

Introduction
Electrocardiographic alternans, a phenomenon of beat-to-beat oscillation in electrocardiographic waveforms, was first described by Hering in 1908 1. Much of the interest in the alternans phenomenon has focused on alternans during the repolarization phase of the cardiac action potential (AP), also known as repolarization alternans (RA). More specifically, RA has been associated with an increased risk for malignant ventricular arrhythmias and sudden cardiac death (SCD) across a wide range of pathophysiological conditions including both ischemic and non-ischemic congestive heart failure with impaired left ventricle ejection fraction (LVEF) and recent myocardial infarction (MI) 2, 3. Cardiac alternans can also be produced in structurally normal hearts under conditions of chronotropic stimulation 4, 5 or significant metabolic stress 6.
Given that several comprehensive review papers 7-9, 10 have been published on the mechanisms of RA and the clinical risk stratification aspects of microvolt T-wave alternans (MTWA) testing, in this manuscript, we have attempted to present a novel framework for how an “appropriate” substrate and an “appropriate” trigger event may synergistically contribute to the mechanisms generating cardiac alternans from the cellular to the whole heart level, and propose novel aspects of the use of RA to guide therapy.
Mechanisms of Alternans in Isolated Myocytes
Two major hypotheses have been developed to explain the alternans phenomenon at the cellular level. The first hypothesis suggests that alternation in sarcolemmal currents, membrane voltage and AP morphology leads to beat-to-beat fluctuations in intracellular calcium concentration. In support of this hypothesis, it has recently been shown that the modulation of sarcolemmal Ca2+ 11 and K+ 12, 13 currents based on changes in AP morphology 14 has a significant effect on the stability of Ca2+ handling processes and the transition to stable alternans 15,16 (Figure 1A). In contrast, the second major hypothesis suggests that alternation of intracellular calcium concentration ([Ca2+]i) is the primary event which then secondarily leads to alternans of membrane voltage and AP morphology 6, 14, 18-23. According to the second hypothesis, [Ca2+]i alternans can result from stress-induced 5, 18 perturbations in any number of Ca2+ transport processes including Ca2+ entry into the cytoplasm 13, recovery of ryanodine receptors (RyRs) from inactivation, triggering of sarcoplasmic reticulum (SR) Ca2+ release 6, 19, SR Ca2+ uptake 24, intra-SR Ca2+ redistribution 25, 26 and linking of intracellular Ca2+ handling to surface membrane voltage 14 alternans 17 (Figure 1B). The mechanisms that give rise to cardiac alternans may reside anywhere along this multi-step process of intracellular calcium cycling. A preponderance of recent data has emerged in support of the second hypothesis, suggesting the primacy of perturbations in Ca2+ handling processes as the fundamental event in the genesis of cellular alternans.
Figure 1. Illustration of [Ca2+]i and AP alternans 16, 17.
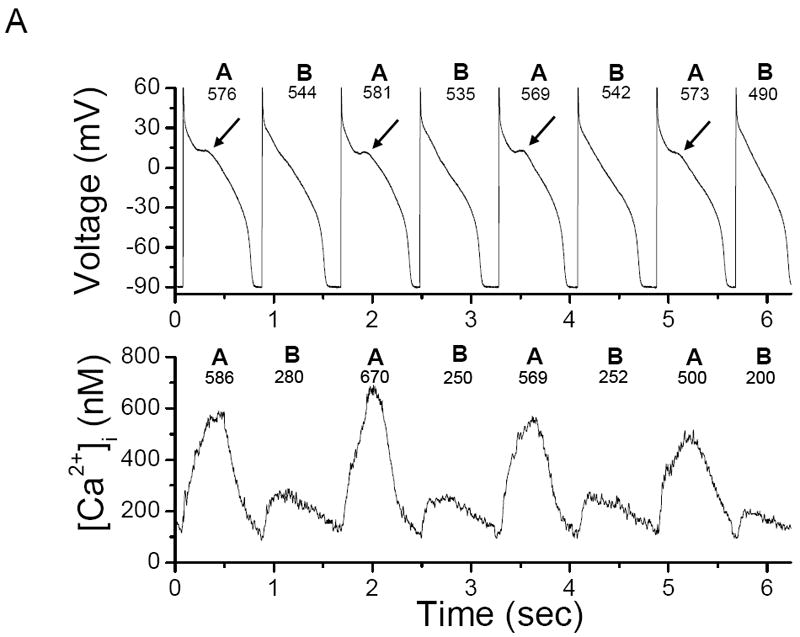
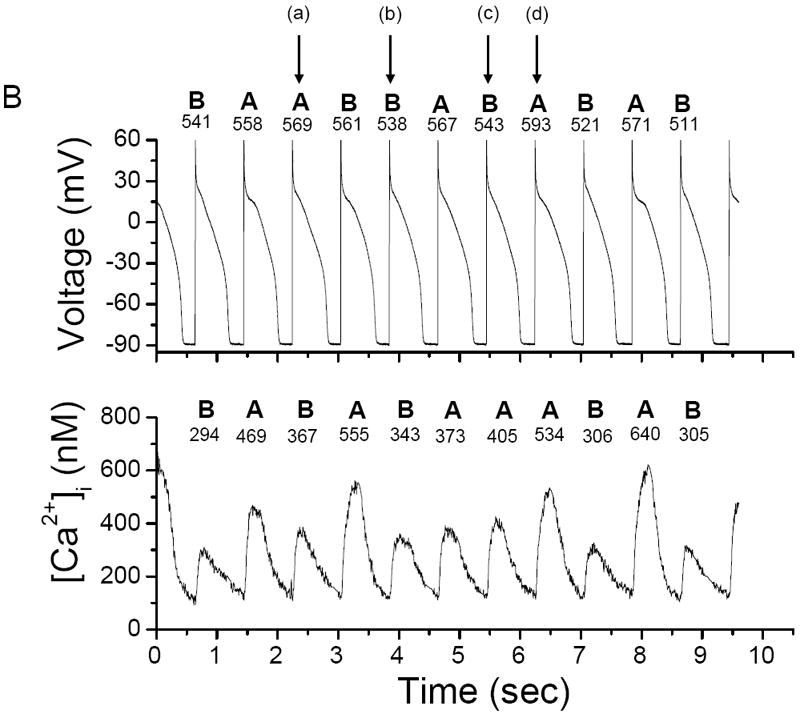
(A) Representative example of concordant [Ca2+]i and AP alternans in a left ventricular canine myocyte stimulated every 0.8 sec. The arrows indicate a sub-threshold early after-depolarization; (B) representative example of phase transitions (a)-(d) between [Ca2+]i and AP alternans obtained during the same data record as Figure 1A. In-phase (concordant) [Ca2+]i and AP alternans leads to out-of-phase (discordant) alternans (a) and (c) and back again to in-phase alternans (b) and (d). “A” and “B” denote large and small [Ca2+]i or long and short APD respectively; the peak [Ca2+]i and APD for these beats are also shown.
Among the many steps involved in calcium cycling, alternation of calcium entry into the cell via incomplete recovery from inactivation of the L-type calcium channel (ICa,L) could theoretically lead to [Ca2+]i alternans 13, 27. However, a number of studies have demonstrated that peak ICa,L is unchanged during alternans 6, 19, 28, 29, and equally importantly, ICa,L has been shown to be unaltered in myocytes from diseased hearts 30, making this a less likely mechanism for the lower alternans threshold observed in the failing heart. Furthermore, alternans of [Ca2+]i can be elicited in a high-frequency stimulated myocyte during AP-clamp with similar AP morphology20, also suggesting that the Ca2+ influx trigger of calcium-induced-calcium-release (CICR) is not the primary event in inducing alternans. The use of small depolarizing pulses 28 to induce alternans may account for alternans encountered at very high stimulation frequencies when most of the L-type Ca2+ channels are unavailable, and thus provide a plausible explanation for the presence of alternans in the normal heart at unusually high stimulation frequencies 4, 5.
Beat-to-beat fluctuations in sarcoplasmic reticulum Ca2+ content have also been implicated as a potential mechanism for alternans. Sarcoplasmic reticulum Ca2+ measurements made during alternans using the indirect approach of measurement of the caffeine-evoked Na+/Ca2+ exchanger (NCX) current have suggested that SR Ca2+ alternates 17, 28, 31. However, others have shown that while [Ca2+]SR exerts a major influence on SR Ca2+ release, beat-to-beat alternation in [Ca2+]SR is not required for [Ca2+]i alternans to occur 6, 29.
The rate of recovery of the RyR from a refractory (adapted or inactivated) state is another step in the calcium cycling machinery that may give rise to chronotropically induced alternans. With increased steepness of the released Ca2+-SR Ca2+ content relationship, as may occur in diseased hearts 30, small changes in [Ca2+]SR should result in large changes in the beat-to-beat [Ca2+]i, even for a constant ICa,L trigger 32, 33. As such, a large [Ca2+]i would be produced when the [Ca2+]SR is relatively high and a disproportionately small [Ca2+]i when the [Ca2+]SR content is relatively low. A large [Ca2+]i would then cause enhanced Ca2+ mediated L-type current inactivation, thus suppressing Ca2+ entry, as well as enhanced Ca2+ extrusion from the myocyte via the NCX 16, 17, all of which results in a lower SR Ca2+ content and hence lower [Ca2+]i on the next beat. The lower [Ca2+]i then results in decreased Ca2+ mediated L-type current inactivation and reduced Ca2+ extrusion through the NCX, leading to increased SR Ca2+ content and a return to the higher [Ca2+]i on the following beat (Figure 2). This sequence sets the stage for concordant cellular alternans between [Ca2+]i and membrane voltage/APD such that both oscillate in-phase (i.e. large [Ca2+]i corresponds to a long APD and vice versa).
Figure 2. Cellular calcium content and alternans threshold.
