

Background: TLR4 signaling requires unknown interactions between TIR domains of TLR4 and its adapters.
Results: We identify three binding sites in the TLR4 TIR domain that are important for TLR4 interactions.
Conclusion: Two binding sites in TLR4 are important for adapter binding and NF-κB activation.
Significance: This work provides new insights in the first steps of TLR activation.
Keywords: Innate Immunity, Mal TIRAP, Molecular Modeling, Mutagenesis Site-specific, MyD88, Structural Biology, Toll IL-1 Receptor (TIR) Domain, Toll-like Receptors (TLR), TRIF
Abstract
Toll-like receptor signaling requires interactions of the Toll/IL-1 receptor (TIR) domains of the receptor and adapter proteins. Using the mammalian protein-protein interaction trap strategy, homology modeling, and site-directed mutagenesis, we identify the interaction surfaces in the TLR4 TIR domain for the TLR4-TLR4, TLR4-MyD88 adapter-like (MAL), and TLR4-TRIF-related adapter molecule (TRAM) interaction. Two binding sites are equally important for TLR4 dimerization and adapter recruitment. In a model based on the crystal structure of the dimeric TLR10 TIR domain, the first binding site mediates TLR4-TLR4 TIR-TIR interaction. Upon dimerization, two identical second binding sites of the TLR4 TIR domain are juxtaposed and form an extended binding platform for both MAL and TRAM. In our mammalian protein-protein interaction trap assay, MAL and TRAM compete for binding to this platform. Our data suggest that adapter binding can stabilize the TLR4 TIR dimerization.
Introduction
Toll-like receptors (TLRs)3 are pathogen recognition receptors that play a crucial role in innate immunity (1–3). TLRs are composed of an extracellular domain with leucine-rich repeats for ligand recognition, a transmembrane helix, and a cytoplasmic part with a Toll-IL-1R (TIR) domain. Ligand-induced receptor dimerization or oligomerization leads to the recruitment of TIR domain-containing adapter proteins for downstream signaling.
Lipopolysaccharide (LPS) binding to the TLR4 complex is followed by interaction with the adapter proteins MAL and TRAM. MAL and TRAM are bridging adapters that recruit the signaling adapters MyD88 and TRIF to the receptor (4–8).The TLR4-MAL-MyD88 complex is formed at the plasma membrane, and localization of MAL at the plasma membrane is facilitated by its phosphatidylinositol 4,5-bisphosphate binding domain (6). The MAL/MyD88 axis leads to activation of MAPKs and of transcription factors, activation protein 1 (AP-1) and NF-κB. A myristoylation site of TRAM directs this adapter protein to the membrane of early endosomes (7). The TLR4-TRAM-TRIF complex is formed in endosomal compartments and leads to activation of the transcription factor IRF3 and interferon production (9).
Interactions between the TIR domains are crucial for TLR signal transduction. Both the receptor and the adapter TIR domains can dimerize or oligomerize; the receptor TIR domains interact with adapter TIR domains, and additionally, the TIR domains of MAL and TRAM interact;, respectively, with the TIR domains of MyD88 (10) and TRIF (11). Although the molecular structures of the TIR domain of TLR1, TLR2, TLR10, IL-1RAPL, MAL, and MyD88 were determined (12–17), the TIR domain interaction mechanism and the exact positions of the interaction interfaces remain unknown.
The TIR domain contains a central fully parallel five-stranded β sheet (βA through βE) surrounded by five α helices (αA through αE). The β sheets and α helices are connected by loops (AA–EE). Various articles demonstrate the importance of the BB loop between the βB strand and the αB helix (18). A P712H mutation in the BB loop of TLR4 in C3H/HeJ mice leads to complete unresponsiveness to LPS (19). Small synthetic peptides based on the BB loop sequence can act as inhibitors of TIR-TIR interactions and of TLR or IL-1 signal transduction (20–24). Peptidomimetics of the BB loop exert a similar inhibitory effect on TLR signaling and have anti-inflammatory properties in vivo (22, 24)
The TLR10 crystal structure was proposed as a good model for TLR TIR-TIR dimerization, with an interface formed by the DD loop, BB loop, and αC helix (15). The BB loops in this dimer interact with the reciprocal BB loop and αC helix, explaining how BB loop peptides and peptidomimetics can inhibit TIR-TIR interactions (15).
In 2002, Ronni et al. (25) published an alanine scan mutagenesis study of the TLR4 TIR domain. Mapping of the mutations on a TLR4 TIR homology model revealed the importance of at least two surface patches, corresponding to the BB loop and to the DD loop and residues in the αC′ helix. Interestingly, none of the mutations in this study showed specificity in their effects for any of the different pathways. This led to the suggestion that pathways diverge downstream of the adapters or that different adapters all bind to the same TLR4 TIR-binding sites.
