

Background: Biodegradation of polyamides is important from the industrial and environmental point of view.
Results: We identified the catalytic residue of nylon hydrolase as Thr-267 and enhanced the protein thermostability by 36 °C (Tm = 88 °C) by introducing mutations at the subunit interfaces of tetramer structure.
Conclusion: We revealed the mechanism of nylon-6 hydrolysis.
Significance: We established an approach to biodegrade polymeric nylon-6.
Keywords: Biodegradation, Hydrolases, Protein Stability, Protein Structure, X-ray Crystallography, Nylon
Abstract
We performed x-ray crystallographic analyses of the 6-aminohexanoate oligomer hydrolase (NylC) from Agromyces sp. at 2.0 Å-resolution. This enzyme is a member of the N-terminal nucleophile hydrolase superfamily that is responsible for the degradation of the nylon-6 industry byproduct. We observed four identical heterodimers (27 kDa + 9 kDa), which resulted from the autoprocessing of the precursor protein (36 kDa) and which constitute the doughnut-shaped quaternary structure. The catalytic residue of NylC was identified as the N-terminal Thr-267 of the 9-kDa subunit. Furthermore, each heterodimer is folded into a single domain, generating a stacked αββα core structure. Amino acid mutations at subunit interfaces of the tetramer were observed to drastically alter the thermostability of the protein. In particular, four mutations (D122G/H130Y/D36A/E263Q) of wild-type NylC from Arthrobacter sp. (plasmid pOAD2-encoding enzyme), with a heat denaturation temperature of Tm = 52 °C, enhanced the protein thermostability by 36 °C (Tm = 88 °C), whereas a single mutation (G111S or L137A) decreased the stability by ∼10 °C. We examined the enzymatic hydrolysis of nylon-6 by the thermostable NylC mutant. Argon cluster secondary ion mass spectrometry analyses of the reaction products revealed that the major peak of nylon-6 (m/z 10,000–25,000) shifted to a smaller range, producing a new peak corresponding to m/z 1500–3000 after the enzyme treatment at 60 °C. In addition, smaller fragments in the soluble fraction were successively hydrolyzed to dimers and monomers. Based on these data, we propose that NylC should be designated as nylon hydrolase (or nylonase). Three potential uses of NylC for industrial and environmental applications are also discussed.
Introduction
Nylons are synthetic polymers that contain recurring amide groups (R-CO-NH-R′) as integral parts of their main polymer chains. The high strength, elasticity, abrasion resistance, chemical resistance, and shape-holding characteristics of nylons over wide temperature ranges make these polymers suitable for the production of fibers and plastics. Currently, the worldwide production of nylons is estimated to be three to four million tons per year. Nylons tend to be partially crystalline, and the degree of crystallinity affects the properties of nylons, such as the melting points, strength, and rigidity. Two forms of crystals (α and γ) have been reported (1). In the α-form, each polymer chain is stabilized by hydrogen bonds with adjacent chains aligned in an antiparallel orientation, whereas the chains are parallel in the γ-form. The α-form of nylon-6 is generally more stable (1).
Nylon-6 is produced by the ring cleavage polymerization of ϵ-caprolactam and consists of more than 100 units of 6-aminohexanoate (Ahx).4 However, during the polymerization reaction, some molecules fail to polymerize and remain oligomers, whereas others undergo head-to-tail condensation to form cyclic oligomers. We have isolated bacterial strains that can degrade Ahx oligomers, which are by-products of nylon-6 production, and use these oligomers as their sole carbon and nitrogen source (2–6). Previous biochemical studies have revealed that three enzymes, NylABC, are responsible for the degradation of the by-products produced during nylon-6 manufacturing. The Ahx cyclic dimer hydrolase (NylA; EC 3.5.2.12), a member of the amidase signature hydrolase family, specifically hydrolyzes one of the two equivalent amide bonds in the Ahx cyclic dimer, generating an Ahx linear dimer (7). The Ahx dimer hydrolase (NylB; EC 3.5.1.46), a member of the penicillin-recognizing family of serine reactive hydrolases, hydrolyzes Ahx oligomers by an exo-type mode (8–12). The Ahx oligomer hydrolase (NylC; EC 3.5.-.-) degrades Ahx cyclic and linear oligomers with a degree of polymerization greater than three by an endo-type mode (5, 13–15).
NylC has been found in Arthrobacter (pOAD2 plasmid-encoded NylC; NylCp2), Agromyces (NylCA), and Kocuria (NylCK) (5). These enzymes are each encoded by a single gene that corresponds to a polypeptide chain of 355 amino acids. However, the post-translational cleavage of the nascent polypeptide between Asn-266 and Thr-267 generates a 27-kDa (α)-subunit and a 9-kDa (β)-subunit (5, 14). The autoprocessing of the inactive precursor to an active enzyme is a specific feature of the N-terminal nucleophile (N-tn) hydrolase family (16–33). NylCA and NylCK have 5 and 15 amino acid substitutions, respectively, relative to the NylCp2 sequence (supplemental Fig. S1), and both have 10–20 °C higher thermostability than NylCp2 (5).
Recently, we reported the crystallization conditions of NylCA that are suitable for x-ray crystallographic analyses (34). In this study, we performed x-ray crystallographic analyses of NylCA, examined its structural/evolutional relationship to proteins registered in the Protein Data Bank (PDB), and estimated the residues responsible for the catalytic function and autoprocessing. We also analyzed the effect of amino acid mutations on the thermostability of NylC. Furthermore, we investigated the possibility of using the thermostable NylC mutant to degrade polymeric nylon-6 at high temperatures, which is expected to increase the reactivity of the polymer.
EXPERIMENTAL PROCEDURES
DNA Preparation and Site-directed Mutagenesis
The plasmids pSKFC4 (NylCp2), pSKRC4 (NylCA), and pSKKC4 (NylCK) contained 1.1-kb genes flanked by BamHI and PstI restriction sites, which were cloned into the expression vector pBluescript II SK(+) (Stratagene, La Jolla, CA.) (5). Escherichia coli JM109-competent cells were prepared by the CaCl2 method (35) and stored at −80 °C before use. To introduce the D36A, G111S, D122G, H130Y, L137A, V225M, and E263Q mutations into the NylCp2 sequence, site-directed mutagenesis was performed using the PrimeSTAR mutagenesis kit (Takara Bio Inc.) with the primers listed in supplemental Table S1. The plasmids that contained the 1.1-kb fragments with mutated NylCp2 were isolated from transformed E. coli JM109 cells. DNA sequencing confirmed that the desired mutations were introduced into the wild-type nylCp2 sequence.
Cultivation, Enzyme Purification, and Enzyme Assay
Cultivation and purification from E. coli clones were performed as reported previously (34). In the NylC activity assays, the enzyme solution (0.1 ml) was mixed with an Ahx cyclic oligomer solution (0.9 ml, 4 mg ml−1 Ahx cyclic oligomer in 20 mm phosphate buffer, pH 7.3, 10% glycerol (buffer A)) and incubated at 30 °C (standard assay condition). An increase in the concentration of the amino group was determined using trinitrobenzene sulfonic acid (5). Kinetic studies were performed under standard assay conditions, with the exception of the different Ahx cyclic oligomer concentrations used.