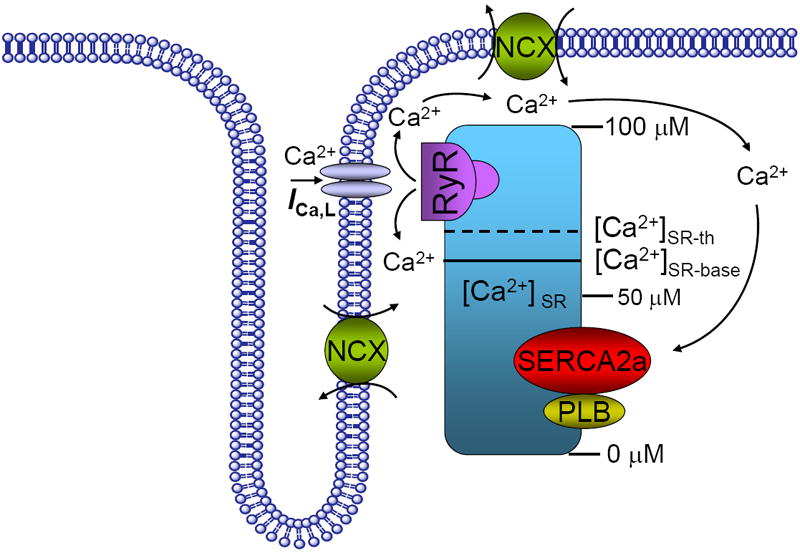
Schematic diagram of the effect of the SR Ca2+ content on a proposed model for cellular alternans. Ca2+ cycling though calcium-induced-calcium-release (CICR) includes the L-type Ca2+ channel, the SR Ca2+ ATPase pump (SERCA2a), phospholamban (PLB), the ryanodine receptor (RyR) channel and the Na+/Ca2+ exchanger (NCX). The solid line shows the SR Ca2+ baseline ([Ca2+]SR-base) and the dashed line shows the threshold SR Ca2+ ([Ca2+]SR-th) content at which Ca2+ release occurs. In the normal heart, CICR is manifested by an operational baseline of [Ca2+]SR that is lower than the threshold to trigger spontaneous Ca2+ release. However, high stimulation frequency or β-adrenergic stimulation results in SR Ca2+ overload that raises the SR Ca2+ baseline level above the threshold such that spontaneous sub-threshold Ca2+ release may occur.
While the use of small depolarizing pulses to induce alternans 28 may differ significantly from the often encountered chronotropic induction of alternans, the biphasic rise in [Ca2+]i 16 has been attributed to an initial steep rise in activation of the RyRs, while the second slower phase has been attributed to wave like propagation. We 16, 17 and others 34 have ascribed this secondary slower phase to secondary RyR openings. In computer simulations, we have shown that in isolated myocytes, elevated SR Ca2+ content results in both aberrant SR Ca2+ release and [Ca2+]i alternans, and also gives rise to an inward depolarizing current that results in spontaneous early after-depolarizations (sEADs) and APD prolongation which correlates directly with the magnitude and timing of the aberrant Ca2+ release. We have also shown the presence of discordant cellular alternans between [Ca2+]i and APD at the myocyte level and the importance of [Ca2+]i in defining the in- or out-of-phase relationship between experimentally obtained [Ca2+]i and AP (Figure 1B).
In aggregate, these findings support the primacy of alternation in [Ca2+]i in driving APD alternans and also in determining the presence of concordance or discordance between [Ca2+]i and AP morphology within the individual myocyte. Furthermore, experimental evidence suggests that the same Ca2+ cycling perturbations which give rise to cellular alternans also play a fundamental role in the pathogenesis of trigger events (i.e. transient β-stimulation bursts), which in concert create the necessary conditions for the establishment of cellular alternans.
Many studies have suggested that RyRs are more likely to be triggered by cytosolic Ca2+ when SR lumenal Ca2+ is elevated 35-37 and that increasing SR Ca2+ content increases spontaneous SR Ca2+ release 38 and delayed after-depolarization (DAD) amplitude towards the threshold to trigger an AP 39-42. Furthermore, triggered activity arising from DADs in response to high stimulation rates 43 or to catecholamines has been demonstrated in normal ventricular myocytes 44, experimental heart failure preparations 45 and cardiomyopathic human hearts 46. These studies provide a plausible justification for the hypothesis that SR Ca2+ “stabilization” at a sub-maximal value is the primary reason for abolishing alternans in studies in which thapsigargin and ryanodine treatment of myocytes markedly suppressed [Ca2+]i and prevented APD alternans 18, and ryanodine treatment alone abolished both tension and AP alternans in papillary muscles 25, 26.
In that context, in the normal heart, CICR is manifest by an operational baseline [Ca2+]SR that is lower than the threshold to trigger spontaneous Ca2+ release. However, high stimulation frequency or β-adrenergic stimulation may trigger SR Ca2+ overload that raises the SR Ca2+ baseline level close to or above the threshold at which spontaneous sub-threshold Ca2+ release may occur. In the diseased heart, although the baseline SR Ca2+ level is decreased, the [Ca2+]SR-threshold for RyR opening is also decreased. Although β-adrenergic responsiveness is impaired in the diseased heart 47, even a moderate residual or transient β-adrenergic responsiveness 45 may trigger spontaneous sub-threshold Ca2+ release at a lower [Ca2+]SR. The lower than normal [Ca2+]SR-threshold for RyR opening in diseased hearts may explain the presence of electrocardiographic alternans at lower heart rates than in normal hearts.
Further justification for the role of SR Ca2+ content in the genesis of alternans comes from the recent study by Xie and Weiss 34 demonstrating that under control conditions, myocytes become susceptible to Ca2+ overload during rapid pacing and that interactions between spontaneous Ca2+ waves and AP-triggered [Ca2+]i produce sub-cellular spatially discordant alternans (SDA) and even more complex sub-cellular [Ca2+]i patterns. Therefore, the genesis 43, 48 and propagation 49 of Ca2+ waves, which are in general associated with increased SR Ca2+ content through increased luminal Ca2+ sensitization of the RyR to cytosolic Ca2+ and perhaps through increased ability of cytosolic Ca2+ to activate adjacent RyR sites, may essentially reset local [Ca2+]SR 34 and give rise to sub-cellular alternans. According to this mechanism, a partially propagated Ca2+ wave triggers a gradient in SR refractoriness when the next AP occurs. In the region of the myocyte through which the Ca2+ wave has already passed, the affected SR is empty and partially refractory, thus minimizing Ca2+ release. In contrast, the region into which the Ca2+ wave has not entered causes the release of a normal amount of SR Ca2+, resulting in a spatially non-uniform [Ca2+]i. On the next beat, both [Ca2+]SR content and excitability of the refractory region will have recovered, producing a large release, therefore perpetuating the presence of sub-cellular SDA.
The presence of sub-cellular spatially discordant [Ca2+]i leads to increased dispersion of sub-cellular electrophysiologic properties and, in the setting of an appropriate trigger, may lead to an arrhythmia at the cellular level. Although sub-cellular SDA is usually preceded by sub-cellular spatially concordant alternans, under certain circumstances sub-cellular SDA may arise spontaneously 34.
In support of the concept of sub-cellular SDA, it is also possible that as SERCA2a, NCX and RyR function is dynamically regulated on a beat-to-beat basis by many metabolic and ionic factors in the microdomain of the SR 50-55, SR Ca2+ uptake and release is also dynamically changing, especially in the diseased heart 34, 45, 56, thus creating differential spatial heterogeneity of thresholds for the onset of alternans in different regions of the myocyte 34, 50. In such cases, small differences of SR Ca2+ content in different parts of the myocyte may exist under basal conditions and these differences may be amplified once the steepness of the relationship between Ca2+ release and SR Ca2+ content begins to rise.
In summary, these data suggest that in the diseased heart, cellular alternans requires a trigger event (such as increased β-stimulation or a Ca2+ wave) and an appropriate sub-cellular substrate to develop. Increasing the probability of RyR opening alone does not produce arrhythmogenic Ca2+ release due to an accompanying decrease in SR Ca2+ content. β-adrenergic stimulation increases SR Ca2+ content and thereby allows the increased RyR open probability to produce Ca2+ release 56. A trigger event alone may be sufficient to induce alternans in the normal heart, however, it requires supra-physiologic heart rates in order to create a heterogeneous (fragmented) sub-cellular Ca2+ release profile. In the diseased heart, however, perturbations in the intracellular calcium cycling machinery create a sufficiently heterogeneous sub-cellular substrate leading to development of alternans at lower heart rates (Figure 3) and predisposing to arrhythmogenesis 57.
Figure 3. Theoretical paradigm for differential mechanisms of alternans in normal and diseased hearts.
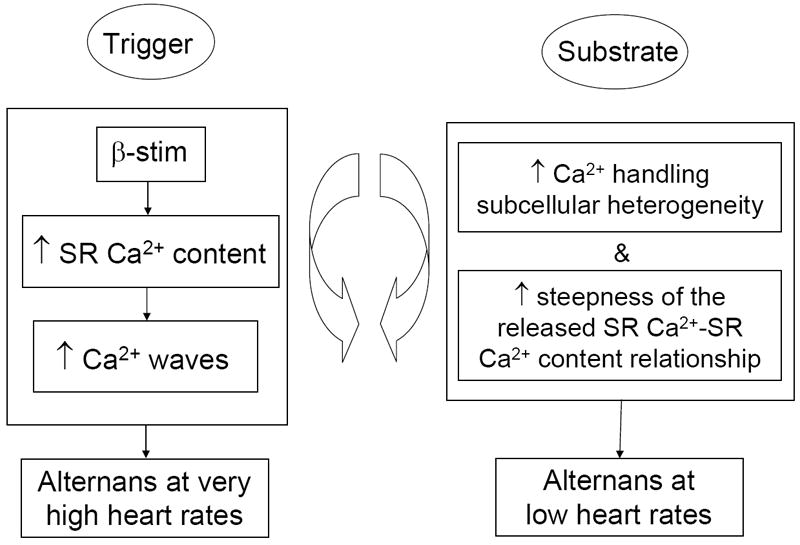
In the diseased heart, cellular alternans requires a trigger event and the appropriate substrate to develop. A trigger event alone is sufficient to induce alternans even in the normal heart, however, it requires supra-physiologic heart rates. In the diseased heart, however, the presence of an appropriate sub-cellular substrate makes more favorable the conditions for alternans development, and thus the lower heart rate alternans onset.
Mechanisms of Alternans in the Intact Heart
In a manner analogous to sub-cellular spatially concordant and discordant [Ca2+]i alternans, APD alternans at the tissue or whole heart level can also be spatially concordant or discordant (for definitions see Table).