The mammalian protein-protein interaction trap (MAPPIT) technique allows studying TIR-TIR interactions in detail in situ in intact living cells (26). In this study, we use this method plus NF-κB and IRF-3 reporter assays in combination with site-directed mutagenesis and homology modeling to determine the specific interaction sites for dimerization or oligomerization and adapter recruitment in the TLR4 TIR domain. We developed an assay in which we can specifically detect the TLR4-TLR4, TLR4-MAL, and TLR4-TRAM TIR-TIR interactions. Mutations in two binding sites simultaneously affect all three interactions. We propose a model based on the TLR10 TIR domain structure, in which TLR TIR dimerization is required for formation of an extended binding platform for both the MAL and TRAM adapters.
EXPERIMENTAL PROCEDURES
Vectors
The pMG2-SVT (SV40 large T protein), pMG2-SH2β, pMG2-MAL, and pMG2-TRAM MAPPIT prey vectors were described earlier (26–28). For generation of the pMG2-TLR4ic MAPPIT prey vector, the TLR4ic DNA fragment from the pCLL-TLR4ic bait (26) was amplified with primers 1 and 2 (supplemental Table 1) and cloned in the pMG2 prey vector with EcoRI and NotI. The pCLG TLR4ic MAPPIT bait vector was generated by recloning the TLR4ic DNA fragment from the pCLL-TLR4ic bait (26) in the MAPPIT bait vector pCLG (29). The pCLG-TLR4ic bait was mutated with primers 5 and 6, just before Gly-663 of the TLR4ic DNA fragment, to introduce an AgeI site that allows recloning of TIR domain mutants into the pMX-FLAG-TLR4-IRES-GFP constructs (see below). The TLR4ic DNA fragment in the pCLL-TLR4ic bait was amplified using primers 3 and 4 and ligated in the pCLG bait vector via restriction sites SacI and NotI. The TLR4ic mutants were generated via the QuickChangeTM site-directed mutagenesis method (Stratagene) with the primers 17–78 listed in supplemental Table 1, except for mutants Q704A, N792A, E798A, and R810S, for which the inserts were made via gene synthesis (Geneart). The pXP2d2-rPAPI-luciferase reporter, originating from the rat pancreatic associated protein I (rPAPI) promoter was previously described by Eyckerman et al. (30).
The pMX-TLR4-IRES-GFP vector was created by ligating FLAG-tagged TLR4 from the pFLAG-CMV1-TLR4 vector (gift from Dr. Luke O'Neill) into the pMX-IRES-GFP vector (gift from Dr. Stefan Constantinescu). The existing NgoMIV site in pMX-IRES-GFP was deleted (primers 7 and 8), and StuI and XhoI sites were inserted by ligating annealed primers 9 and 10 into the BamHI/NotI opened vector. FLAG-tagged TLR4 was inserted in the construct via XmnI (compatible with StuI) and XhoI. A new NgoMIV (compatible with AgeI) site was inserted with primers 11 and 12 just before Gly-663 of TLR4 to allow exchange of TLR4ic mutants from the pCLG-TLR4ic MAPPIT bait vector. The pMX-mMD-2 and pMX-mCD14 plasmids were the kind gifts from Dr. Shinichiroh Saitoh (31). The pNFconluc reporter was a gift from Dr. Alain Israel. The pFLAG-CMV1-hMD2 plasmid was a kind gift of Dr. D. Golenbock. The GAL4/IRF3, GAL4DBD, and p55 UASG Luc plasmids were the kind gifts of Dr. T. Fujita (32). The pE-CMV1-TLR4 vector was created by insertion of a SacII site (primers 13 and 14) in the pFLAG-CMV1-TLR4 vector and subsequent ligation of an annealed primer pair coding for the E-tag (primers 15 and 16) via restriction sites SacII and ClaI. Inserts originating from the pMX-FLAG-TLR4-IRES-GFP mutant vectors were cloned into pE-CMV1-TLR4 with ClaI/XhoI.
pMET7-FLAG-TRAM was generated by amplification of full-length TRAM from HEK293 cDNA, via primers 79 and 80. After BsteII/XhoI digestion, the fragment was cloned in the pMet7 vector.
MAPPIT Analysis
HEK293T cells were maintained in a 5% CO2-humidified atmosphere at 37 °C and were grown in DMEM with 10% fetal bovine serum and 100 μg/ml gentamicin (Invitrogen). Typically, 104 cells were seeded the day before transfection in a black 96-well plate (Nunc). Eight replicate wells were transfected overnight with 50 ng of bait, 30 ng of prey, and 10 ng of pXP2d2-rPAPI-luciferase reporter vector per well using a standard calcium phosphate precipitation procedure. One day after transfection, four wells were stimulated with 100 ng/ml leptin, although the other four wells were left unstimulated. The next day, the cells were lysed in 50 μl of CCLR buffer (25 mm Tris phosphate, pH 7.8, 10 mm DTT, 10 mm 1,2-diaminocyclohexane-N,N,N′,N′-tetraacetic acid, 50% glycerol, 5% Triton X-100), and just before measuring 35 μl of luciferin substrate buffer was added (40 mm Tricine, 2.14 mm (MgCO3)4Mg(OH)2·5H2O, 5.34 mm MgSO4·7H2O, 66.6 mm DTT, 0.2 mm EDTA, 270 μm coenzyme A (Sigma), 530 μm ATP (Sigma), 470 μm luciferin (Duchefa)). Luciferase activity was measured in quadruplicate using a chemiluminescence reader (TopCount, Packard Instrument Co. and PerkinElmer Life Sciences). The MAPPIT signal was expressed as fold induction (leptin stimulated/unstimulated) of luciferase activity.