Nylon Degradation Tests
Nylon-6 that was mechanically disintegrated to a powder was a generous gift from Toyobo Co. (Tsuruga, Japan). To analyze the particle sizes, the nylon sample was magnified 80-fold with a scanning electron microscope (Hitachi, model TM-1000). The diameter (maximum length) of 157 particles observed in five electron microphotographs was measured. The diameters ranged from 68 to 720 μm (average ± S.D., 270 ± 140 μm). To increase the reactivity of the nylon sample, the nylon-6 powder (10 mg) was autoclaved in plastic tubes (Eppendorf Co.) containing buffer A (180 μl) at 120 °C for 20 min prior to the initiation of the enzyme reaction. Hydrolytic reactions were initiated by the addition of the thermostable NylCp2-G122Y130A36Q263 mutant enzyme (1 mg ml−1, 20 μl) to each tube and incubated at 60 °C for 2 h. The gas cluster secondary ion mass spectrometry (SIMS) equipment (developed in the project “Development of System and Technology for Advanced Measurement and Analysis,” Japan Science and Technology Agency, 2006–2010) was used to analyze the reaction products (36). Enzyme reactions were performed in triplicate. The reaction products (containing both the insoluble and the soluble fractions) from the first tube were spotted onto a silicon plate (1-cm2 square plate). Mass-to-charge ratio (m/z) ranges of 5,000–35,000 and 0–1,500 were analyzed. The solid fractions (∼10 mg) that were recovered from the reaction products (the second tube) were washed with distilled water, lyophilized, and dissolved in trifluoroethanol (0.2 ml). Subsequently, a fraction (0.02 ml) was spotted onto a silicon plate (1-cm2 square plate). The m/z range of 1,500–15,000 was analyzed. The soluble fraction (1 μl) from the third tube was spotted onto a thin layer plate and developed by a solvent mixture containing 1-propanol:water:ethyl acetate:ammonia (24:12:4:1.3). The degradation products were detected by spraying the plate with a 0.2% ninhydrin solution (in n-butyl alcohol saturated with water) (5).
Crystallographic Analysis
Crystallization and Diffraction Data Collection
For analysis of NylCA, plate-like crystals (0.8 × 0.4 × 0.3 mm) were obtained by the sitting-drop vapor diffusion method. Droplets were prepared by mixing 2 μl of purified NylCA solution (10 mg ml−1 protein in buffer A) and 2 μl of reservoir solution (1.0 m sodium citrate as a precipitant in 0.1 m HEPES buffer (pH 7.5), 0.2 m NaCl) and were equilibrated against 100 μl of reservoir solution at 10 °C for 1 week. The crystal belonged to the space group I222, with unit cell parameters a = 155.86 Å, b = 214.45 Å, and c = 478.80 Å (Table 1).
TABLE 1.
Data collection statistics and refinement statistics
Values in parentheses are for the outer resolution shell.
| Native (NylCA) | K2PtCl4 derivative (NylCA) | ||
|---|---|---|---|
| Data collection statistics | |||
| Space group | I222 | I222 | |
| Unit cell (Å) | |||
| a | 155.86 | 155.99 | |
| b | 214.45 | 214.72 | |
| c | 478.80 | 477.82 | |
| Wavelength (Å) | 1.0000 | 1.0000 | |
| Resolution (outer shell) (Å) | 30-2.00 (2.07-2.00) | 30-2.20 (2.28-2.20) | |
| Total reflections | 1,789,588 | 2,786,883 | |
| Unique reflections (outer shell) | 501,977 (38,621) | 398,011 (36,644) | |
| Completeness (outer shell) (%) | 94.3 (73.0) | 99.0 (91.7) | |
| Rmerge (outer shell) (%) | 11.1 (25.4) | 6.4 (46.2) | |
| <I / σ(I)> (outer shell) | 10.6 (2.32) | 26.1 (2.87) | |
| Multiplicity | 3.6 (2.0) | 7.0 (4.8) | |
| Refinement statistics | |||
| Resolution range (outer shell) (Å) | 50-2.00 (2.07-2.00) | ||
| Rworka (outer shell) (%) | 22.3 (31.9) | ||
| Rfreea (outer shell) (%) | 25.2 (33.5) | ||
| No. of protein atoms | 37,819 | ||
| No. of water molecules | 3,006 | ||
| r.m.s. deviations from ideal values | |||
| r.m.s. bond distances (Å) | 0.006 | ||
| r.m.s. bond angles (°) | 0.9 | ||
| Dihedral angles (°) | 22.3 | ||
| Improper angles (°) | 0.74 | ||
| Ramachandran plot | |||
| Favored (%) | 96.2 | ||
| Allowed (%) | 99.9 | ||
| Outliers | 4 | ||
a R = Σhkl ‖Fobs| − k|Fcalc‖ (Σhkl|Fobs|)−1; k = scaling factor.
For native crystals, the crystals were soaked for 24 h in a cryoprotectant solution (1.0 m sodium citrate, 0.1 m HEPES (pH 7.5), 0.2 m NaCl, 25% glycerol). Heavy atom derivatives of NylCA were prepared by soaking the crystals for 72 h in cryoprotectant solution containing 5 mm K2PtCl4. Cryo-cooling was performed by blowing cold nitrogen steam onto the crystals at 100 K.
The diffraction data sets were collected at the SPring-8 (Hyogo, Japan) beamline BL38B1 equipped with a Rigaku Jupiter CCD detector system. The following parameters were chosen for data collection: wavelength, 1.0000 Å; crystal to detector distance, 180 mm; oscillation range per image, 0.5° (for native and platinum derivative). Indexing, integration, and the scaling of reflections were performed using the HKL2000 program package (37). Diffraction data were collected from native NylCA crystals and from the K2PtCl4-derivative to resolutions of 2.00 Å and 2.20 Å, respectively (Table 1).
Phase Determination, Model Building, and Crystallographic Refinement
The NylC structure was determined by the single wavelength anomalous diffraction method using the K2PtCl4 derivative data. The platinum substructure for the derivative crystal was solved at a resolution of 2.2 Å by the program BnP (38) using the anomalous signal of the platinum atoms. Initial phase parameters were determined using the program SHARP (39). The electron density map was automatically traced with ARP/wARP. Models for the untraced regions, with the exception of the amino acids at positions 1–18 and 261–266, were constructed by manual model building using XFIT (40). Rigid body refinement was performed using the coordinates of the initial model to fit the unit cell of the NylCA crystal followed by positional and B-factor refinement with the program CNS (41). Non-crystallographic symmetry restraints were applied throughout refinement with the exception of the residues 25–40 and 131–137 in which significant structural differences among chains were detected.
After several cycles of manual model rebuilding by XFIT, R-factor and Rfree were determined to be 18.1 and 19.5%. No residue was found in the outlying regions. The results of the crystal structure analysis are summarized in Table 1. The figures of the three-dimensional structural models were generated with the program MolFeat (version 3.6, FiatLux Co., Tokyo, Japan).