Table.
Definitions of cardiac alternans
|
Early work has demonstrated significant variation across species in the ability to induce alternans and has also demonstrated that APD alternans is more easily induced at lower temperatures 58, which tend to prolong APD, therefore suggesting a primary role for membrane voltage dynamics in alternans at the tissue level. Subsequently, Weiss et al9 in computer simulations have shown that at the cellular level, steep APD restitution (the relationship between APD and the previous diastolic interval) slope and [Ca2+]i cycling dynamics cause the APD and [Ca2+]i to alternate. They have also demonstrated that at the tissue level, additional factors, such as conduction velocity restitution and ectopic beats, promote spatially discordant alternans. However, despite the demonstration that sustained APD alternans occurs when the APD restitution slope is >1 at a given cycle length, experimental evidence indicates that the onset of APD alternans is primarily attributable to an instability in [Ca2+]i cycling dynamics rather than steep APD restitution 9, 59. Voltage clamp experiments in isolated myocytes 20 have demonstrated that [Ca2+]i exhibits alternans despite a constant beat-to-beat AP (voltage) waveform, suggesting that APD alternans is typically driven by [Ca2+]i dynamics and not by voltage dynamics (i.e. steep APD restitution slope). In both isolated ventricular myocytes 18 and intact tissue 60, the onset of APD alternans occurred at a constant cycle length at which APD restitution slope was still considerably <1 and interventions that suppressed [Ca2+]SR cycling eliminated AP alternans irrespective of the APD restitution 18. As such, studies from both isolated myocytes and intact tissue suggest a primary role for perturbations in calcium cycling processes in the genesis of APD alternans in the whole heart.
However, whether the same mechanisms give rise to APD alternans under all circumstances, and whether the presence of alternans are necessarily always reflective of a proarrhythmic substrate, remains an area of controversy 57. The presence of discordant alternans and ventricular arrhythmias in a pacing-induced model in the guinea pig 22, a species believed to be highly resistant to alternans 58, suggests that chronotropically induced alternans may generate a non-specific pro-arrhythmic substrate. In contrast to pacing-induced alternans, discordant alternans induced in the setting of acute ischemia 61-63 or heart failure 64 appears to be primarily due to sub-cellular [Ca2+] cycling perturbations and is believed to represent a truly arrhythmogenic substrate. These data further support the hypothesis that the further the diseased state of the heart, the higher the probability of inducing alternans with progressively smaller trigger events (i.e. at lower heart rates resulting from small, transient bursts of β-adrenergic stimulation).
Regardless of the method used to induce alternans, the emergence of discordant APD alternans (reflecting two adjacent areas of the myocardium that oscillate with opposite phase) appears to be a fundamental step in the development of an arrhythmogenic substrate. Studies in normal hearts using optical mapping techniques have shown that discordant AP alternans is associated with a state of marked cardiac electrical instability, as evidenced by the fact that ventricular fibrillation is always preceded by discordant, but never by concordant, APD alternans 5. This unstable electrical substrate is consistently induced at a critical heart rate threshold and is largely independent of the pacing site 5, suggesting that it is caused by heterogeneities of cellular repolarization properties and not heterogeneous propagation delay. Interestingly, in this study, alternans most commonly involved the slope of the AP plateau and the onset of final repolarization, timing during CICR that coincides with the timing of aberrant RyR release during alternans observed by our group 16 and others 19.
Recently a two-photon confocal imaging study in the intact rat ventricle 65 has shown that the spatial distribution of [Ca2+]i alternans within the myocyte is time-dependent. Specifically, areas that mark the boundaries between regions of the myocyte that are out of phase during alternans can drift within the myocyte. These phase-mismatched myocyte regions are essentially driven by the myocyte membrane potential, defined by a spatial average potential of all myocytes within the electrotonic space constant, and thus providing a spatial constraint to the region of discordant alternans. Furthermore, the same study 65 has shown that rapid pacing synchronized Ca2+ waves in a sufficient mass of neighboring myocytes to cause DADs at the tissue level. In contrast, sporadic Ca2+ waves in individual myocytes at slow rates had no effect on membrane potential due to source-sink mismatch. Therefore, sub-cellular heterogeneities in [Ca2+] likely play an important role in the genesis of triggered activity (i.e. EADs and DADs) which may trigger the onset of an arrhythmia in presence of an appropriate substrate 34, 65, 66. It is also conceivable that if myocytes in a region of tissue synchronously develop Ca2+ waves 65, the amplitude and the phase of APD alternans in that region may change relative to the surrounding tissue, thus increasing dispersion of APD and directly contributing to the development of the arrhythmogenic substrate. It should be noted, however, that the precise relationship between discordant sub-cellular [Ca2+] alternans and APD alternans in the whole heart remains to be fully elucidated and the presence of bidirectional coupling (between [Ca2+]i and membrane voltage) 57 adds significant complexity to these dynamic interactions.
Building on the premise that sub-cellular [Ca2+] alternans contributes to APD alternans at the tissue level, it is conceivable that following cardiac “injury”, during the remodeling phase of the heart, the compensatory increase in β-adrenergic stimulation results in progressively increased SR Ca2+ content and a higher probability of inducing alternans. Although in end-stage heart failure the loss of β-adrenergic responsiveness is almost complete 47, in moderate cardiomyopathy, it is likely that residual β-adrenergic responsiveness results in higher [Ca2+]SR content and spontaneous SR Ca2+ release 45. As the heart transitions from the compensatory phase to clinical heart failure, cardiac remodeling progresses to the point that the slope of the released SR Ca2+ -SR Ca2+ content relationship is steep enough that despite the loss of β-adrenergic responsiveness 47, transient/residual β-adrenergic responsiveness 45 may result in higher [Ca2+]SR content, increased incidence of fractionated and aberrant SR Ca2+ release and Ca2+ waves, and higher probability of alternans occurrence.
In summary, it appears that AP alternans begins in a localized area in the heart and gives rise to micro-volt level alternans on the surface electrocardiogram 67. When this region of AP alternans extends to a significant portion of the myocardium (such that it is large enough to overcome the 3-dimensional current sink problem) and becomes sufficiently synchronous, it can then be seen on the surface electrocardiogram as milli-volt level alternans 68. Localized alternation in APD in turn is associated with delayed recovery on an every other beat basis, resulting in spatial dispersion of recovery, wavebreak and setting the stage for the development of re-entry and arrhythmia on-set (Figure 4) 22, 69, 70.
Figure 4. Functional relationship of alternans and re-entry.

Localized action potential alternans is manifested as repolarization alternans on the electrocardiogram. Localized regions of tissue exhibiting action potential alternans are associated with delayed recovery on an every other beat basis. These tissue areas of delayed recovery may lead to wavebreak and the development of reentry.
Clinical Relevance of Repolarization Alternans
Repolarization Alternans and Arrhythmia Susceptibility
The paradigm that repolarization alternans arises from perturbations in calcium cycling within the individual myocyte and the critical role of both substrate and triggers in the pathophysiology of RA, as delineated in the preceding discussion, has important clinical implications. To date, RA has been most commonly encountered in the clinical setting through the use of MTWA testing to predict the risk of ventricular tachyarrhythmic events (VTE) and SCD 71. A positive MTWA test result has been associated with a significantly heightened risk for SCD during medium and long-term follow-up across a wide range of clinical settings including ischemic 72 and non-ischemic 73 cardiomopathy and structural heart disease with preserved left ventricle EF 74.
More recently, prospective studies assessing the prognostic utility of MTWA testing in cohorts where a large percentage of patients are implanted with prophylactic implantable cardioverter-defibrillators (ICDs) 75, 76 have suggested that MTWA testing is not as good a predictor of “appropriate” ICD therapy as it is a predictor of VTE/SCD in patients without ICDs. This observation has been attributed to the fact that many “appropriate” ICD therapies treat arrhythmias that would have self-terminated or that ICDs may induce arrhythmias that they subsequently treat 77-79. To overcome this cofounding factor, we have recently shown that in a pooled cohort of 2883 patients without ICDs, a negative MTWA test in patients with LVEF ≤ 35% predicts a very low annual risk for SCD, while a positive MTWA test predicts a significantly heightened risk of SCD, both in patients with LVEF ≤ or > 35% 80. If confirmed in prospective studies, these findings may have important implications for refining primary prevention ICD treatment algorithms.
These clinical observations also demonstrate that an increased magnitude of RA is closely associated with the substrate that gives rise to malignant ventricular arrhythmias 9, 59, 81 and that clinical heart failure significantly lowers the heart rate threshold to induce ventricular alternans 64, 82. Other lines of evidence suggest that RA may also play an important role in the pathogenesis of atrial arrhythmias 83, a setting where the paradigm of substrate and triggers (i.e. pulmonary vein potentials) may have particular relevance. However, these observations do not necessarily prove that RA plays a causative role in the genesis of arrhythmias or that suppressing RA would be a viable therapeutic target. Although differentiating association from causation in the clinical setting can be challenging, several lines of clinical evidence do lend support to a causative role for RA in the genesis of cardiac arrhythmias. Analysis of ambulatory body-surface electrograms (Holter monitors) from patients with various forms of heart disease has demonstrated a sharp upsurge in both alternans and non-alternans periodicities (measured by time-domain techniques) within the minutes prior to spontaneous VTE 84, 85. These studies demonstrate that non-alternans periodicities such as T-wave lability, a T-wave oscillation pattern that does not follow an alternans-like pattern, may also precede VTEs 86, 87. However, in contrast to clinical MTWA testing utilizing frequency-domain techniques, the medium and long-term prognostic significance of heightened non-alternans periodicities has not been as well validated.
Analysis of intracardiac electrograms from ICD leads has also demonstrated a sharp increase in RA magnitude immediately prior to spontaneous ventricular arrhythmias 88-90. However, a similar upsurge in RA has not been observed prior to induced ventricular arrhythmias or preceding inappropriate ICD shocks 88, suggesting that the presence of increased RA magnitude is not just a by-product of a ventricular arrhythmia or a consequence of an ICD shock. Simultaneous measurement of RA from body-surface and intracardiac electrograms by our group 91 and others 92 has shown a high degree of correlation suggesting that these measurements are detecting the same electrical phenomenon.