Determination of MAPPIT Bait Surface Expression via a Leptin-SEAP Binding Assay
A chimeric mouse leptin-alkaline phosphatase fusion protein (leptin-SEAP) was produced in HEK293T cells as described before (33, 34). The cell supernatant containing leptin-SEAP was used to determine the expression of the MAPPIT baits as follows. The day before transfection, 3 × 105cells/well were seeded in 6-well plates. 2 μg of pCLG-TLR4ic wild type or mutant bait vector was transfected per well. The cells were detached 48 h post-transfection with PBS buffer containing 1% fetal calf serum and 0.5 mm EDTA. Subsequently, the cells were incubated for 2 h at 4 °C in DMEM with 1/20 leptin-SEAP supernatant or with 1/20 leptin-SEAP and an excess of leptin (1 μg/ml) to determine aspecific leptin-SEAP binding. After three washing steps with DMEM containing 10% fetal calf serum and 0.01% Tween, the cells were lysed in PhosphaLight (Tropix) dilution buffer and treated at 65 °C for 30 min to inactivate endogenous alkaline phosphatases. Alkaline phosphatase activity was measured using the CSPD substrate (PhosphaLight, Tropix) according to the manufacturer's specifications in a TopCount chemiluminescence counter (Packard Instrument Co.).
TLR4 NF-κB Reporter Assays
The day before transfection, 8 × 103 HEK293 cells were seeded in black 96-well plates. Cells were transfected with 16 ng of pMX-FLAG-TLR4-IRES-GFP WT or mutant vector, 16 ng of pMX-mMD-2 plasmid, 16 ng of pMX-mCD-14 plasmid, and 4 ng of pNFconcluc reporter. One day after transfection, the cells were treated with 0, 10, 100, or 1000 ng/ml LPS (standard LPS, Escherichia coli K12, InvivoGen). After 24 h, the cells were lysed, and luciferase activity was measured in triplicate as described above.
TLR4 IRF3 Reporter Assays
The day before transfection, 8 × 103 HEK293 cells were seeded in black 96-well plates. Cells were transfected with 10 ng of pMX-FLAG-TLR4-IRES-GFP WT or mutant vector, 10 ng of pFLAG-CMV1-hMD2, 10 ng of pEF6-FLAG-hCD-14, 0.25 ng of GAL4/IRF3 or GAL4/DBD, and 7.5 ng of IRF3 reporter (p55 UASG Luc) per well. After 48 h, the cells were lysed, and luciferase activity was measured in triplicate as described above.
Western Blot Analysis of TLR4 Surface Expression
2 × 106 HEK293T cells were seeded in 60-cm2 Petri dishes and transfected the next day with 10 μg of pMX-FLAG-TLR4 wild type or mutant. 48 h after transfection, the cells were lysed in RIPA buffer (200 mm NaCl, 50 mm Tris-HCl, pH 8.0, 0.05% SDS, 2 mm EDTA, 1% Nonidet P-40, 0.5% sodium deoxycholate, CompleteTM protease inhibitor mixture (Roche Applied Science)), and the FLAG-tagged proteins were captured overnight on anti-FLAG® M2 magnetic beads (Sigma) at 4 °C. The beads were washed three times with RIPA and subsequently heated at 95 °C during 10 min in 1× loading buffer (40 mm Tris-HCl, pH 6.8, 2% SDS, 8% glycerol, 0.01% bromphenol blue, 2.5% β-mercaptoethanol). The samples were electrophoresed on a 7.5% SDS-polyacrylamide gel and electroblotted onto a nitrocellulose membrane. FLAG-tagged proteins were revealed using monoclonal anti-FLAG antibody M2 (Sigma), and secondary anti-mouse DyLight 680 antibody (Thermo Scientific) was used for detection, and Western blot analysis was performed using the Odyssey Infrared Imaging System (Li-Cor).
