figure 6).
Figure 6.

Scaffold hopping of the benzisothiazole dioxide. Regarding synthesis of A: (a)i- BuLi, THF, Me(CH2)4Me, rt to - 23°C; 1 h, 23°C ;ii- isobutylchloroformate, 23°C to rt; overnight, rt; iii- H2O;(b) Pd(OAc)2, Dioxane, CsF, methylboronic acid , dicyclohexylphosphanyl-biphenyl-2-yl)- dimethyl-amine, 12 h,90°C (c) NBS, (BzO)2, CCl4, reflux, 1 d, 49%; (d) NHBocMs, Cs2CO3, DMF, 35–70 C, 6 h, 81%; (e) ˚ AcOH, reflux, 1–2 d, 94%; (f) POCl3,DMF, reflux, 10 min, 70%. Regarding synthesis of B: (g) i-ClSO3H, 120°C ii-NH4OH; (h) CrO3, H2SO4; (i) H2/Pd; (j) CH3SO2Cl, pyridine; (j) PCl5, 150°
The library design module was applied on a reaction basis where the scaffold A was varied by different isocaynates and hydrazines that were retrieved from Scifinder such that they are commercially available. The constructed virtual library was screened rapidly using the pharmacophore in a way to remove those bulky substituents that will not fit into the binding site. The refined library was used to conduct the optimization study using the E-novo protocol as mentioned in the methodology. One of the top ranked-hits was checked for stability in the binding site using molecular dynamics where it showed a stable sigma-pi interaction between the ligand and Valine414 (Figure 7). The simple synthetic feasibility of this hit triggered us to verify it experimentally where it showed 70% inhibition at 10uM concentration on genotype 4.
Figure 7.
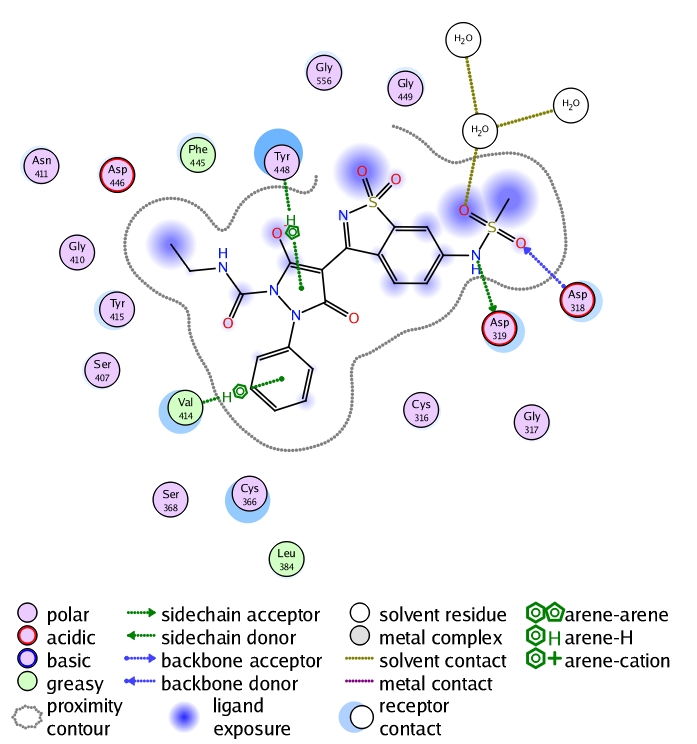
2D interaction analysis of the hit chosen for experimental validation. It is clear that Val414 can form sigmapi interaction with the phenyl substituent.
Conclusion:
In this study, we provided a novel workflow that can be used to optimize an inhibitor activity on another genotype that shows mutation in the binding site. This workflow was applied on HCV NS5b polymerase enzyme of genotype 4 to optimize benzisothiazole dioxide inhibitors on it. A focused library created using reaction enumeration was screened using structure-based pharmacophore followed by core-constrained docking and scoring using MM-GBSA. This tweaked protocol was used to identify an optimized inhibitor for this genotype.
Supplementary material
Footnotes
Citation:Mahmoud et al, Bioinformation 7(7): 328-333 (2011)
References
- 1.CM Lange, et al. Aliment Pharmacol Ther. 2010;32:14. doi: 10.1111/j.1365-2036.2010.04317.x. [DOI] [PubMed] [Google Scholar]
- 2.A Parfieniuk, et al. World J Gastroenterol. 2007;13:5673. doi: 10.3748/wjg.v13.i43.5673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.AJ Thompson, JC Mc Hutchison. J Viral Hepat. 2009;16:377. doi: 10.1111/j.1365-2893.2009.01124.x. [DOI] [PubMed] [Google Scholar]
- 4.F Legrand-Abravanel, et al. Expert Opin Investig Drugs. 2010;19:963. doi: 10.1517/13543784.2010.500285. [DOI] [PubMed] [Google Scholar]
- 5.H Li, ST Shi. Future Med Chem. 2010;2:121. doi: 10.4155/fmc.09.148. [DOI] [PubMed] [Google Scholar]
- 6.RR Deore, JW Chern. Curr Med Chem. 2010;17:3806. doi: 10.2174/092986710793205471. [DOI] [PubMed] [Google Scholar]
- 7.WJ Watkins, et al. Curr Opin Drug Discov Devel. 2010;13:441. [PubMed] [Google Scholar]
- 8.RF Schinazi, et al. Handb Exp Pharmacol. 2009;189:25. [Google Scholar]
- 9.R Flisiak, A Parfieniuk. Expert Opin Investig Drugs. 2010;19:63. [Google Scholar]
- 10.BC Pearce, et al. J Chem Inf Model. 2009;49:1797. doi: 10.1021/ci900073k. [DOI] [PubMed] [Google Scholar]
- 11.A Fiser, A Sali. Methods Enzymol. 2003;374:461. doi: 10.1016/S0076-6879(03)74020-8. [DOI] [PubMed] [Google Scholar]
- 12. http://www.uniprot.org/uniprot/O39929.
- 13. http://www.uniprot.org/blast/?about=O39929[2418-3008.
- 14.G Wolber, T Langer. J Chem Inf Model. 2005;45:160. doi: 10.1021/ci049885e. [DOI] [PubMed] [Google Scholar]
- 15.S Boyd, et al. Chemistry World. 2006;3:69. [Google Scholar]
- 16.SH Kim, et al. Bioorg Med Chem Lett. 2008;18:4181. doi: 10.1016/j.bmcl.2008.05.083. [DOI] [PubMed] [Google Scholar]
- 17.T Cheeseright, et al. Expert Opinion on Drug Discovery. 2007;2:131. doi: 10.1517/17460441.2.1.131. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.