The mechanism(s) linking RA and arrhythmogenesis have been explored by Kuo et al93 who have shown that increased dispersion of repolarization (DR) is an important condition for the development of reentrant arrhythmias and Chinushi et al 94, 95 who have shown that increased DR is associated with VTE and concordant or discordant alternans (DR is greater at sites of discordant vs. concordant alternans). Numerous experimental 22, 23, 96, 97 and computational 98-100 studies have demonstrated that APD alternans can provide the substrate for reentry and support the notion that beyond medium and long-term prognosis, heightened RA is also an important short-term predictor of arrhythmia susceptibility. Although the presence of discordant APD alternans leading to wavebreak and reentry (also known as the multiple wavelet hypothesis) has emerged as a major model to explain the pathogenesis of VTE, it is important to note that other overlapping models have also been proposed including the focal source hypothesis, in which wavebreak represents a distant epiphenomenon and is not necessarily required to sustain VF. Evidence to support both types of fibrillatory activity may be seen in the same heart and both may be relevant clinically 101 and the extent to which these competing models may have clinical therapeutic implications remains to be defined.
In aggregate, clinical data suggest that the heart either passes through a state of heightened RA on the way to VT/VF or heightened RA occurs in close conjunction with developing VTE 22, 23. In either scenario, these findings suggest that detecting significantly elevated levels of RA may serve as an important short-term predictor of impending arrhythmias and also raise the possibility of using upstream therapies to abort VT/VF prior to arrhythmia onset.
Therapeutic Implications
The ability to detect heightened levels of RA from implantable intracardiac devices opens the door to the possibility of delivering upstream therapy to suppress RA and prevent the development of a favorable substrate for arrhythmogenesis. Upstream therapy also has the important potential benefit of preventing the need for ICD shocks, which have an adverse impact on quality of life and may also have a detrimental effect on heart failure disease progression102.
The concept of upstream therapy depends on the ability to detect RA with a high degree of sensitivity. Repolarization alternans in vivo is known to be a spatially and temporally heterogeneous phenomenon 103 and therefore, any attempt to suppress RA is predicated on the ability to accurately detect alternans regardless of where in the heart it originates. Our group has recently identified a novel lead configuration for the optimal spatio-temporal detection of intra-cardiac repolarization alternans 91. To examine which intracardiac lead combination is most sensitive for RA detection, in Figure 5 we plot the probability that a far-field bipolar intracardiac lead configuration is positive for RA, given that at least one intracardiac far-field lead is positive, for each of a right-ventricular (RV), coronary sinus (CS), left-ventricular (LV), epicardial (EPI), and triangular RV-CS far-field intracardiac lead configuration. When an intracardiac lead is positive, the probability that a triangular RV-CS lead is positive is 85.5%, greater than any other intracardiac lead, suggesting that this lead configuration may provide an optimal approach for intracardiac RA detection. The use of an RV-CS lead configuration also has important clinical applicability since many currently utilized intracardiac devices already have RV and CS leads (i.e. cardiac resynchronization therapy platforms).
Figure 5. Intracardiac RA detection.

Probability that a far-field bipolar intracardiac lead is positive for RA, given that at least one intracardiac far-field lead is positive for RA, for each of the RV, CS, LV, EPI, and triangular RV-CS far-field intracardiac lead configurations 91. The RV-CS positive percentage was significantly (*) larger than for the RV configuration (p=0.040), the CS configuration (p=0.004), and the LV configuration (p=0.035), but not for the EPI configuration (p=0.270).
Following detection of heightened RA, electrical therapy has been proposed as a means of suppressing RA and preempting the development of a potentially arrhythmogenic substrate. The use of electrical therapy for this purpose draws on the experience with the use of pace termination of ventricular arrhythmias. Electrical therapy as a means of terminating ventricular arrhythmias 104-113 may result in one of several outcomes: (i) termination of reentry and VTE, (ii) changes in the shape and/or position of the center of the activity and induction of different reentrant waveforms or a focal pattern of repetitive activation, (iii) changes in the “exit” pathway or in the direction of the activity, and (iv) resetting of the activity and persistence of the same reentry.
In an analogous manner, it is conceivable that appropriately delivered pacing stimuli may suppress/terminate RA and abort reentry, thus preventing VT/VF. Although detecting an upsurge in magnitude of RA immediately prior to the onset of ventricular arrhythmias is suggestive of a causative role for RA in arrhythmogenesis, definitive proof of causation requires clear demonstration that suppressing RA prevents arrhythmias. Preclinical studies have demonstrated the feasibility of suppressing RA with dynamic pacing protocols that can be modulated based on real-time measurements of APD114, 115 with the result of suppressing RA and re-stabilizing the myocardial substrate. However, reproducing these findings in the whole heart has been limited by the inherent spatial and temporal variability of RA as it occurs in situ.
Our group has recently developed a method for in situ dynamic control of RA in a swine model 116. This method is based on the premise that adaptive sub-threshold pacing impulses delivered during the absolute refractory period may be capable of controlling RA. In this model, RA is induced via an R-wave triggered pacing protocol which delivers impulses on an every other beat basis and hence leads to a significant rise in RA magnitude as detected by an increase in Kscore (for a description of the use of the Kscore to quantify RA magnitude, please see reference 117). The increase in Kscore is detectable from both intracardiac (right ventricle, left ventricle, coronary sinus) and body-surface (lead II) electrodes. Following induction of significant RA, triggered pacing stimuli delivered from a remote location on alternate beats can be used to suppress RA. In this example, RA is induced by pacing from the RV12 electrode on even beats (Figure 6, panel B) and then suppressed by pacing from RV56 on odd beats (Figure 6, panel C). Other permutations of even and odd beat pacing and changes in the polarity of triggered impulses can be used to induce and suppress alternans with a high degree of fidelity (Figure 6, panels D-F).
Figure 6. Intracardiac RA control.
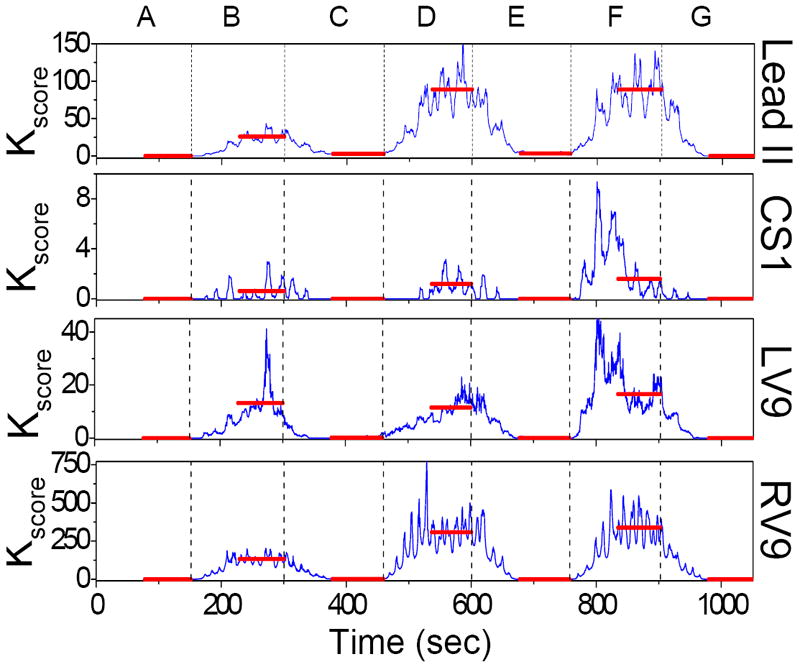
Repolarization alternans (RA) control during which RA is induced via RV12 triggered pacing (-7 mA, 30 ms width, 30 ms coupling) and suppressed via RV56 triggered pacing (-7 mA, 30 ms width, 30 ms coupling). A: baseline, B: RV12 even beats, C: RV12 even & RV56 odd, D: RV12 even & RV56 odd with polarity flip, E: RV12 even & RV56 even with polarity flip, F: RV12 even & RV56 even, G: RV12 every beat. Interventions B, D, and F induce RA, while C and E suppress RA. Transitions occur at times marked by dashed vertical black lines, while the red horizontal lines during each intervention indicate the mean Kscore (see reference 117 for a description of the method used to estimate Kscore).
Extension of these findings raises the possibility of incorporating adaptive pacing protocols into implantable devices such that if the device detects an unstable myocardial substrate (as evidenced by heightened RA magnitude), the adaptive pacing protocol would be activated to deliver electrical therapy to re-stabilize the electrical substrate so that even if a trigger event occurred (i.e. a PVC), that trigger would no longer encounter a vulnerable electrical substrate and the onset of arrhythmia would be prevented. The adaptive pacing protocol could be terminated when the RA magnitude falls below a predetermined threshold.
Beyond adaptive pacing protocols, detection of RA by implantable devices may also be coupled to other forms of suppressive therapy. For instance, there is significant interest in coupling micro-electromechanical systems (MEMS) to implantable devices to facilitate localized delivery of pharmacologic agents for treating various aspects of chronic heart failure (i.e. neurohormonal antagonists, diuretics, anti-arrhythmic agents) 118. Several classes of pharmacologic agents have been demonstrated to suppress RA and prevent ventricular arrhythmias including β-blockers 119, 120 and certain sodium channel blockers such as ranolazine 121. It’s conceivable that timely and potentially localized delivery of such agents may be capable of suppressing RA and re-stabilizing the electrical substrate. It should be noted, however, that the hypothesis that suppression of RA in vivo can be used to preempt arrhythmia on-set remains to be proven. An important proof of concept study recently demonstrated that the use of SERCA2a adenoviral gene transfer in a guinea pig model resulted in a 4-fold reduction in susceptibility to alternans-mediated ventricular arrhythmias 122, and has opened the door to other potential therapeutic approaches for the suppression of RA and prevention of VTEs.
The potential to couple detection of elevated RA magnitude and delivery of therapy within an implantable device offers a real opportunity for developing iterative and closed-loop systems to prevent arrhythmias.