AlphaScreenTM Analysis
AlphaScreenTM experiments were performed according to the manufacturer's protocol (PerkinElmer Life Sciences). Cell culture conditions and transfections were comparable with those described in the leptin-SEAP assay. 1 μg of pE-CMV1-TLR4 plasmid encoding for E-tagged TLR4 wild type and mutants and 1 μg of pMET7-FLAG-TRAM plasmid were co-transfected in HEK293T cells. 48 h after transfection, cells were lysed in 50 mm Tris-HCl, pH 7.5, 125 mm NaCl, 5% glycerol, 0.2% Nonidet P-40, 1.5 mm MgCl2, 25 mm NaF, 1 mm Na3VO4, Complete protease inhibitor mixture (Roche Applied Science). The lysates were incubated for 2 h at room temperature with biotinylated anti-E-tag antibody (Amersham Biosciences) and subsequently for 2 h with the AlphaScreenTM FLAGTM (M2) detection kit (PerkinElmer Life Sciences) acceptor beads. Subsequently, streptavidin donor beads were added, and the lysates were further incubated for an additional 30 min. The lysates were transferred into 384-well plates and measured in triplicate using the EnVision plate reader (PerkinElmer Life Sciences). The anti-E-tag antibody (Amersham Biosciences) was biotinylated with sulfo-NHS-biotin (Pierce) according to the manufacturer's guidelines.
Expression of the E-tagged TLR4 and FLAG-tagged TRAM was verified via Western blot analysis as described above. E-tagged proteins were revealed using monoclonal anti-E-tag antibody M2 (Amersham Biosciences) and secondary anti-mouse-DyLight 680 antibody (Thermo Scientific).
Homology Modeling
Homology models for the monomeric human TLR4 TIR domain (residues 674–815) were built using modeler version 9.7 (35) based on a MAFFT (36) alignment of the 10 human TLRs and mammalian orthologues. The loop model procedure was used, and 400 models were built. The final model was selected based on the DOPE (Discrete Optimized Protein Energy) and molpdf (molecular probability density function) scores. Homology models for a TLR4 TIR dimer were built in a similar way using the TLR10 TIR dimer structure as a template, with symmetry restraints between the interacting TIR domains. Models were visualized and pictures generated using University of California San Francisco chimera (37). % residue solvent accessibility (% Rsa) in supplemental Table 2 was calculated as (water accessibility of the residue in the TLR4 TIR model·100/water accessibility of the residue type in an extended Ala-Xaa-Ala tripeptide) using NACCESS (47).
RESULTS
MAPPIT Is Able to Detect Mutants That Disrupt TIR-TIR Interactions
Despite the available structural knowledge on the TIR domains, theoretical docking models, and mutagenesis data, it remains unclear how TIR domains exactly interact with each other. MAPPIT combined with site-directed mutagenesis allows distinguishing between residues that specifically affect receptor-receptor interaction or receptor-adapter interaction. We used MAPPIT to study the role of potential binding sites of the TLR4 TIR domain for its dimerization or oligomerization or for its interaction with MAL or TRAM. The MAPPIT principle and configuration used in this study are outlined in supplemental Fig. 1.
The pCLG-TLR4ic MAPPIT bait contains the intracellular domain of human TLR4 (TLR4ic) as bait. The MAPPIT preys consist of human TLR4ic, MAL, or TRAM coupled to the C terminus of a gp130 receptor tail. The pCLG-TLR4ic bait was transiently co-transfected in HEK293T cells with the TLR4ic, MAL, or TRAM prey (Fig. 1). In parallel, a pCLG bait that lacks the TLR4ic and an irrelevant prey, the SVT antigen coupled to the C terminus of a gp130 receptor tail, were used as negative controls. The SH2β prey containing SH2β adapter protein 1 coupled to the C terminus of a gp130 receptor tail was used as a positive control. SH2β adapter protein 1 binds to JAK2 and leads to STAT3 activation with every MAPPIT bait with proper surface expression. The cell surface expression of all pCLG-TLR4ic bait mutants was further tested in a leptin-SEAP assay, in which leptin labeled with alkaline phosphatase binds to the extracellular part of the bait receptor (34). All MAPPIT assays described in this study are paralleled by these controls (supplemental Table 2). Via MAPPIT, we can specifically detect the TLR4ic-TLR4ic, TLR4ic-TRAM, and TLR4ic-MAL interaction (Fig. 1).
FIGURE 1.
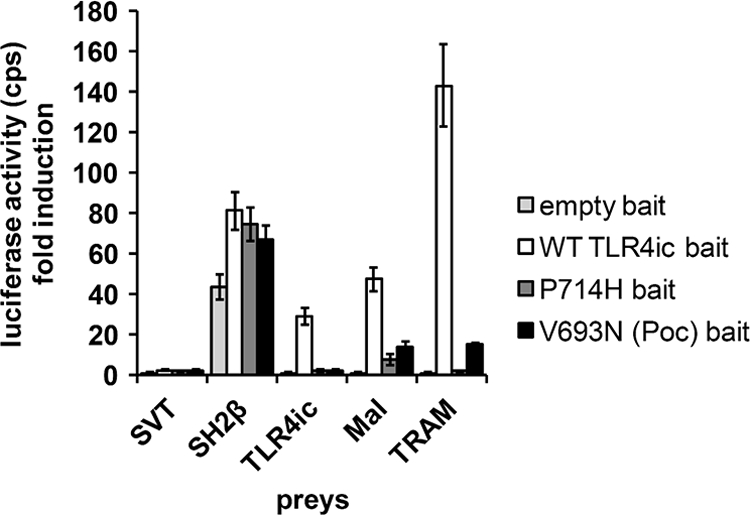
MAPPIT detects disruptive TLR4ic mutants. HEK293T cells were transiently co-transfected with plasmids encoding the empty bait, WT TLR4ic bait, P714H, or V693N mutant bait and the prey constructs encoding for the SVT (negative control), SH2β (positive control), TLR4ic, MAL, and TRAM preys. The transfected cells were either stimulated for 24 h with leptin (100 ng/ml) or were left untreated. Luciferase readouts were performed in quadruplicate. The interaction of TLR4ic with TLR4ic, MAL, or TRAM is detectable and abolished by the P714H and V693N mutations.