CD Analysis
Circular dichroism (CD) spectra at far-UV wavelengths (200–250 nm) were measured using a spectropolarimeter (JASCO, model J-720WI). A cuvette with a path length of 1 mm was used for far-UV CD measurements. The results are expressed as the mean residue molar ellipticity, [θ], which is defined as [θ] = 100 (θobs − θback) l−1 c−1, where θobs is the observed ellipticity in degrees, θback is the observed ellipticity in degrees in the absence of enzyme (as background), c is the molar concentration of the residue, and l is the length of the light path (in centimeters). The temperature was controlled at 25 °C with a JASCO PTC-348WI Peltier system. For thermal transition experiments, the temperature was shifted from 25 to 95 °C with a JASCO PTC-348WI Peltier system at 1 °C min−1, and CD measurements were performed at 220 nm. The protein concentration used in the far-UV CD measurements was 0.1 mg ml−1.
RESULTS AND DISCUSSION
Quaternary Structure
The asymmetric unit contains 15 molecules (molecules A–O), each composed of an α- and a β-subunit. One molecule (O) is part of a tetramer in which the four heterodimers are related by crystallographic 222 symmetry. Two molecules in dimer (M/N) form another tetramer that is related by a crystallographic two-fold axis, and the rest of the 12 molecules comprise three tetramers (A/B/C/D, E/F/G/H, and I/J/K/L) that are related by non-crystallographic 222 symmetries. The root mean square (r.m.s.) deviations of the superimposed Cα atoms for all 15 molecules were calculated to be within the range of 0.13–0.38 Å by secondary structure matching (supplemental Table S2) (42), demonstrating that the overall structures are almost identical. Based on these results, we concluded that NylCA adopts a doughnut-shaped quaternary structure in which four heterodimer molecules are mutually related by a perpendicular crystallographic and/or non-crystallographic two-fold axis (Fig. 1). Contacts at the A/B and C/D interfaces (3,121 Å2) were observed to be more extensive than those at the B/C and A/D interfaces (1,451 Å2).
FIGURE 1.

Stereo view of quaternary structure of NylCA. A, the quaternary structure is shown, with different colors highlighting the individual molecules A (green), B (blue), C (red), and D (yellow). The catalytic residue Thr-267 (the N terminus of the β-subunit) is shown as a space-filling model (red). B, an enlarged view of molecule A and its interfaces with the adjacent molecules B and D. Six residues selected for mutagenesis (Asp-36 (Ala-36 in NylCK), Ser-111, Gly-122, Tyr-130, Ala-137, and Met-225) are shown as space-filling models. Because of the poor electron density distribution of the C-terminal region in the α-subunit, including Glu-263 (Gln-263 in NylCK), the adjacent Pro-260 is shown as a space-filling model (magenta). C, surface structure of NylCA. The α-subunit and β-subunit in a single heterodimer (molecule A) are highlighted in dark green and light green, respectively. The other three heterodimers are shown in different shades of gray.
Subunit Structure and Function Relationship with Other N-tn Family Enzymes
Each heterodimer contains 10 helices, H1–H10, which are α-helices with the exception of H7 (310 helix), and 18 β-strands. The two subunits (α and β) obtained by intracleavage at Asn-266/Thr-267 are folded into a single domain, generating a stacked αββα core structure (Fig. 2A, supplemental Fig. S2). Namely, the central antiparallel β-sheet composed of five β-strands (β3, β4a, β5, β6, β7) and another antiparallel β-sheet composed of five β-strands (β4b, β12, β13, β16, β18) are packed against each other and connected by a common long β-strand (β4). These two sheets are flanked on one side by the α-helices H4 and H5 and on the other by α-helices H9 and H10. The αββα fold is typically conserved throughout the N-tn hydrolase superfamily (16–33). However, our data demonstrate that NylC differs from most N-tn hydrolases in the directionality and connectivity of its secondary structure elements.
FIGURE 2.

Stereo view of subunit structure and catalytic center of NylCA. A, the overall structure of the heterodimer (molecule A) is shown as a ribbon diagram. Ten helices (H1–H10) and eighteen β-strands (S1–S18) are colored in green and orange, respectively. H1–H6 and H8–H10 are α-helices. H7 is a 310 helix. B, the structure around the catalytic residue Thr-267 of NylCA (green) is superimposed onto the structure of DmpA (PDB ID code, 1B65; magenta) and BapA (PDB ID code, 3N33; orange). Possible hydrogen bonds in NylCA are indicated as dotted lines with the distances listed in angstroms.
To search for proteins that are evolutionary related to NylC, we performed a homology search based on the NylCA structure for PDB using the DALI program (43). The following four proteins (including one hypothetical protein) that exhibited high z-scores (z-score >14) were identified: l-aminopeptidase d-Ala-esterase/amidase from Ochrobactrum anthropi (DmpA; z-score = 26.3; PDB ID code: 1B65) (18), hypothetical d-aminopeptidase (z-score = 25.9; PDB ID code, 2DRH), β-peptidyl aminopeptidase from Sphingosinicella xenopeptidilytica (BapA; z-score = 25.2; PDB ID code: 3N33) (20), and ornithine acetyltransferase from Streptomyces clavuligerus (OAT; z-score = 14.1; PDB ID code: 1VZ6) (21) (supplemental Table S3). In contrast, the DALI z-scores of the other proteins in the PDB were determined to be below 8.
DmpA is an aminopeptidase that hydrolyzes peptides, with a preference for N-terminal residues in an l-configuration (l-Ala-Gly-Gly) and also amide or ester derivatives of d-Ala (19). BapA hydrolyzes β-oligopeptides and mixed β/α-oligopeptides with a β-amino acid residue at the N terminus (20). OAT is involved in the arginine biosynthetic pathway because it catalyzes the transfer of an acetyl group from N-acetylornithine to glutamate (21, 22). In contrast, NylCp2 hydrolyzes Ahx cyclic and linear oligomers (degree of polymerization >3) but has no detectable activity with the 66 peptides tested (supplemental Table S4), including d,l-Ala-Gly-Gly (14, 15). Thus, NylC exhibits distinct substrate specificity from that of DmpA/BapA/OAT.
DmpA/OAT are considered to have a unique fold (designated as DOM-fold), which is different from that of most N-tn hydrolases (17). Based on fundamental topology differences, Cheng and Grishin (17) proposed that the functional similarities between DmpA/OAT and the other N-tn hydrolases result from convergent evolution rather than from having a common evolutionary origin. The fold of NylC is similar to that of DmpA/BapA, and the structurally superimposable regions comprise 259 residues (DmpA) and 264 residues (BapA) (supplemental Table S3). The r.m.s. deviations of the superimposed Cα atoms were calculated to be 2.0 Å (DmpA) and 2.0 Å (BapA) by secondary structure matching. However, the following major structural differences were found among the family enzymes.
In contrast to the single domain structure of NylC/DmpA/BapA, OAT is composed of two domains (supplemental Fig. S3). NylC is superimposable with OAT domain 1 located at positions 1–258. OAT domain 2 is located at the C-terminal region and is composed of 124 residues that have a successive order of secondary structure (β10-H9-H10-H11-β11-β12-H12-β13-β14-H13). DmpA and BapA are longer than NylC at the C-terminal region of the β-subunit by 34 and 30 residues, respectively (supplemental Fig. S4).