Conclusions
The generally accepted paradigm of requiring both substrate and triggers for the genesis of ventricular arrhythmias 123 lends significant complexity to understanding the underlying mechanisms that give rise to life threatening arrhythmias and SCD. Electrocardiographic alternans-type oscillations represent a response of the ventricle at the first sub-harmonic of the driving frequency (the mean heart rate) which might be viewed as the first bifurcation in the pathway to ventricular fibrillation 124. This review presents a contemporary view of the mechanisms underlying [Ca2+]i and AP alternans in the normal and diseased heart.
The prevailing hypothesis of repolarization alternans is that dynamic sub-cellular perturbations in intracellular Ca2+ homeostatic mechanisms occurring on a beat-to-beat basis give rise to [Ca2+]i alternans, which in turn gives rise to APD alternans and electrocardiographic alternans. At the whole heart level, the transition from concordant to discordant APD alternans is associated with a state of significantly heightened cardiac electrical instability due to the fact that discordant APD alternans leads to increased spatial dispersion of refractoriness and wavefront fractionation and eventually to the onset of reentrant arrhythmias. Enhanced understanding of the pathophysiologic processes which give rise to alternans at the cellular and whole heart level may have important implications for pharmacologic and/or electrical therapeutic approaches to prevent ventricular arrhythmias and sudden cardiac death.
Acknowledgments
Funding Sources The work was supported by a Scientist Development Grant (#0635127N), by National Institute of Aging (NIA) grant 1R21AG035128 and NIH grant 1RO1HL103961. This work was also supported by a Fellowship and a Science Award from the Center for Integration of Medicine and Innovative Technology (CIMIT), the Deane Institute for Integrative Research in Atrial Fibrillation and Stroke and the Cardiovascular Research Society.
Footnotes
Conflict of Interest Disclosures None
References
- 1.Hering HE. Das Wesen des Herzalternans. Munchen Med Wchenshr. 1908;4:1417–1421. [Google Scholar]
- 2.Armoundas AA, Tomaselli GF, Esperer HD. Pathophysiological basis and clinical application of T Wave alternans. JACC. 2002;40:207–217. doi: 10.1016/s0735-1097(02)01960-5. [DOI] [PubMed] [Google Scholar]
- 3.Armoundas AA, Hohnloser SH, Ikeda T, Cohen RJ. Can microvolt T-wave alternans testing reduce unnecessary defibrillator implantation? Nat Clin Pract Cardiovasc Med. 2005;2:522–528. doi: 10.1038/ncpcardio0323. [DOI] [PubMed] [Google Scholar]
- 4.Turitto G, Caref EB, El-Attar G, Hellal M, Mohamed A, Pedalino RP, El-Sherif N. Optimal target heart rate for exercise-induced T-wave alternans. ANE. 2001;6:123–128. doi: 10.1111/j.1542-474X.2001.tb00096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Laurita KR, Pastore JM, Rosenbaum DS. How restitution, repolarization, and alternans form arrhythmogenic substrates: insights from high-resolution optical mapping. In: Zipes DP, Jalife J, editors. Cardiac Electrophysiology: From Cell to Bedside. 2. Philadelphia, PA: W.B.Saunders; 1999. pp. 239–248. [Google Scholar]
- 6.Hüser J, Wang YG, Sheehan KA, Cifuentes F, Lipsius SL, Blatter LA. Functional coupling between glycolysis and excitation-contraction coupling underlies alternans in cat heart cells. J Physiol (Lond) 2000;524(Pt 3):795–806. doi: 10.1111/j.1469-7793.2000.00795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clusin WT. Mechanisms of calcium transient and action potential alternans in cardiac cells and tissues. Am J Physiol Heart Circ Physiol. 2008;294:H1–H10. doi: 10.1152/ajpheart.00802.2007. [DOI] [PubMed] [Google Scholar]
- 8.Narayan SM. T-wave alternans and the susceptibility to ventricular arrhythmias. J Am Coll Cardiol. 2006;47:269–281. doi: 10.1016/j.jacc.2005.08.066. [DOI] [PubMed] [Google Scholar]
- 9.Weiss JN, Karma A, Shiferaw Y, Chen PS, Garfinkel A, Qu Z. From pulsus to pulseless: the saga of cardiac alternans. Circ Res. 2006;98:1244–1253. doi: 10.1161/01.RES.0000224540.97431.f0. [DOI] [PubMed] [Google Scholar]
- 10.Cutler MJ, Rosenbaum DS. Risk stratification for sudden cardiac death: is there a clinical role for T wave alternans? Heart Rhythm. 2009;6(8 Suppl):S56–61. doi: 10.1016/j.hrthm.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mahajan A, Sato D, Shiferaw Y, Baher A, Xie LH, Peralta R, Olcese R, Garfinkel A, Qu Z, Weiss JN. Modifying L-type calcium current kinetics: consequences for cardiac excitation and arrhythmia dynamics. Biophys J. 2008;94:411–423. doi: 10.1529/biophysj.106.98590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hua F, Johns DC, Gilmour RF., Jr Suppression of electrical alternans by overexpression of HERG in canine ventricular myocytes. Am J Physiol Heart Circ Physiol. 2004;286:H2342–2351. doi: 10.1152/ajpheart.00793.2003. [DOI] [PubMed] [Google Scholar]
- 13.Fox JJ, McHarg JL, Gilmour RF., Jr Ionic mechanism of electrical alternans. Am J Physiol Heart Circ Physiol. 2002;282:H516–530. doi: 10.1152/ajpheart.00612.2001. [DOI] [PubMed] [Google Scholar]
- 14.Jordan PN, Christini DJ. Action potential morphology influences intracellular calcium handling stability and the occurrence of alternans. Biophys J. 2006;90:672–680. doi: 10.1529/biophysj.105.071340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allen DG, Orchard CH. Myocardial contractile function during ischemia and hypoxia. Circ Res. 1987;60:153–168. doi: 10.1161/01.res.60.2.153. [DOI] [PubMed] [Google Scholar]
- 16.Armoundas AA. Mechanism of abnormal sarcoplasmic reticulum calcium release in canine left-ventricular myocytes results in cellular alternans. IEEE Trans Biomed Eng. 2009;56:220–228. doi: 10.1109/TBME.2008.2003283. [DOI] [PubMed] [Google Scholar]
- 17.Armoundas AA. Discordant calcium transient and action potential alternans in a canine left-ventricular myocyte. IEEE Trans Biomed Eng. 2009;56:2340–2344. doi: 10.1109/TBME.2009.2023671. [DOI] [PubMed] [Google Scholar]
- 18.Goldhaber JI, Xie LH, Duong T, Motter C, Khuu K, Weiss JN. Action potential duration restitution and alternans in rabbit ventricular myocytes: the key role of intracellular calcium cycling. Circ Res. 2005;96:459–466. doi: 10.1161/01.RES.0000156891.66893.83. [DOI] [PubMed] [Google Scholar]
- 19.Diaz ME, Eisner DA, O’Neill SC. Depressed ryanodine receptor activity increases variability and duration of the systolic Ca2+ transient in rat ventricular myocytes. Circ Res. 2002;91:585–593. doi: 10.1161/01.res.0000035527.53514.c2. [DOI] [PubMed] [Google Scholar]
- 20.Chudin E, Goldhaber J, Garfinkel A, Weiss J, Kogan B. Intracellular Ca(2+) dynamics and the stability of ventricular tachycardia. Biophys J. 1999;77:2930–2941. doi: 10.1016/S0006-3495(99)77126-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kockskamper J, Zima AV, Blatter LA. Modulation of sarcoplasmic reticulum Ca2+ release by glycolysis in cat atrial myocytes. J Physiol. 2005;564(Pt 3):697–714. doi: 10.1113/jphysiol.2004.078782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pastore JM, Girouard SD, Laurita KR, Akar FG, Rosenbaum DS. Mechanism linking T-wave alternans to the genesis of cardiac fibrillation. Circulation. 1999;99:1385–1394. doi: 10.1161/01.cir.99.10.1385. [DOI] [PubMed] [Google Scholar]
- 23.Pastore JM, Rosenbaum DS. Role of structural barriers in the mechanism of alternans-induced reentry. Circ Res. 2000;87:1157–1163. doi: 10.1161/01.res.87.12.1157. [DOI] [PubMed] [Google Scholar]
- 24.Kameyama M, Hirayama Y, Saitoh H, Maruyama M, Atarashi H, Takano T. Possible contribution of the sarcoplasmic reticulum Ca(2+) pump function to electrical and mechanical alternans. J Electrocardiol. 2003;36:125–135. doi: 10.1054/jelc.2003.50021. [DOI] [PubMed] [Google Scholar]
- 25.Kihara Y, Morgan JP. Abnormal Cai2+ handling is the primary cause of mechanical alternans: study in ferret ventricular muscles. Am J Physiol. 1991;261(6 Pt 2):H1746–1755. doi: 10.1152/ajpheart.1991.261.6.H1746. [DOI] [PubMed] [Google Scholar]
- 26.Lab MJ, Lee JA. Changes in intracellular calcium during mechanical alternans in isolated ferret ventricular muscle. Circ Res. 1990;66:585–595. doi: 10.1161/01.res.66.3.585. [DOI] [PubMed] [Google Scholar]
- 27.Shiferaw Y, Watanabe MA, Garfinkel A, Weiss JN, Karma A. Model of intracellular calcium cycling in ventricular myocytes. Biophys J. 2003;85:3666–3686. doi: 10.1016/S0006-3495(03)74784-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diaz ME, O’Neill SC, Eisner DA. Sarcoplasmic reticulum calcium content fluctuation is the key to cardiac alternans. Circ Res. 2004;94:650–656. doi: 10.1161/01.RES.0000119923.64774.72. [DOI] [PubMed] [Google Scholar]