In this work, we study the effect of mutations in potential binding sites in the TLR4 TIR domain on its dimerization and its interaction with MAL and TRAM in MAPPIT. As a proof of concept for these mutagenesis studies, we tested the effect of the P714H and the Poc mutation on the different MAPPIT assays. Both mutations abolish TLR4 signaling (17, 18). The MAPPIT data, presented in Fig. 2B, show that these mutations in the TLR4ic bait disrupt all three interactions (Fig. 1). This shows that the MAPPIT technique allows detecting mutants that disrupt interactions of TLR4ic with TLR4ic and the adapter proteins.
FIGURE 2.

Identification of three candidate binding sites in a homology model of the TLR4 TIR domain. The model is shown in three orientations. The suggested candidate binding sites are indicated by a dashed line. A, residue conservation of TLR4. Residues are colored according to the ClustalX (45) score in an alignment of 29 TLR4 orthologues. Red, highly conserved; blue, less conserved. B, indication of the alanine scanning mutagenesis data of Ronni et al. (25). Residues are colored according to the NF-κB signal versus the WT in that study. Blue, 100% of WT; red, 0% of WT; black, not mutated in that study. C, secondary structure elements, box 1–3 motifs and position of binding sites I–III in the human TLR4 sequence. Binding sites I–III residues are labeled 1–3 below the sequence. The box 1–3 motifs of the TIR domain as originally defined by Slack et al. (46) are underlined.
Identification of Three Putative TIR-TIR Interaction Surfaces in the TLR4 TIR Domain
For our mutagenesis study, we first selected potential binding sites in the TLR4 TIR domain. Via alanine scanning mutagenesis, Ronni et al. (25) identified mutations of the TLR4 TIR domain that affect different TLR4 signaling pathways. We mapped the effect of these mutations on a homology domain for the TLR4 TIR domain. This map was compared with the residue conservation in 29 TLR4 orthologues. Fig. 2A shows that mutations that affect the NF-κB signaling pathway in the study of Ronni et al. (25) cluster in three surface areas in the TLR4 TIR domain. These surface areas correspond with the areas with higher residue conservation (Fig. 2B). We hypothesized that these three areas may form three binding sites, I–III, for TIR-TIR interactions. In the model, potential binding sites I and II form two contiguous surfaces that form an 80° angle. Both surfaces contain residues of the BB loop. Pro-714 is found at the edge between both surfaces. Potential binding site III is found opposite the potential binding site I. In this study, we test the effect of mutations in the three potential binding sites on dimerization of the TLR4 TIR domain and on interaction of the TLR4 TIR domain with the TIR domain of MAL and TRAM. The position of the mutations and of different structural elements in TLR4 is shown in Fig. 2C.
MAPPIT Data Suggest That BSI and BSII Play a Crucial Role in TLR4ic Oligomerization and Adapter Binding, whereas BSIII Is Specifically Involved in TLR4ic Oligomerization
We tested the effects of mutations in potential binding sites I–III of TLR4 on the TLR4ic-TLR4ic, TLR4ic-MAL, and TLR4ic-TRAM interactions in our MAPPIT assays. Mutations in BSI that negatively affected TLR4ic-TLR4ic interaction also disrupted the interaction of TLR4ic with adapter proteins MAL and TRAM (Fig. 3A). Similar results were obtained mutating BSII (Fig. 3B). Mutations in BSIII specifically impair TLR4ic-TLR4ic binding (Fig. 3C). The effect of the mutations on the different interactions is summarized in supplemental Table 2. Solvent-exposed control mutations, which are not located in one of the binding sites and are not predicted to disturb the protein structure, do not affect any of the interactions. The mutations involved in binding were mapped on the TLR4 TIR model (Fig. 4). These models suggest that all three binding sites are important for TLR4 oligomerization (Fig. 4, left panel) and that BSI and BSII are both also necessary for adapter binding (Fig. 4, middle and right panels).
FIGURE 3.