Helix H8, which is located at the C-terminal region of the α-subunit, is unique in NylC. H8 and its linked loop region is flipped out to the surface of the adjacent molecules upon autoprocessing (Fig. 1C). In DmpA/OAT, the corresponding helix is absent. Furthermore, in BapA, the corresponding region (positions 236–245), which contains β14 and a loop, is folded within the same subunit molecule without flipping out to the adjacent molecule.
The N-terminal 26 residues, including H1, generate unique structure specifically found in NylC. Moreover, in OAT, N-terminal 48 residues, including H1-H2-β1-β2, are absent (supplemental Fig. S4).
A loop flanked by β-strand β18 and helix H10 in NylC (positions 317–333) spatially share different positions in Dmp (positions 299–318) and in BapA (positions 299–316). In OAT, the corresponding loop is absent, and β9 and H18 are combined with a single residue (Val-235) at the junction (supplemental Fig. S4).
In NylC, a long β-strand (β4a, β4b) constitutes a part of the central two β-sheets (Fig. 2A). However, in DmpA/BapA/OAT, the corresponding β-strand is divided into two β-strands (e.g. β2, β3 in DmpA), which are connected by a short loop and constitute each β-sheet (supplemental Fig. S4).
Residues Responsible for Autoprocessing and Catalytic Function
In DmpA, autoprocessing occurs between Gly-249 and Ser-250. Additionally, the newly generated N-terminal Ser-250 of the β-subunit plays the roles of both the nucleophile (hydroxyl group) and the general base (α-amino group) in catalytic reactions (18). In NylC, Thr-267 participates in a hydrogen-bonding network with Asn-219, Asp-306, and Gly-307 (backbone-N). The positions of these four residues are spatially similar to those of Ser-250, Asn-218, Ser-288, and Gly-289 (backbone-N) in DmpA, respectively (Fig. 2B). The catalytic nucleophile (Ser-250) and the surrounding residues (Asn-207, Ser-288, and Gly-289 (backbone-N)) are also conserved in BapA. These results indicate that Thr-267 is most likely responsible for the catalytic function of NylC. Assuming that auto-proteolysis of the NylC precursor proceeds in manner similar to that proposed for other N-tn hydrolases (23–27), the reaction would be initiated by nucleophilic attack of Thr-267-Oγ to Asn-266-Cα carbon, generating a tetrahedral intermediate. This intermediate would rearrange into an ester intermediate (N–O acyl shift) that is subsequently hydrolyzed by an adjacent water molecule, producing the active enzyme.
Structural Alterations Induced by Autoprocessing
The poor electron density distribution in the carboxyl-terminal region of the α-subunit (positions 261–267) of the active NylCA enzyme has prohibited the determination of a structural model. The C-terminal region of the α-subunit has not been identified by x-ray crystallographic analyses of DmpA/BapA/OAT (supplemental Fig. S4). In a three-dimensional model of NylCA, the distance between Pro-260-Cα and Thr-267-Cα is estimated to be 32.6 Å (supplemental Fig. S2). The large distance suggests that the local structural alteration that occurs is accompanied by autoprocessing. Namely, the terminal region of the α-subunit, including helix H8 and Pro-260, at each subunit interface flips out toward the adjacent molecules (Fig. 1C). We speculate that the structural alteration is important for generating the catalytic center responsible for the hydrolysis of incoming substrates (6-aminohexanoate-oligomers). Structural alterations induced by autoprocessing have been reported for other N-tn hydrolases (25).
Effect of Amino Acid Substitutions on Protein Stability
Thermal denaturation experiments using CD showed that the Tm of NylCA and NylCK are 60 and 67 °C, respectively (Fig. 3, Table 2). These values are 8–15 °C higher than the Tm of NylCp2 (52 °C). We have suggested that at least one among the five alterations (G111S, D122G, H130Y, L137A, V225M) in NylCA contributes to the increase in the thermostability of NylCp2. Moreover, at least one of the 10 alterations (D36A, A41V, M50T, I60V, A62S, T230G, V231I, V257L, E263Q, G354A) in NylCK is estimated to contribute to the further increase in thermostability (supplemental Fig. S1) (5). The three-dimensional structure of NylCA shows that most amino acid residues that differ among the three NylC enzymes were found to be located at the interfaces between the subunits (Fig. 1B). To examine the effect of the amino acid substitutions on the thermostability of proteins, we individually replaced the residues in NylCp2 with the corresponding NylCA or NylCK sequences.
FIGURE 3.

Thermostability of NylC mutants. A, thermal transition curves of the various NylC mutant enzymes. CD measurements were performed at 220 nm from 25 to 95 °C (1 °C min−1). The results are expressed as the mean residue molar ellipticity [θ]. Protein concentrations of 0.1 mg ml−1 were used. deg, degree. B, the cumulative effects of amino acid mutations on the melting temperatures of denaturation (Tm) are shown.
TABLE 2.
Effect of amino acid substitutions on thermostability
| Enzyme | Amino acid substitutions in NylCp2 sequence | Tm |
|---|---|---|
| °C | ||
| NylCp2 | 52 | |
| NylCA | G111S/D122G/H130Y/L137A/V225M | 60 |
| NylCK | D36A/A41V/M50T/I60V/A62S | 67 |
| G111S/D122G/H130Y/L137A/V225M | ||
| T230G/V231I/V257L/E263Q/G354A | ||
| Single mutant | ||
| NylCp2-S111 | G111S | 43 |
| NylCp2-G122 | D122G | 76 |
| NylCp2-Y130 | H130Y | 63 |
| NylCp2-A137 | L137A | 41 |
| NylCp2-M225 | V225M | Not expressed |
| Double mutant | ||
| G122Y130 | D122G/H130Y | 81 |
| Triple mutant | ||
| G122Y130A36 | D122G/H130Y/D36A | 84 |
| G122Y130Q263 | D122G/H130Y/E263Q | 84 |
| G122Y130M225 | D122G/H130Y/V225M | 83 |
| S111G122Y130 | G111S/D122G/H130Y | 78 |
| G122Y130A137 | D122G/H130Y/L137A | 54 |
| Quadruple mutant | ||
| G122Y130A36Q263 | D122G/H130Y/D36A/E263Q | 88 |
| S111G122Y130A137 | G111S/D122G/H130Y/L137A | 51 |
A single amino acid mutation of either D122G or H130Y, which are located at the A/D interface, increased the Tm of NylCp2 by 24 and 11 °C, respectively (Fig. 3, Table 2). However a single substitution of either G111S or L137A at the same interface decreases the stability by ∼10 °C. Even more drastic effects were observed with the combinations of these mutations. D122G/H130Y double mutations in NylCp2 enhanced the thermostability to 81 °C. Far-UV CD spectra showed very little change, even at 75 °C, for the G122Y130 mutant, whereas the parental NylCp2 and NylCA enzymes denatured at 75 °C (supplemental Fig. S5). The addition of two further mutations, D36A/E263Q derived from NylCK, to the G122Y130 mutant enhanced the Tm to 88 °C (G122Y130A36Q263 mutant; Fig. 3). Notably, we observed that more than 90% of the enzyme activity was retained even after incubation of the G122Y130A36Q263 mutant for 30 min at 70 °C. In contrast, an L137A mutation drastically decreased thermostability in the G122Y130 and S111G122Y130 mutants by 27 °C. Similarly, a V225M single mutation decreased the protein stability and/or the expression level in cells because no protein was detected in the cell extracts that were prepared by expressing the M225-mutant NylCp2. However, the stabilization effect is altered by the combination of other amino acid alterations close to Met-225 because the V225M mutation, in the context of the S111G122Y130A137 quadruple mutant, recapitulates the sequence of NylCA and improves its stability by 9 °C. Additionally, the V225M mutation in the G122Y130 mutant improved the stability by 2 °C (see G122Y130M225). These results demonstrate that subunit interactions that generate the quaternary structure drastically affect the thermostability of the enzyme (47 °C by five mutations; Fig. 3, Table 2).