- 29.Picht E, DeSantiago J, Blatter LA, Bers DM. Cardiac alternans do not rely on diastolic sarcoplasmic reticulum calcium content fluctuations. Circ Res. 2006;99:740–748. doi: 10.1161/01.RES.0000244002.88813.91. [DOI] [PubMed] [Google Scholar]
- 30.Belevych AE, Terentyev D, Viatchenko-Karpinski S, Terentyeva R, Sridhar A, Nishijima Y, Wilson LD, Cardounel AJ, Laurita KR, Carnes CA, Billman GE, Gyorke S. Redox modification of ryanodine receptors underlies calcium alternans in a canine model of sudden cardiac death. Cardiovasc Res. 2009;84:387–395. doi: 10.1093/cvr/cvp246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eisner DA, Choi HS, Diaz ME, O’Neill SC, Trafford AW. Integrative analysis of calcium cycling in cardiac muscle. Circ Res. 2000;87:1087–1094. doi: 10.1161/01.res.87.12.1087. [DOI] [PubMed] [Google Scholar]
- 32.Bassani JW, Yuan W, Bers DM. Fractional SR Ca release is regulated by trigger Ca and SR Ca content in cardiac myocytes. Am J Physiol. 1995;268(5 Pt 1):C1313–1319. doi: 10.1152/ajpcell.1995.268.5.C1313. [DOI] [PubMed] [Google Scholar]
- 33.Trafford AW, Diaz ME, Sibbring GC, Eisner DA. Modulation of CICR has no maintained effect on systolic Ca2+: simultaneous measurements of sarcoplasmic reticulum and sarcolemmal Ca2+ fluxes in rat ventricular myocytes. J Physiol. 2000;522(Pt 2):259–270. doi: 10.1111/j.1469-7793.2000.t01-2-00259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xie LH, Weiss JN. Arrhythmogenic consequences of intracellular calcium waves. Am J Physiol Heart Circ Physiol. 2009;297:H997–H1002. doi: 10.1152/ajpheart.00390.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Y, Kranias EG, Mignery GA, Bers DM. Protein kinase A phosphorylation of the ryanodine receptor does not affect calcium sparks in mouse ventricular myocytes. Circ Res. 2002;90:309–316. doi: 10.1161/hh0302.105660. [DOI] [PubMed] [Google Scholar]
- 36.Gyorke I, Gyorke S. Regulation of the cardiac ryanodine receptor channel by luminal Ca2+ involves luminal Ca2+ sensing sites. Biophys J. 1998;75:2801–2810. doi: 10.1016/S0006-3495(98)77723-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ikemoto N, Ronjat M, Meszaros LG, Koshita M. Postulated role of calsequestrin in the regulation of calcium release from sarcoplasmic reticulum. Biochemistry. 1989;28:6764–6771. doi: 10.1021/bi00442a033. [DOI] [PubMed] [Google Scholar]
- 38.Satoh H, Blatter LA, Bers DM. Effects of [Ca2+]i, SR Ca2+ load, and rest on Ca2+ spark frequency in ventricular myocytes. Am J Physiol. 1997;272(2 Pt 2):H657–668. doi: 10.1152/ajpheart.1997.272.2.H657. [DOI] [PubMed] [Google Scholar]
- 39.Kass RS, Tsien RW. Fluctuations in membrane current driven by intracellular calcium in cardiac Purkinje fibers. Biophys J. 1982;38:259–269. doi: 10.1016/S0006-3495(82)84557-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Orchard CH, Eisner DA, Allen DG. Oscillations of intracellular Ca2+ in mammalian cardiac muscle. Nature. 1983;304:735–738. doi: 10.1038/304735a0. [DOI] [PubMed] [Google Scholar]
- 41.Stern MD, Kort AA, Bhatnagar GM, Lakatta EG. Scattered-light intensity fluctuations in diastolic rat cardiac muscle caused by spontaneous Ca++-dependent cellular mechanical oscillations. J Gen Physiol. 1983;82:119–153. doi: 10.1085/jgp.82.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wier WG, Kort AA, Stern MD, Lakatta EG, Marban E. Cellular calcium fluctuations in mammalian heart: direct evidence from noise analysis of aequorin signals in Purkinje fibers. Proc Natl Acad Sci U S A. 1983;80:7367–7371. doi: 10.1073/pnas.80.23.7367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pogwizd SM, Bers DM. Cellular basis of triggered arrhythmias in heart failure. Trends Cardiovasc Med. 2004;14:61–66. doi: 10.1016/j.tcm.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 44.Priori SG, Corr PB. Mechanisms underlying early and delayed afterdepolarizations induced by catecholamines. Am J Physiol. 1990;258(6 Pt 2):H1796–1805. doi: 10.1152/ajpheart.1990.258.6.H1796. [DOI] [PubMed] [Google Scholar]
- 45.Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure: Roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circ Res. 2001;88:1159–1167. doi: 10.1161/hh1101.091193. [DOI] [PubMed] [Google Scholar]
- 46.Gilmour RF, Jr, Heger JJ, Prystowsky EN, Zipes DP. Cellular electrophysiologic abnormalities of diseased human ventricular myocardium. Am J Cardiol. 1983;51:137–144. doi: 10.1016/s0002-9149(83)80024-1. [DOI] [PubMed] [Google Scholar]
- 47.Bristow MR, Ginsburg R, Minobe W, Cubicciotti RS, Sageman WS, Lurie K, Billingham ME, Harrison DC, Stinson EB. Decreased catecholamine sensitivity and beta-adrenergic-receptor density in failing human hearts. N Engl J Med. 1982;307(4):205–211. doi: 10.1056/NEJM198207223070401. [DOI] [PubMed] [Google Scholar]
- 48.Venetucci LA, Trafford AW, O’Neill SC, Eisner DA. The sarcoplasmic reticulum and arrhythmogenic calcium release. Cardiovasc Res. 2008;77:285–292. doi: 10.1093/cvr/cvm009. [DOI] [PubMed] [Google Scholar]
- 49.Lukyanenko V, Subramanian S, Gyorke I, Wiesner TF, Gyorke S. The role of luminal Ca2+ in the generation of Ca2+ waves in rat ventricular myocytes. J Physiol. 1999;518(Pt 1):173–186. doi: 10.1111/j.1469-7793.1999.0173r.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aistrup GL, Shiferaw Y, Kapur S, Kadish AH, Wasserstrom JA. Mechanisms underlying the formation and dynamics of subcellular calcium alternans in the intact rat heart. Circ Res. 2009;104:639–649. doi: 10.1161/CIRCRESAHA.108.181909. [DOI] [PubMed] [Google Scholar]
- 51.Cannell MB, Cheng H, Lederer WJ. Spatial non-uniformities in [Ca2+]i during excitation-contraction coupling in cardiac myocytes. Biophys J. 1994;67:1942–1956. doi: 10.1016/S0006-3495(94)80677-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cherednichenko G, Zima AV, Feng W, Schaefer S, Blatter LA, Pessah IN. NADH oxidase activity of rat cardiac sarcoplasmic reticulum regulates calcium-induced calcium release. Circ Res. 2004;94:478–486. doi: 10.1161/01.RES.0000115554.65513.7C. [DOI] [PubMed] [Google Scholar]
- 53.Zima AV, Kockskamper J, Mejia-Alvarez R, Blatter LA. Pyruvate modulates cardiac sarcoplasmic reticulum Ca2+ release in rats via mitochondria-dependent and - independent mechanisms. J Physiol. 2003;550(Pt 3):765–783. doi: 10.1113/jphysiol.2003.040345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Valdivia HH, Kaplan JH, Ellis-Davies GC, Lederer WJ. Rapid adaptation of cardiac ryanodine receptors: modulation by Mg2+ and phosphorylation. Science. 1995;267:1997–2000. doi: 10.1126/science.7701323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu L, Mann G, Meissner G. Regulation of cardiac Ca2+ release channel (ryanodine receptor) by Ca2+, H+, Mg2+, and adenine nucleotides under normal and simulated ischemic conditions. Circ Res. 1996;79:1100–1109. doi: 10.1161/01.res.79.6.1100. [DOI] [PubMed] [Google Scholar]
- 56.Venetucci LA, Trafford AW, Eisner DA. Increasing ryanodine receptor open probability alone does not produce arrhythmogenic calcium waves: threshold sarcoplasmic reticulum calcium content is required. Circ Res. 2007;100:105–111. doi: 10.1161/01.RES.0000252828.17939.00. [DOI] [PubMed] [Google Scholar]
- 57.Choi BR, Salama G. Simultaneous maps of optical action potentials and calcium transients in guinea-pig hearts: mechanisms underlying concordant alternans. J Physiol. 2000;529(Pt 1):171–188. doi: 10.1111/j.1469-7793.2000.00171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Spear JF, Moore EN. A comparison of alternation in myocardial action potentials and contractility. Am J Physiol. 1971;220:1708–1716. doi: 10.1152/ajplegacy.1971.220.6.1708. [DOI] [PubMed] [Google Scholar]
- 59.Narayan SM, Bayer JD, Lalani G, Trayanova NA. Action potential dynamics explain arrhythmic vulnerability in human heart failure: a clinical and modeling study implicating abnormal calcium handling. J Am Coll Cardiol. 2008;52:1782–1792. doi: 10.1016/j.jacc.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pruvot EJ, Katra RP, Rosenbaum DS, Laurita KR. Role of calcium cycling versus restitution in the mechanism of repolarization alternans. Circ Res. 2004;94:1083–1090. doi: 10.1161/01.RES.0000125629.72053.95. [DOI] [PubMed] [Google Scholar]
- 61.Lee HC, Mohabir R, Smith N, Franz MR, Clusin WT. Effect of ischemia on calcium-dependent fluorescence transients in rabbit hearts containing indo 1. Correlation with monophasic action potentials and contraction. Circulation. 1988;78:1047–1059. doi: 10.1161/01.cir.78.4.1047. [DOI] [PubMed] [Google Scholar]