A–C, MAPPIT analysis of TLR4ic mutants. HEK293T cells were transiently co-transfected with plasmids encoding for the TLR4ic bait wild type and mutant constructs and the prey constructs encoding for proteins TLR4ic, MAL, and TRAM, combined with the pXP2d2-rPAP-luci reporter. The MAPPIT results are expressed in % of TLR4icWT interaction. A, mutants in BSI; B, mutants in BSII; C, mutants in BSIII.
FIGURE 4.

Model of the TLR4 TIR domain with the mutants that abrogate the interaction indicated in red. The numbers refer to the binding sites. Left panel, mutations in the three conserved regions of the TLR4 TIR domain affect its dimerization in MAPPIT; middle panel, binding sites I and II mutations strongly affect TLR4ic-MAL interaction, although mutations in BSIII have less effect; right panel, binding sites I and II mutations strongly affect TLR4ic-TRAM interaction, although mutations in BSIII have no effect.
The Y794A and E796A/E798A mutations have more drastic effects than the other eight mutations in binding site III. The aromatic ring of Tyr-794 is buried, and only the hydroxyl group is really available for protein interactions. Similarly, the solvent accessibility of the Glu-796 and the Glu-798 side chains in our model(s) is rather low. Both mutations may affect the structure of the protein, as Y794F, E796A, and E798A mutations and five other mutations in binding site III do not have strong effects on MAL or TRAM binding.
The AlphaScreenTM technology (PerkinElmer Life Sciences) was used for validation of the MAPPIT data for the TLR4ic-TRAM interaction. Full size E-tagged TLR4 WT or mutant was co-expressed with FLAG-tagged TRAM, and TLR4-TRAM protein interactions were detected using the AlphaScreenTM FLAGTM (M2) detection kit (PerkinElmer Life Sciences). In line with the MAPPIT data, mutations in BSI and -II weaken the TLR4-TRAM interaction, although mutations in BSIII have no effect on the TLR4-TRAM interaction (Fig. 5 and supplemental Table 2). Western blotting showed a good expression of most E-tagged TLR4 constructs, and small expression variability seemed not to influence the AlphaScreenTM results.
FIGURE 5.

Top, AlphaScreenTM analysis of the interaction between TLR4ic mutants and TRAM. HEK293T cells were transiently co-transfected with plasmids encoding the E-tagged TLR4 WT or mutants and FLAG-tagged TRAM. The transfected cells were lysed after 48 h, and protein interactions were detected with the AlphaScreen FLAGTM (M2) detection kit (PerkinElmer Life Sciences). The results are displayed as fold induction, namely relative to the counts of an irrelevant E-tagged construct. Bottom, expression levels of the E-tagged TLR4 constructs were detected on Western blot; TLR4 is the lower band on the blot. In the 1st lane (negative control (neg. ctrl)), the control lysate was loaded; the order of the other loaded samples aligns with the order of the samples on the graph.
NF-κB and IRF-3 Signaling Studies Confirm the Importance of BSI and BSII for Signal Transduction
The alanine scanning mutagenesis data of TLR4 of Ronni et al. (25) were an important guide in our definition of the three potential binding sites. However, many of the mutations mapped in Fig. 2A are combined mutations of several consecutive exposed and buried residues. Moreover, in the study of Ronni et al. (25), the TLR4 TIR domain was coupled to the external part of the CD4 receptor, which can only dimerize, but a role for oligomerization of the full-length TLR4 cannot be excluded (4).
We therefore opted to test the effect of our mutations in full-length TLR4. HEK293 cells were transiently co-transfected with a pMX-mMD-2, pMX-mCD14, and a pMX-TLR4-IRES-GFP WT or mutant vector together with pNFconluc reporter plasmid. This assay allowed us to study NF-κB signaling of TLR4 mutants in an LPS dose-dependent manner (Fig. 6A). The effect of the TLR4 mutations on IRF-3 activation was determined via an IRF-3/Saccharomyces cerevisiae GAL4 DNA binding domain fusion protein that induces a UASG/luciferase reporter upon IRF-3 activation (Fig. 6B) (32).
FIGURE 6.

A, LPS-induced NF-κB activation of TLR4 mutant HEK293 cells were transiently co-transfected with a pMX-mMD-2, pMX-mCD14, and pMX-TLR4-IRES-GFP WT or mutant vector together with the pNFconluc reporter plasmid. After 24 h, the cells were stimulated overnight with 0, 10, 100, or 1000 ng/ml LPS. Luciferase measurements were performed in triplicate. B, effect of TLR4 mutant expression on IRF3/GAL4DBD activation. Data are presented as % of wild type TLR4 and present the median of three separate transfection experiments. All tested mutants affect the IRF3/DBD activation.
Our data show that binding site I and II are indispensable for NF-κB signaling (Fig. 6A). Overall, the mutants disrupting the interaction of TLR4ic with MAL and TRAM negatively affect NF-κB signaling. Most mutations in binding site III that specifically abrogate TLR4ic-TLR4ic interaction in MAPPIT have no strong effect on NF-κB signaling (supplemental Table 2). In contrast, our data suggest that mutations in binding sites I–III all affect IRF-3 activation (Fig. 6B). Binding site III may thus have a specific role in the TRIF-dependent pathway.