Molecular Basis of Protein Stabilization
Based on the three-dimensional structure of NylCA, we estimate that the following structural effects occurring at the subunit interfaces A/D (D122G, H130Y, L137A, G111S, and D36A) and A/B (V225M and E263Q) can cause changes in the thermostability of NylCp2.
D122G
In NylCA, Lys-159-NH3+ (in molecule A) is located 2.79 Å apart from Glu-115-COO− (in molecule D) (Fig. 4A). Therefore, the electrostatic effect between the two residues will enhance subunit binding around Gly-122 located on helix H4. However, in NylCp2, the amino acid residue at position 122 is replaced with Asp, and the close proximity of the acidic residue Asp-122 (close to Glu-115) will reduce this stabilization effect.
FIGURE 4.

Possible interactions at the subunit interfaces. A–D, the possible interactions at the subunit interfaces around Gly-122 (A), Asp-36/Tyr-130 (B), Ser-111/Ala-137 (C), and Met-225 (D) are shown. Molecule A, molecule B, and molecule D in the quaternary structure are colored in green, blue, and yellow, respectively. Possible hydrogen bonds and contacts between two atoms are indicated as dotted lines with the distances listed in angstroms.
H130Y and L137A
Tyr-130 (in molecule A) is located at a loop region (Arg-127–Ala-135) between helix H4 and β-strand β7 (supplemental Fig. S4). Tyr-130-Oη forms a hydrogen bond with Glu-126-Oϵ located at the end of helix H4 (Fig. 4B). Mutating Tyr-130 to His may destroy the hydrogen bonding and destabilize the loop. Therefore, a H130Y substitution in NylCp2 will contribute to an increase in thermostability by stabilizing the loop region. In contrast, L137A substitution (in NylCp2, G122Y130 and S111G122Y130) occurring at the same loop region resulted in the drastic decrease in the protein stability (Fig. 3B). Therefore, hydrophobic effect by Leu-137 should be involved in the stabilization of the loop region. Ala-137/Leu-137 affects the subunit interaction cooperatively with Gly-111/Ser-111 (Fig. 4C).
G111S
Hydrophilic Ser-111-Oγ (in molecule A) is surrounded by four hydrophobic residues (Tyr-98, Ala-137, Leu-139 (in molecule D) and Tyr-112 (in molecule A)) (Fig. 4C). Therefore, substitution from Ser-111 to Gly-111 (aliphatic small residue) should improve the hydrophobic stabilization effect at the A/D interface. In contrast, its reverse G111S substitution (in NylCp2, G122Y130 and G122Y130A137) actually reduces the thermostability (Fig. 3B).
D36A
Asp-36 (in molecule D) is located at helix H2, which is close to Glu-126 (in molecule A) (4.40 Å), whereas Asp-36 is replaced with Ala-36 in NylCK (Fig. 4B). Therefore, the D36A mutation will reduce electrostatic repulsion with Glu-126. Actually, D36A substitution (in G122Y130 and G122Y130Q263) improves the thermostability (Fig. 3B).
V225M
Met-225 (in molecule A) does not directly contact with any adjacent subunits (Leu-9-Cδ1 in molecule B is the nearest position; 5.29 Å). Met-225 is close to Gln-299 in the same subunit, which interacts with Arg-296-Nη2 (3.01 Å) (in molecule B) (Fig. 4D). Moreover, the position of Gln-299 should be stabilized by interaction with His-245-Nϵ2 (2.81 Å). Therefore, it is likely that amino acid substitution at position 225 indirectly affects the subunit interaction. Actually, V225M substitution (in G122Y130 and S111G122Y130A137) improves the thermostability (Fig. 3B), probably by generating a new interaction between Met-Sδ and Gln-299-Nϵ1 (3.56 Å) (Fig. 4D). However, it should be also noted that V225M substitution decreases the expression level and/or stability of NylCp2 in the cell, as described above. Therefore, the total stabilization effects should be dependent on the combined interactions with the surrounding residues.
E263Q
Glu-263 is located at the terminal region of the α-subunit (Val-261–Asn-266), which had a poor electron density distribution in the x-ray diffraction study of NylCA (Fig. 1). In NylCK, the acidic amino acid residue Glu-263 is replaced with neutral Gln, suggesting that altering the electrostatic environment induced by the E263Q mutation assists in improving subunit interactions.
Enzymatic Hydrolysis of Polymeric Nylon-6
Although the melting point of nylon-6 (220–225 °C) is much higher than the thermostability of the enzyme, performing the reaction at a high temperature should have the following advantages for the hydrolysis of nylon. 1) Hydrogen bonding between the polymer chains is partially weakened, allowing the enzyme to attack the polymer chains that are exposed to the solvent; and 2) the release of the cleaved fragments from the solid phase generates new sites for the subsequent reaction.
The mutated sites in the G122Y130A36Q263 quadruple mutant are located at the interface with other subunits and apart from catalytic Thr-267 (e.g. 26.6 Å to Gly-122 at Cα). Kinetic studies of the G122Y130A36Q263 mutant with the Ahx cyclic oligomer determined that the kcat and Km values are 2.8 ± 0.11 s−1 and 0.72 ± 0.06 mg ml−1, respectively. When compared with the kinetic parameters of wild-type NylCp2 (kcat = 6.5 ± 0.29 s−1; Km = 3.7 ± 0.27 mg ml−1) (5), these mutations decreased the turnover of the product but improved the affinity of the enzyme for the substrate. As a result, the calculated kcat/Km of the G122Y130A36Q263 mutant (3.9 s−1 ml mg−1) is 2-fold higher than that of NylCp2 (1.8 s−1 ml mg−1). Based on the high thermostability and increasing catalytic activity of the G122Y130A36Q263 mutant, we considered this mutant to be suitable for testing the hydrolysis of polymeric nylon-6.
To increase the efficiency of nylon degradation, pellets of nylon-6 plastic were mechanically disintegrated to powder (diameter = 0.27 ± 0.14 mm, see “Experimental Procedures”). The powder was resuspended in buffer A (50 mg ml−1), and the suspension was pretreated at 120 °C for 20 min. The enzyme reaction was performed using the G122Y130A36Q263 mutant (0.1 mg ml−1) at 60 °C for 2 h.