- 62.Wu Y, Clusin WT. Calcium transient alternans in blood-perfused ischemic hearts: observations with fluorescent indicator fura red. Am J Physiol. 1997;273(5 Pt 2):H2161–2169. doi: 10.1152/ajpheart.1997.273.5.H2161. [DOI] [PubMed] [Google Scholar]
- 63.Konta T, Ikeda K, Yamaki M, Nakamura K, Honma K, Kubota I, Yasui S. Significance of discordant ST alternans in ventricular fibrillation. Circulation. 1990;82:2185–2189. doi: 10.1161/01.cir.82.6.2185. [DOI] [PubMed] [Google Scholar]
- 64.Bayer JD, Narayan SM, Lalani GG, Trayanova NA. Rate-dependent action potential alternans in human heart failure implicates abnormal intracellular calcium handling. Heart Rhythm. 2010;7:1093–1101. doi: 10.1016/j.hrthm.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fujiwara K, Tanaka H, Mani H, Nakagami T, Takamatsu T. Burst emergence of intracellular Ca2+ waves evokes arrhythmogenic oscillatory depolarization via the Na+- Ca2+ exchanger: simultaneous confocal recording of membrane potential and intracellular Ca2+ in the heart. Circ Res. 2008;103:509–518. doi: 10.1161/CIRCRESAHA.108.176677. [DOI] [PubMed] [Google Scholar]
- 66.Wasserstrom JA, Sharma R, Kapur S, Kelly JE, Kadish AH, Balke CW, Aistrup GL. Multiple defects in intracellular calcium cycling in whole failing rat heart. Circ Heart Fail. 2009;2:223–232. doi: 10.1161/CIRCHEARTFAILURE.108.811539. [DOI] [PubMed] [Google Scholar]
- 67.Smith JM, Clancy EA, Valeri CR, Ruskin JN, Cohen RJ. Electrical alternans and cardiac electrical instability. Circulation. 1988;77:110–121. doi: 10.1161/01.cir.77.1.110. [DOI] [PubMed] [Google Scholar]
- 68.Lewis T. Notes upon alternation of the heart. Q J Med. 1910;4:141–144. [Google Scholar]
- 69.Armoundas AA, Tomaselli GF, Esperer HD. Pathophysiological basis and clinical application of T-wave alternans. J Am Coll Cardiol. 2002;40:207–217. doi: 10.1016/s0735-1097(02)01960-5. [DOI] [PubMed] [Google Scholar]
- 70.Restivo M, Caref EB, Kozhevnikov DO, El-Sherif N. Spatial dispersion of repolarization is a key factor in the arrhythmogenicity of long QT syndrome. J Cardiovasc Electrophysiol. 2004;15:323–331. doi: 10.1046/j.1540-8167.2004.03493.x. [DOI] [PubMed] [Google Scholar]
- 71.Bloomfield DM, Hohnloser SH, Cohen RJ. Interpretation and classification of microvolt T wave alternans tests. J Cardiovasc Electrophysiol. 2002;13:502–512. doi: 10.1046/j.1540-8167.2002.00502.x. [DOI] [PubMed] [Google Scholar]
- 72.Chow T, Kereiakes DJ, Bartone C, Booth T, Schloss EJ, Waller T, Chung ES, Menon S, Nallamothu BK, Chan PS. Prognostic utility of microvolt T-wave alternans in risk stratification of patients with ischemic cardiomyopathy. J Am Coll Cardiol. 2006;47:1820–1827. doi: 10.1016/j.jacc.2005.11.079. [DOI] [PubMed] [Google Scholar]
- 73.Salerno-Uriarte JA, De Ferrari GM, Klersy C, Pedretti RF, Tritto M, Sallusti L, Libero L, Pettinati G, Molon G, Curnis A, Occhetta E, Morandi F, Ferrero P, Accardi F. Prognostic value of T-wave alternans in patients with heart failure due to nonischemic cardiomyopathy: results of the ALPHA Study. J Am Coll Cardiol. 2007;50:1896–1904. doi: 10.1016/j.jacc.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 74.Ikeda T, Yoshino H, Sugi K, Tanno K, Shimizu H, Watanabe J, Kasamaki Y, Yoshida A, Kato T. Predictive value of microvolt T-wave alternans for sudden cardiac death in patients with preserved cardiac function after acute myocardial infarction: results of a collaborative cohort study. J Am Coll Cardiol. 2006;48:2268–2274. doi: 10.1016/j.jacc.2006.06.075. [DOI] [PubMed] [Google Scholar]
- 75.Chow T, Kereiakes DJ, Onufer J, Woelfel A, Gursoy S, Peterson BJ, Brown ML, Pu W, Benditt DG. Does microvolt T-wave alternans testing predict ventricular tachyarrhythmias in patients with ischemic cardiomyopathy and prophylactic defibrillators? The MASTER (Microvolt T Wave Alternans Testing for Risk Stratification of Post-Myocardial Infarction Patients) trial. J Am Coll Cardiol. 2008;52:1607–1615. doi: 10.1016/j.jacc.2008.08.018. [DOI] [PubMed] [Google Scholar]
- 76.Gold MR, Ip JH, Costantini O, Poole JE, McNulty S, Mark DB, Lee KL, Bardy GH. Role of microvolt T-wave alternans in assessment of arrhythmia vulnerability among patients with heart failure and systolic dysfunction: primary results from the T-wave alternans sudden cardiac death in heart failure trial substudy. Circulation. 2008;118:2022–2028. doi: 10.1161/CIRCULATIONAHA.107.748962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hohnloser SH, Ikeda T, Cohen RJ. Evidence regarding clinical use of microvolt T-wave alternans. Heart Rhythm. 2009;6(3 Suppl):S36–44. doi: 10.1016/j.hrthm.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 78.Ellenbogen KA, Levine JH, Berger RD, Daubert JP, Winters SL, Greenstein E, Shalaby A, Schaechter A, Subacius H, Kadish A. Are implantable cardioverter defibrillator shocks a surrogate for sudden cardiac death in patients with nonischemic cardiomyopathy? Circulation. 2006;113:776–782. doi: 10.1161/CIRCULATIONAHA.105.561571. [DOI] [PubMed] [Google Scholar]
- 79.Germano JJ, Reynolds M, Essebag V, Josephson ME. Frequency and causes of implantable cardioverter-defibrillator therapies: is device therapy proarrhythmic? Am J Cardiol. 2006;97:1255–1261. doi: 10.1016/j.amjcard.2005.11.048. [DOI] [PubMed] [Google Scholar]
- 80.Merchant FM, Zheng H, Ikeda T, Pedretti RFE, Salerno-Uriate JA, Chow T, Chan PS, Bartone C, Hohnloser SH, Ruskin JN, Cohen RJ, Armoundas AA. Clinical utility of microvolt T-wave alternans testing in identifying patients at high or low risk of sudden cardiac death. Heart Rhythm. 2011;8:S206. doi: 10.1016/j.hrthm.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ng GA, Brack KE, Patel VH, Coote JH. Autonomic modulation of electrical restitution, alternans and ventricular fibrillation initiation in the isolated heart. Cardiovasc Res. 2007;73:750–760. doi: 10.1016/j.cardiores.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 82.Wilson LD, Jeyaraj D, Wan X, Hoeker GS, Said TH, Gittinger M, Laurita KR, Rosenbaum DS. Heart failure enhances susceptibility to arrhythmogenic cardiac alternans. Heart Rhythm. 2009;6:251–259. doi: 10.1016/j.hrthm.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Narayan SM, Bode F, Karasik PL, Franz MR. Alternans of atrial action potentials during atrial flutter as a precursor to atrial fibrillation. Circulation. 2002;106:1968–1973. doi: 10.1161/01.cir.0000037062.35762.b4. [DOI] [PubMed] [Google Scholar]
- 84.Swerdlow C, Chow T, Das M, Gillis AM, Zhou X, Abeyratne A, Ghanem RN. Intracardiac electrogram T-wave alternans/variability increases before spontaneous ventricular tachyarrhythmias in implantable cardioverter-defibrillator patients: a prospective, multi-center study. Circulation. 2011;123:1052–1060. doi: 10.1161/CIRCULATIONAHA.110.986364. [DOI] [PubMed] [Google Scholar]
- 85.Shusterman V, Goldberg A, London B. Upsurge in T-wave alternans and nonalternating repolarization instability precedes spontaneous initiation of ventricular tachyarrhythmias in humans. Circulation. 2006;113:2880–2887. doi: 10.1161/CIRCULATIONAHA.105.607895. [DOI] [PubMed] [Google Scholar]
- 86.Nemec J, Kim JJ, Gabris B, Salama G. Calcium oscillations and T-wave lability precede ventricular arrhythmias in acquired long QT type 2. Heart Rhythm. 7:1686–1694. doi: 10.1016/j.hrthm.2010.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nearing BD, Verrier RL. Progressive increases in complexity of T-wave oscillations herald ischemia-induced ventricular fibrillation. Circ Res. 2002;91:727–732. doi: 10.1161/01.res.0000038887.17976.33. [DOI] [PubMed] [Google Scholar]
- 88.Kim JW, Pak HN, Park JH, Nam GB, Kim SK, Lee HS, Jang JK, Choi JI, Kim YH. Defibrillator electrogram T wave alternans as a predictor of spontaneous ventricular tachyarrhythmias in defibrillator recipients. Circ J. 2009;73:55–62. doi: 10.1253/circj.cj-08-0311. [DOI] [PubMed] [Google Scholar]
- 89.Armoundas AA, Albert CM, Cohen RJ, Mela T. Utility of Implantable Cardioverter Defibrillator Electrograms to Estimate Repolarization Alternans Preceding a Tachyarrhythmic Event. J Cardiovasc Electrophysiol. 2004;15:594–597. doi: 10.1046/j.1540-8167.2004.03411.x. [DOI] [PubMed] [Google Scholar]
- 90.Swerdlow CD, Zhou X, Voroshilovsky O, Abeyratne A, Gillberg J. High amplitude T-wave alternans precedes spontaneous ventricular tachycardia or fibrillation in ICD electrograms. Heart Rhythm. 2008;5:670–676. doi: 10.1016/j.hrthm.2008.02.018. [DOI] [PubMed] [Google Scholar]