MAL and TRAM Bind to an Identical or Overlapping Binding Site
The MAPPIT results indicate that MAL and TRAM interaction with TLR4ic are both disrupted by mutations in binding site I and II. This suggests that MAL and TRAM require the same or at least overlapping binding sites. We set up an assay to detect competitive binding between the two adapter proteins. For this purpose, we co-expressed FLAG-tagged TRAM or MAL together with the TLR4ic bait and the MAL or TRAM prey, respectively. Co-transfection of the competitive adapter results in disruption of the MAPPIT interaction, in line with our hypothesis that MAL and TRAM bind to an identical or overlapping binding site in TLR4ic (Fig. 7). The TIR domain is required for strong competition (data not shown). On average, co-transfection of TRAM leads to 57% inhibition, although a TIR-less variant of Tram has no effect. Co-transfection of MAL leads to 85% inhibition, although a TIR-less MAL variant has a partial inhibitory effect (35% inhibition).
FIGURE 7.

Competition between MAL and TRAM in MAPPIT. HEK293T cells were transiently co-transfected with plasmids encoding for the Mock or TLR4ic bait and the SVT (negative control), SH2β (positive control), and MAL (A) or TRAM prey (B). A, comparison of TLR4ic-MAL interaction with and without co-transfection of plasmid encoding for TRAM. Dashed arrow, TRAM overexpression specifically inhibits the MAPPIT signal for the TLR4ic-Mal interaction. B, comparison of the TLR4ic-TRAM interaction with and without co-transfection of plasmid encoding for MAL. Dashed arrow, MAL overexpression specifically inhibits the MAPPIT signal for the TLR4ic-TRAM interaction.
Dimerization of TLR4ic Increases after Co-transfection of TRAM
Our MAPPIT data show that binding site I and II, which according to our model (Fig. 1) are positioned 80° away from each other, are both important for adapter recruitment and for TLR4ic dimerization. This suggests that dimerization and adapter recruitment have a synergistic effect in TIR-TIR interactions. To test this hypothesis, a MAPPIT assay was developed in which we co-expressed E-tagged TRAM together with the TLR4ic bait and prey. Fig. 8A shows that co-expression of TRAM resulted in an increase of the luciferase induction, supporting the assumption that the adapter TIR domains support or stabilize dimerization of the TLR4ic. Co-expression of MAL or a TRAM mutant that lacks its TIR domain does not increase the luciferase activity (data not shown).
FIGURE 8.

A, adapter-induced dimerization of TLR4ic in MAPPIT. HEK293T cells were transiently co-transfected with plasmids encoding for the mock or TLR4ic bait and the SVT (negative control), SH2β (positive control), and TLR4ic prey. Dashed arrow, TLR4ic-TLR4ic interaction is specifically enhanced by co-transfection of TRAM. B, rescue experiment of the TLR4ic-TLR4ic interaction of TLR4ic-binding site III mutants. 200 ng of plasmid encoding for E-tagged TRAM or empty vector (Mock) was co-transfected together with TLR4ic WT or mutant bait vector and TLR4ic prey vector. The leftmost interaction is a TLR4-independent MAPPIT interaction of the pCLG-CT2 bait with the pMG2-CT1 prey, used as a control. TLR4ic-TLR4ic interactions disrupted by mutations in binding site III are rescued by co-expression of the TRAM adapter protein.
Mutations in binding site III only affect receptor dimerization but still allow adapter binding and do not strongly affect TLR4 NF-κB activation. We tested whether the inhibitory effect of site III mutations on the TLR4ic-TLR4ic interaction could be negated by co-expression of TRAM. Fig. 8B shows that co-expression of TRAM specifically rescues TLR4ic-TLR4ic interaction of these mutants.
Mapping of Multiple Data on a Dimer Model for TLR4 TIR Domain Suggests Dimerization via Binding Site II and Adapter Binding to Binding Site I
The dimeric TLR10 TIR domain crystal structure was presented as a good model for TLR TIR domain dimerization (15). We therefore built a homology model for the TLR4 TIR domain dimer based on this structure. We mapped our MAPPIT data, amino acid conservation, and alanine scanning data of Ronni et al. (25) on this model.
The TLR4 dimer interface corresponds with binding site II, and this dimerization juxtaposes the binding sites I of both TIR domains. Both binding sites I thus form an extended platform where mutations that disrupt TLR4-TLR4 and TLR4-adapter TIR TIR interactions cluster (Fig. 9, A–D). The mutations in this platform also affect different signaling pathways, and the platform shows higher residue conservation (Fig. 9, E and F). Interestingly, the conserved platform seems to be large enough to bind two adapters as discussed below (Fig. 9G).
FIGURE 9.