SIMS measures the time of flight of the secondary ions generated by the bombardment of a sample with primary ion beams and sensitively detects the alterations in mass size occurring at the surface of solid particles (36, 44). We used gas cluster SIMS equipment, in which the primary ions are argon cluster ions with a kinetic energy per atom controlled to be lower than 4 eV. This low energy of primary ions reduces the internal cleavage of polymer molecules, increasing the detection of the intact ions (36, 44). We observed a wide peak with an m/z range of 10,000–25,000 (top peak: 14,500) for untreated nylon-6, but the major peak was shifted to a smaller range with an m/z range of 8,000–23,000 (top peak: 13,000) for the reaction products (Fig. 5A). Moreover, SIMS analyses of the reaction products (dissolved in trifluoroethanol) revealed a new peak corresponding to an m/z range of 1,500–3,000 (top peak: 2,000; Fig. 5B). The presence of the specific fragments in the solid fraction demonstrated that NylC hydrolyzes nylon, but the fragments that were produced were still bound to polymer chains through hydrogen bonding (Fig. 6). These fragments corresponded to oligomers with 13–25 monomeric units, assuming an electric charge (z) = 1. In contrast, smaller fragments (<10 monomeric units) released from the solid fraction should be readily hydrolyzed to dimers and monomers (Fig. 5C). Consistent with this hypothesis, TLC analysis showed that the dimers and monomers are detected in the soluble fractions (Fig. 5D). Therefore, we confirmed that the dimers are converted to monomers by a subsequent NylB reaction.
FIGURE 5.

Nylon degradation tests using argon cluster SIMS and TLC. Nylon-6 powder (10 mg) was pretreated in triplicate in buffer A (180 μl) at 120 °C for 20 min. The NylCp2-G122Y130A36Q263 mutant (1 mg ml−1, 20 μl) was then added and incubated at 60 °C for 2 h. The experiment was performed in triplicate. A, the reaction products (both the soluble and the insoluble fractions) were spotted onto a silicon plate (1 cm2). The m/z range of 5,000–35,000 was analyzed. Red line, pretreated nylon-6 at 120 °C in buffer A; blue line, enzyme-treated nylon-6; black line, enzyme alone. B, the solid fraction was washed with distilled water, lyophilized, and dissolved in trifluoroethanol (0.2 ml), and a fraction (0.02 ml) was spotted onto a silicon plate (1 cm2). The m/z range of 1,500–15,000 was analyzed. C, the reaction products (both the soluble and the insoluble fractions) were spotted onto a silicon plate (1 cm2). The m/z range of 0–1,500 was analyzed. The positions of Ahx monomer to heptamer (marked by 1–7; m/z = 113, 226, 339, 452, 565, 678, 791) and those of the potassium-bound forms (marked by 3K–7K) are shown. D, the soluble fractions (1 μl) were spotted onto a thin layer plate and developed, and the degradation products were detected by the ninhydrin reaction (5). Slot 1, 6-aminohexanoate; slot 2, 6-aminohexanoate-dimer; slot 3, pretreated nylon-6 at 120 °C in buffer A; and slot 4, enzyme-treated nylon-6.
FIGURE 6.
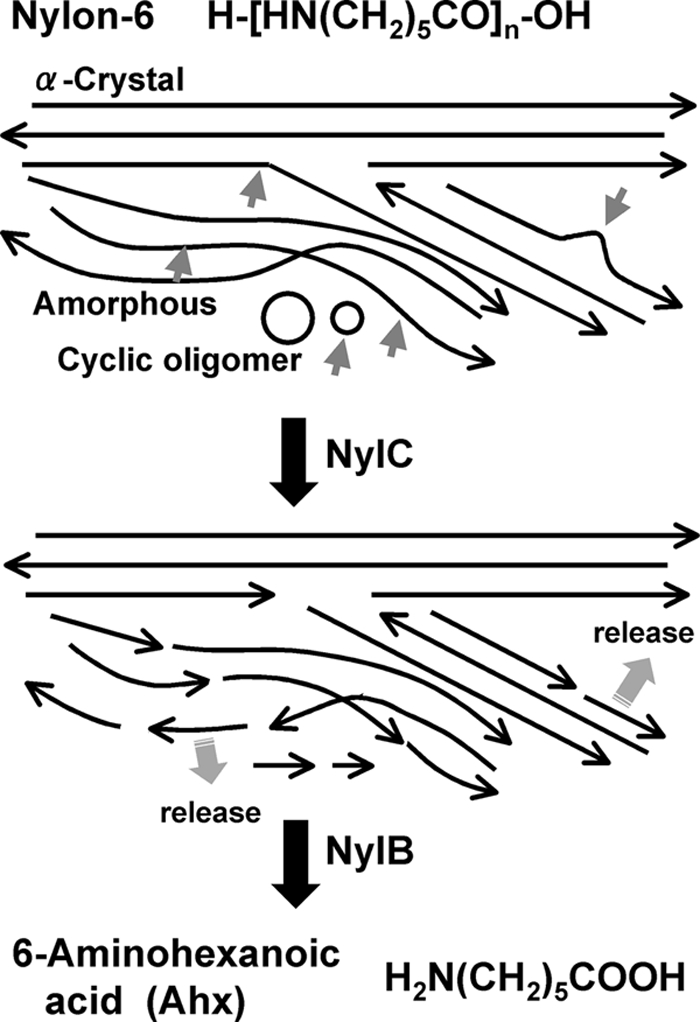
Mode of nylon-6 degradation. The polymer chains are stabilized by hydrogen bonds with adjacent chains aligned in the reverse orientation (α-Crystal) (1). Some chains constitute amorphous regions. The circles indicate the cyclic oligomers attached to the polymers. Long arrows indicate the direction of the polymer chains. Short arrows indicate cleavage by the enzyme. The 6-aminohexanoate-oligomers were converted to 6-aminohexanoate by the subsequent NylB reaction.
The original nylon sample exhibited five major peaks (Fig. 5C, 3K–7K), which are estimated to be potassium ion-bound forms, [HN(CH2)5CO]nK+ for Ahx trimer to heptamer (m/z = 378, 491, 604, 717, and 830), because the sample includes potassium ions in the reaction mixture and the cation-bound forms generally display a higher intensity than the unbound forms in the SIMS analysis (36). However, these peaks disappeared in the NylC-treated sample. Additionally, TLC analysis of the untreated nylon did not detect any spots by ninhydrin reaction. However, after NylC treatment, spots indicative of dimers and monomers were produced (Fig. 5D). From these results, we conclude that the Ahx cyclic oligomers were attached to the nylon sample and that these compounds were hydrolyzed by NylC (Fig. 6).
Potential Use in Industrial and Environmental Applications
Because SIMS analyses of the reaction products demonstrate that the thermostable NylC hydrolyzes polymeric nylon-6, we propose that NylC should be designated as nylon hydrolase (or nylonase) (EC 3.5.-.-). Notably, there is potential for the use of nylon hydrolysis in industrial and environmental applications, although the catalytic function and thermostability of NylC, as well as the pretreatment conditions of nylons for efficient hydrolysis, warrant further improvement.
Tools for Evaluation of Biodegradability of Polyamides
Ordinary biodegradability tests of polymers are performed in activated sludge (for 1 month) or in soil (for 4 months). Because the extent of hydrolysis of the polyamides by NylC is related to the biodegradability of the polyamide, an NylC reaction followed by SIMS analysis is appropriate for the prescreening of “biodegradable polyamides.”