- 91.Weiss EH, Merchant FM, d’Avila A, Foley L, Reddy VY, Singh JP, Mela T, Ruskin JN, Armoundas AA. A novel lead configuration for optimal spatio-temporal detection of intracardiac repolarization alternans. Circ Arrhythm Electrophysiol. 2011:407–417. doi: 10.1161/CIRCEP.109.934208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Paz O, Zhou X, Gillberg J, Tseng HJ, Gang E, Swerdlow C. Detection of T-wave alternans using an implantable cardioverter-defibrillator. Heart Rhythm. 2006;3:791–797. doi: 10.1016/j.hrthm.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 93.Kuo CS, Amlie JP, Munakata K, Reddy CP, Surawicz B. Dispersion of monophasic action potential durations and activation times during atrial pacing, ventricular pacing, and ventricular premature stimulation in canine ventricles. Cardiovasc Res. 1983;17:152–161. doi: 10.1093/cvr/17.3.152. [DOI] [PubMed] [Google Scholar]
- 94.Chinushi M, Restivo M, Caref EB, El-Sherif N. Electrophysiological basis of arrhythmogenicity of QT/T alternans in the long-QT syndrome: tridimensional analysis of the kinetics of cardiac repolarization. Circ Res. 1998;83:614–628. doi: 10.1161/01.res.83.6.614. [DOI] [PubMed] [Google Scholar]
- 95.Chinushi M, Kozhevnikov D, Caref EB, Restivo M, El-Sherif N. Mechanism of discordant T wave alternans in the in vivo heart. J Cardiovasc Electrophysiol. 2003;14:632–638. doi: 10.1046/j.1540-8167.2003.03028.x. [DOI] [PubMed] [Google Scholar]
- 96.Tachibana H, Kubota I, Yamaki M, Watanabe T, Tomoike H. Discordant S-T alternans contributes to formation of reentry: a possible mechanism of reperfusion arrhythmia. Am J Physiol. 1998;275(1 Pt 2):H116–121. doi: 10.1152/ajpheart.1998.275.1.H116. [DOI] [PubMed] [Google Scholar]
- 97.Shimizu W, Antzelevitch C. Cellular and ionic basis for T-wave alternans under long-QT conditions. Circulation. 1999;99:1499–1507. doi: 10.1161/01.cir.99.11.1499. [DOI] [PubMed] [Google Scholar]
- 98.Fox JJ, Riccio ML, Hua F, Bodenschatz E, Gilmour RF., Jr Spatiotemporal transition to conduction block in canine ventricle. Circ Res. 2002;90:289–296. doi: 10.1161/hh0302.104723. [DOI] [PubMed] [Google Scholar]
- 99.Qu Z, Garfinkel A, Chen PS, Weiss JN. Mechanisms of discordant alternans and induction of reentry in simulated cardiac tissue. Circulation. 2000;102(14):1664–1670. doi: 10.1161/01.cir.102.14.1664. [DOI] [PubMed] [Google Scholar]
- 100.Watanabe MA, Fenton FH, Evans SJ, Hastings HM, Karma A. Mechanisms for discordant alternans. J Cardiovasc Electrophysiol. 2001;12:196–206. doi: 10.1046/j.1540-8167.2001.00196.x. [DOI] [PubMed] [Google Scholar]
- 101.Weiss JN, Chen PS, Wu TJ, Siegerman C, Garfinkel A. Ventricular fibrillation: new insights into mechanisms. Ann N Y Acad Sci. 2004;1015:122–132. doi: 10.1196/annals.1302.010. [DOI] [PubMed] [Google Scholar]
- 102.Cevik C, Perez-Verdia A, Nugent K. Implantable cardioverter defibrillators and their role in heart failure progression. Europace. 2009;11:710–715. doi: 10.1093/europace/eup091. [DOI] [PubMed] [Google Scholar]
- 103.Selvaraj RJ, Picton P, Nanthakumar K, Mak S, Chauhan VS. Endocardial and epicardial repolarization alternans in human cardiomyopathy: evidence for spatiotemporal heterogeneity and correlation with body surface T-wave alternans. J Am Coll Cardiol. 2007;49:338–346. doi: 10.1016/j.jacc.2006.08.056. [DOI] [PubMed] [Google Scholar]
- 104.MacLean WA, Plumb VJ, Waldo AL. Transient entrainment and interruption of ventricular tachycardia. Pacing Clin Electrophysiol. 1981;4:358–366. doi: 10.1111/j.1540-8159.1981.tb03713.x. [DOI] [PubMed] [Google Scholar]
- 105.Almendral JM, Rosenthal ME, Stamato NJ, Marchlinski FE, Buxton AE, Frame LH, Miller JM, Josephson ME. Analysis of the resetting phenomenon in sustained uniform ventricular tachycardia: incidence and relation to termination. J Am Coll Cardiol. 1986;8:294–300. doi: 10.1016/s0735-1097(86)80043-2. [DOI] [PubMed] [Google Scholar]
- 106.Naccarelli GV, Zipes DP, Rahilly GT, Heger JJ, Prystowsky EN. Influence of tachycardia cycle length and antiarrhythmic drugs on pacing termination and acceleration of ventricular tachycardia. Am Heart J. 1983;105:1–5. doi: 10.1016/0002-8703(83)90269-7. [DOI] [PubMed] [Google Scholar]
- 107.Ruffy R, Friday KJ, Southworth WF. Termination of ventricular tachycardia by single extrastimulation during the ventricular effective refractory period. Circulation. 1983;67:457–459. doi: 10.1161/01.cir.67.2.457. [DOI] [PubMed] [Google Scholar]
- 108.Garan H, Ruskin JN. Reproducible termination of ventricular tachycardia by a single extrastimulus within the reentry circuit during the ventricular effective refractory period. Am Heart J. 1988;116(2 Pt 1):546–550. doi: 10.1016/0002-8703(88)90630-8. [DOI] [PubMed] [Google Scholar]
- 109.Fisher JD, Kim SG, Furman S, Matos JA. Role of implantable pacemakers in control of recurrent ventricular tachycardia. Am J Cardiol. 1982;49:194–206. doi: 10.1016/0002-9149(82)90294-6. [DOI] [PubMed] [Google Scholar]
- 110.Fisher JD, Kim SG, Matos JA, Ostrow E. Comparative effectiveness of pacing techniques for termination of well-tolerated sustained ventricular tachycardia. Pacing Clin Electrophysiol. 1983;6(5 Pt 1):915–922. doi: 10.1111/j.1540-8159.1983.tb04413.x. [DOI] [PubMed] [Google Scholar]
- 111.Bonometti C, Hwang C, Hough D, Lee JJ, Fishbein MC, Karagueuzian HS, Chen PS. Interaction between strong electrical stimulation and reentrant wavefronts in canine ventricular fibrillation. Circ Res. 1995;77:407–416. doi: 10.1161/01.res.77.2.407. [DOI] [PubMed] [Google Scholar]
- 112.Davidenko JM, Salomonsz R, Pertsov AM, Baxter WT, Jalife J. Effects of pacing on stationary reentrant activity. Theoretical and experimental study. Circ Res. 1995;77:1166–1179. doi: 10.1161/01.res.77.6.1166. [DOI] [PubMed] [Google Scholar]
- 113.Kamjoo K, Uchida T, Ikeda T, Fishbein MC, Garfinkel A, Weiss JN, Karagueuzian HS, Chen PS. Importance of location and timing of electrical stimuli in terminating sustained functional reentry in isolated swine ventricular tissues: evidence in support of a small reentrant circuit. Circulation. 1997;96:2048–2060. doi: 10.1161/01.cir.96.6.2048. [DOI] [PubMed] [Google Scholar]
- 114.Hall GM, Gauthier DJ. Experimental control of cardiac muscle alternans. Phys Rev Lett. 2002;88:198102. doi: 10.1103/PhysRevLett.88.198102. [DOI] [PubMed] [Google Scholar]
- 115.Christini DJ, Riccio ML, Culianu CA, Fox JJ, Karma A, Gilmour RF., Jr Control of electrical alternans in canine cardiac purkinje fibers. Phys Rev Lett. 2006;96:104101. doi: 10.1103/PhysRevLett.96.104101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Weiss EH, Merchant F, Laferriere S, Mela T, Armoundas AA. A Novel Intracardiac Pacing Method to Induce and Suppress Repolarization Alternans. Circulation. 2009;120(Suppl):S710. [Google Scholar]
- 117.Rosenbaum DS, Jackson LE, Smith JM, Garan H, Ruskin JN, Cohen RJ. Electrical alternans and vulnerability to ventricular arrhythmias. N Engl J Med. 1994;330:235–241. doi: 10.1056/NEJM199401273300402. [DOI] [PubMed] [Google Scholar]
- 118.Merchant FM, Dec GW, Singh JP. Implantable sensors for heart failure. Circ Arrhythm Electrophysiol. 2010;3:657–667. doi: 10.1161/CIRCEP.110.959502. [DOI] [PubMed] [Google Scholar]
- 119.Rashba EJ, Cooklin M, MacMurdy K, Kavesh N, Kirk M, Sarang S, Peters RW, Shorofsky SR, Gold MR. Effects of selective autonomic blockade on T-wave alternans in humans. Circulation. 2002;105:837–842. doi: 10.1161/hc0702.104127. [DOI] [PubMed] [Google Scholar]
- 120.Klingenheben T, Gronefeld G, Li YG, Hohnloser SH. Effect of metoprolol and d,l-sotalol on microvolt-level T-wave alternans. Results of a prospective, double-blind, randomized study. J Am Coll Cardiol. 2001;38:2013–2019. doi: 10.1016/s0735-1097(01)01661-8. [DOI] [PubMed] [Google Scholar]
- 121.Nieminen T, Nanbu DY, Datti IP, Vaz GR, Tavares CA, Pegler JR, Nearing BD, Belardinelli L, Verrier RL. Antifibrillatory Effect of Ranolazine during Severe Coronary Stenosis in the Intact Porcine Model. Heart Rhythm. doi: 10.1016/j.hrthm.2010.11.029. [DOI] [PubMed] [Google Scholar]
- 122.Cutler MJ, Wan X, Laurita KR, Hajjar RJ, Rosenbaum DS. Targeted SERCA2a gene expression identifies molecular mechanism and therapeutic target for arrhythmogenic cardiac alternans. Circ Arrhythm Electrophysiol. 2009;2:686–694. doi: 10.1161/CIRCEP.109.863118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jimenez RA, Myerburg RJ. Sudden cardiac death. Magnitude of the problem, substrate/trigger interaction, and populations at high risk. Cardiol Clin. 1993;11:1–9. [PubMed] [Google Scholar]
- 124.Ritzenberg AL, Adam DR, Cohen RJ. Period multupling-evidence for nonlinear behaviour of the canine heart. Nature. 1984;307:159–161. doi: 10.1038/307159a0. [DOI] [PubMed] [Google Scholar]