Improvement of Surface Structures of Nylon Fibers
Suitable methods to improve the surface structures of nylon remain poorly developed. With NylC, the partial enzymatic hydrolysis of nylon surfaces can be used to change the smoothness of nylon fibers.
Recycling of Nylons
Monomers (Ahx) obtained from nylon oligomers are aerobically metabolized in nylon oligomer-degrading strains (2–6). Suitable fermentation or biotransformation processes could enable the conversion of Ahx to other metabolites, such as organic acids or alcohols. Alternatively, Ahx may be a reusable reagent for the production of nylon-6 after conversion to ϵ-caprolactam by intramolecular dehydration. Therefore, the development of these processes could enable the recycling of nylons and decrease the environmental waste caused by the accumulation of man-made compounds.
Supplementary Material
Acknowledgment
Nylon-6 was a generous gift from Toyobo Co. Ltd. (Tsuruga, Japan).
This work was supported in part by a grant-in-aid for scientific research (Japan Society for Promotion of Science) and grants from the Global Center of Excellence Program and the Japan Aerospace Exploration Agency project.

This article contains supplemental Tables S1–S4 and Figs. S1–S5.
The atomic coordinates and structure factors (code 3AXG) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- Ahx
- 6-aminohexanoate
- N-tn
- N-terminal nucleophile
- NylA
- 6-aminohexanoate cyclic dimer hydrolase
- NylB
- 6-aminohexanoate-linear dimer hydrolase
- NylC
- 6-aminohexanoate oligomer hydrolase, or nylon hydrolase
- r.m.s.
- root mean square
- DmpA
- l-aminopeptidase d-Ala-esterase/amidase
- BapA
- β-peptidyl aminopeptidase
- OAT
- ornithine acetyltransferase
- SIMS
- secondary ion mass spectrometry
- S111
- G111S
- G122
- D122G
- Y130
- H130Y
- A137
- L137A
- M225
- V225M
- G122Y130
- D122G/H130Y
- G122Y130A36
- D122G/H130Y/D36A
- G122Y130Q263
- D122G/H130Y/E263Q
- G122Y130M225
- D122G/H130Y/V225M
- S111G122Y130
- G111S/D122G/H130Y
- G122Y130A137
- D122G/H130Y/L137A
- G122Y130A36Q263
- D122G/H130Y/D36A/E263Q
- S111G122Y130A137
- G111S/D122G/H130Y/L137A.
REFERENCES
- 1. Dasgupta S., Hammond W. B., Goddard W. A., 3rd (1996) Crystal structures and properties of nylon polymers from theory. J. Am. Chem. Soc. 118, 12291–12301 [Google Scholar]
- 2. Negoro S. (2000) Biodegradation of nylon oligomers. Appl. Microbiol. Biotechnol. 54, 461–466 [DOI] [PubMed] [Google Scholar]
- 3. Okada H., Negoro S., Kimura H., Nakamura S. (1983) Evolutionary adaptation of plasmid-encoded enzymes for degrading nylon oligomers. Nature 306, 203–206 [DOI] [PubMed] [Google Scholar]
- 4. Kato K., Ohtsuki K., Koda Y., Maekawa T., Yomo T., Negoro S., Urabe I. (1995) A plasmid encoding enzymes for nylon oligomer degradation: nucleotide sequence and analysis of pOAD2. Microbiology 141, 2585–2590 [DOI] [PubMed] [Google Scholar]
- 5. Yasuhira K., Tanaka Y., Shibata H., Kawashima Y., Ohara A., Kato D., Takeo M., Negoro S. (2007) 6-Aminohexanoate oligomer hydrolases from the alkalophilic bacteria Agromyces sp. strain KY5R and Kocuria sp. strain KY2. Appl. Environ. Microbiol. 73, 7099–7102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yasuhira K., Uedo Y., Takeo M., Kato D., Negoro S. (2007) Genetic organization of nylon oligomer-degrading enzymes from alkalophilic bacterium, Agromyces sp. KY5R. J. Biosci. Bioeng. 104, 521–524 [DOI] [PubMed] [Google Scholar]
- 7. Yasuhira K., Shibata N., Mongami G., Uedo Y., Atsumi Y., Kawashima Y., Hibino A., Tanaka Y., Lee Y. H., Kato D., Takeo M., Higuchi Y., Negoro S. (2010) X-ray crystallographic analysis of the 6-aminohexanoate cyclic dimer hydrolase: catalytic mechanism and evolution of an enzyme responsible for nylon-6 byproduct degradation. J. Biol. Chem. 285, 1239–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Negoro S., Ohki T., Shibata N., Mizuno N., Wakitani Y., Tsurukame J., Matsumoto K., Kawamoto I., Takeo M., Higuchi Y. (2005) X-ray crystallographic analysis of 6-aminohexanoate-dimer hydrolase: molecular basis for the birth of a nylon oligomer-degrading enzyme. J. Biol. Chem. 280, 39644–39652 [DOI] [PubMed] [Google Scholar]
- 9. Ohki T., Wakitani Y., Takeo M., Yasuhira K., Shibata N., Higuchi Y., Negoro S. (2006) Mutational analysis of 6-aminohexanoate-dimer hydrolase: relationship between nylon oligomer hydrolytic and esterolytic activities. FEBS Lett. 580, 5054–5058 [DOI] [PubMed] [Google Scholar]
- 10. Negoro S., Ohki T., Shibata N., Sasa K., Hayashi H., Nakano H., Yasuhira K., Kato D., Takeo M., Higuchi Y. (2007) Nylon-oligomer-degrading enzyme/substrate complex: catalytic mechanism of 6-aminohexanoate-dimer hydrolase. J. Mol. Biol. 370, 142–156 [DOI] [PubMed] [Google Scholar]
- 11. Kawashima Y., Ohki T., Shibata N., Higuchi Y., Wakitani Y., Matsuura Y., Nakata Y., Takeo M., Kato D., Negoro S. (2009) Molecular design of a nylon-6 byproduct-degrading enzyme from a carboxylesterase with a β-lactamase fold. FEBS J. 276, 2547–2556 [DOI] [PubMed] [Google Scholar]
- 12. Ohki T., Shibata N., Higuchi Y., Kawashima Y., Takeo M., Kato D., Negoro S. (2009) Two alternative modes for optimizing nylon-6 byproduct hydrolytic activity from a carboxylesterase with a β-lactamase fold: X-ray crystallographic analysis of directly evolved 6-aminohexanoate-dimer hydrolase. Protein Sci. 18, 1662–1673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Negoro S., Kakudo S., Urabe I., Okada H. (1992) A new nylon oligomer degradation gene (nylC) on plasmid pOAD2 from a Flavobacterium sp. J. Bacteriol. 174, 7948–7953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kakudo S., Negoro S., Urabe I., Okada H. (1993) Nylon oligomer degradation gene, nylC, on plasmid pOAD2 from a Flavobacterium strain encodes endo-type 6-aminohexanoate oligomer hydrolase: purification and characterization of the nylC gene product. Appl. Environ. Microbiol. 59, 3978–3980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kakudo S., Negoro S., Urabe I., Okada H. (1995) Characterization of endo-type 6-aminohexanoate-oligomer hydrolase from Flavobacterium sp. J. Ferment. Bioeng. 80, 12–17 [Google Scholar]
- 16. Oinonen C., Rouvinen J. (2000) Structural comparison of Ntn-hydrolases. Protein Sci. 9, 2329–2337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cheng H., Grishin N. V. (2005) DOM-fold: a structure with crossing loops found in DmpA, ornithine acetyltransferase, and molybdenum cofactor-binding domain. Protein Sci. 14, 1902–1910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bompard-Gilles C., Villeret V., Davies G. J., Fanuel L., Joris B., Frère J. M., Van Beeumen J. (2000) A new variant of the Ntn hydrolase fold revealed by the crystal structure of l-aminopeptidase d-Ala-esterase/amidase from Ochrobactrum anthropi. Structure 8, 153–162 [DOI] [PubMed] [Google Scholar]
- 19. Fanuel L., Goffin C., Cheggour A., Devreese B., Van Driessche G., Joris B., Van Beeumen J., Frère J. M. (1999) The DmpA aminopeptidase from Ochrobactrum anthropi LMG7991 is the prototype of a new terminal nucleophile hydrolase family. Biochem. J. 341, 147–155 [PMC free article] [PubMed] [Google Scholar]
- 20. Geueke B., Heck T., Limbach M., Nesatyy V., Seebach D., Kohler H. P. (2006) Bacterial β-peptidyl aminopeptidases with unique substrate specificities for β-oligopeptides and mixed β,α-oligopeptides. FEBS J. 273, 5261–5272 [DOI] [PubMed] [Google Scholar]
- 21. Elkins J. M., Kershaw N. J., Schofield C. J. (2005) X-ray crystal structure of ornithine acetyltransferase from the clavulanic acid biosynthesis gene cluster. Biochem. J. 385, 565–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sankaranarayanan R., Cherney M. M., Garen C., Garen G., Niu C., Yuan M., James M. N. (2010) The molecular structure of ornithine acetyltransferase from Mycobacterium tuberculosis bound to ornithine, a competitive inhibitor. J. Mol. Biol. 397, 979–990 [DOI] [PubMed] [Google Scholar]
- 23. Okada T., Suzuki H., Wada K., Kumagai H., Fukuyama K. (2006) Crystal structures of γ-glutamyltranspeptidase from Escherichia coli, a key enzyme in glutathione metabolism, and its reaction intermediate. Proc. Natl. Acad. Sci. U.S.A. 103, 6471–6476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Marc F., Weigel P., Legrain C., Glansdorff N., Sakanyan V. (2001) An invariant threonine is involved in self-catalyzed cleavage of the precursor protein for ornithine acetyltransferase. J. Biol. Chem. 276, 25404–25410 [DOI] [PubMed] [Google Scholar]
- 25. Okada T., Suzuki H., Wada K., Kumagai H., Fukuyama K. (2007) Crystal structure of the γ-glutamyltranspeptidase precursor protein from Escherichia coli: structural changes upon autocatalytic processing and implications for the maturation mechanism. J. Biol. Chem. 282, 2433–2439 [DOI] [PubMed] [Google Scholar]
- 26. Tikkanen R., Riikonen A., Oinonen C., Rouvinen R., Peltonen L. (1996) Functional analyses of active site residues of human lysosomal aspartylglucosaminidase: implications for catalytic mechanism and autocatalytic activation. EMBO J. 15, 2954–2960 [PMC free article] [PubMed] [Google Scholar]
- 27. Guan C., Liu Y., Shao Y., Cui T., Liao W., Ewel A., Whitaker R., Paulus H. (1998) Characterization and functional analysis of the cis-autoproteolysis active center of glycosylasparaginase. J. Biol. Chem. 273, 9695–9702 [DOI] [PubMed] [Google Scholar]
- 28. Schmidtke G., Kraft R., Kostka S., Henklein P., Frömmel C., Löwe J., Huber R., Kloetzel P. M., Schmidt M. (1996) Analysis of mammalian 20 S proteasome biogenesis: the maturation of β-subunits is an ordered two-step mechanism involving autocatalysis. EMBO J. 15, 6887–6898 [PMC free article] [PubMed] [Google Scholar]
- 29. Guo H. C., Xu Q., Buckley D., Guan C. (1998) Crystal structures of Flavobacterium glycosylasparaginase: an N-terminal nucleophile hydrolase activated by intramolecular proteolysis. J. Biol. Chem. 273, 20205–20212 [DOI] [PubMed] [Google Scholar]
- 30. Saarela J., Oinonen C., Jalanko A., Rouvinen J., Peltonen L. (2004) Autoproteolytic activation of human aspartylglucosaminidase. Biochem. J. 378, 363–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Michalska K., Brzezinski K., Jaskolski M. (2005) Crystal structure of isoaspartyl aminopeptidase in complex with L-aspartate. J. Biol. Chem. 280, 28484–28491 [DOI] [PubMed] [Google Scholar]
- 32. Kim Y., Yoon K., Khang Y., Turley S., Hol W. G. (2000) The 2.0 Å crystal structure of cephalosporin acylase. Structure 8, 1059–1068 [DOI] [PubMed] [Google Scholar]
- 33. Michalska K., Hernandez-Santoyo A., Jaskolski M. (2008) The mechanism of autocatalytic activation of plant-type l-asparaginases. J. Biol. Chem. 283, 13388–13397 [DOI] [PubMed] [Google Scholar]
- 34. Yasuhira K., Shibata N., Tanaka Y., Kumagai N., Tanaka Y., Nagai K., Kato D., Takeo M., Negoro S., Higuchi Y. (2011) Crystallization and X-ray diffraction analysis of nylon-oligomer hydrolase (NylC) from Agromyces sp. KY5R. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 67, 892–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sambrook J., Russell D. W. (2001) Molecular Cloning: A Laboratory Manual, 3rd Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 36. Mochiji K., Hashinokuchi M., Moritani K., Toyoda N. (2009) Matrix-free detection of intact ions from proteins in argon cluster secondary ion mass spectrometry. Rapid Commun Mass Spectrom. 23, 648–652 [DOI] [PubMed] [Google Scholar]
- 37. Otwinowski Z., Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 38. Weeks C. M., Blessing R. H., Miller R., Mungee R., Potter S. A., Rappleye J., Smith G. D., Xu H., Furey W. (2002) Towards automated protein structure determination: BnP, the SnB-PHASES interface. Z. Kristallogr. 217, 686–693 [Google Scholar]
- 39. de La Fortelle E., Bricogne G. (1997) Maximum-likelihood heavy atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods Methods Enzymol. 276, 472–494 [DOI] [PubMed] [Google Scholar]
- 40. McRee D. E. (1993). Practical Protein Crystallography, Academic Press, San Diego, CA [Google Scholar]
- 41. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 42. Krissinel E., Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 [DOI] [PubMed] [Google Scholar]
- 43. Holm L., Sander C. (1993) Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 233, 123–138 [DOI] [PubMed] [Google Scholar]
- 44. Moritani K., Mukai G., Hashinokuchi M., Mochiji K. (2009) Site-specific fragmentation of polystyrene molecule using size-selected argon gas cluster ion beam. Appl. Phys. Express. 2, 046001 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.