Model of the TLR4 TIR domain dimer based on the TLR10 TIR domain structure (15). A, C, and E, top view showing the adjacent binding sites I. B, D, F, and G, side view, showing the position of binding site II. The green line in E represents the conserved platform formed by two adjacent binding sites I. The green line in D and F indicates the approximate docking site of the MAL (D) and TRAM (F) TIR domain in the model presented by Núñez Miguel et al. (38). A and B, effect of a mutation on the TLR4 TIR-TRAM interaction. Residues are colored according to the MAPPIT signal in the TLR4 TIR-TRAM MAPPIT assay. Red, 0% of WT (binding is completely disrupted); yellow, 50% of WT; green, 100% of WT (binding is unaffected). C and D, residue conservation of TLR4. Residues are colored according to the ClustalX (45) score in an alignment of 29 TLR4 orthologues. Red, highly conserved; blue, less conserved. E and F, indication of the alanine scanning mutagenesis data of Ronni et al. (25). Residues are colored according to the NF-κB signal versus the WT in that study. Blue, 100% of WT; red, 0% of WT; black, not mutated in that study. G, effect of a mutation on the TLR4 TIR-TLR4 TIR interaction. Residues are colored according to the MAPPIT signal in the TLR4 TIR-TLR4 TIR MAPPIT assay. Red, 0% of WT (binding is completely disrupted); yellow, 50% of WT; green, 100% of WT (binding is unaffected). The position of binding site I–III is indicated by dashed lines, and a possible binding mode for adapters MAL and TRAM is suggested and compared with the R, S, and A interfaces as postulated by Xu et al. (12).
Núñez Miguel et al. (38) presented a similar TLR4 TIR dimer model but predicted different binding sites for MAL and TRAM based on in silico docking. These predicted binding sites for MAL and TRAM do not show increased conservation, and several mutations in this area, such as the Q755A and W757A mutation, hardly affect adapter binding or signaling pathways as demonstrated in our study and in the study by Ronni et al. (25) (Fig. 9, B, D, and F). We therefore favor a model where two adjacent binding sites I, as defined in this study, form the binding site for MAL and TRAM, as this is more in line with experimental data and residue conservation.
DISCUSSION
A crucial event in TLR signaling is the complex formation between the TIR domains of the dimerized or oligomerized receptor and the TIR domain-containing adapter proteins. The structures of different TIR domains have been determined, and the importance of different structural elements as the BB-loop has been demonstrated (1, 39). Different models for TIR-TIR interactions have been proposed, either based on structural data or on signaling data. However, the structure of a TLR-adapter complex has not been reported, and the architecture of TLR complexes is not yet completely understood. MAPPIT allows studying the separate TIR-TIR interactions, which makes it possible to detect residues or regions that affect one of the interactions. We established MAPPIT assays to detect TLR4ic-TLR4ic, TLR4ic-MAL, and TLR4ic-TRAM interactions, and we were able to show that mutations specifically involved in one of the interactions disturb the MAPPIT signal. Based on homology modeling, sequence conservation, and the results of an alanine scanning experiment of Ronni et al. (25), we selected three possible binding sites in the TLR4 TIR domain.
Mutations in all three possible binding sites abrogate TLR4 TIR-TLR4 TIR interactions, whereas adapter binding is only affected by mutations in binding site I and II. Only potential binding sites I and II in the TLR4 TIR domain are critical for LPS-induced NF-κB signaling, whereas an IRF3/GAL4-based assay suggests that binding site III may be important for IRF-3 activation. Binding site I contains residues of the αA and αB helix and of the BB and BC loop. Binding site II contains residues of the BB loop, DD loop, and αC helix.
Binding site II approximately corresponds with the interface found in the dimer of the TLR10 TIR domain crystal structure (15), which was proposed as a good model for TLR TIR dimerization (15). In a model for a TLR4 dimer based on this structure, binding site II forms the interface between two TLR4 TIR domains. The BB loops of two TLR TIR domains interact at the TLR TIR interface but are at the same time exposed, forming a possible interaction site for adapter recruitment (15). The exposed BB loops are part of the adjacent binding sites I of both TLR4 TIR domains. These adjacent binding sites I form a more extended platform, which may be important for adapter binding. An important role for this platform in TLR4 signaling is supported by our MAPPIT data, by residue conservation, and by the alanine scan data of Ronni et al. (25) (Fig. 9).
This extended platform may allow binding of two adapter molecules. This would be consistent with the existence of three types of interfaces, as postulated by Xu et al. (12), when reporting the first TIR domain crystal structures. Binding site II between two TLR4 TIR domains would correspond to the “R” face. The extended platform formed by two binding sites I of TLR4 forms the “S” face between the TIR domains of the TLR and the adapters. These adapter molecules presumably bind as dimers that interact via the “A” face (Fig. 9G).
Núñez Miguel l et al. (38) presented a similar TLR4 TIR model based on the dimer structure of the TLR10 TIR domain. These authors used in silico protein-protein docking of homology models to predict the binding site for the adapters TRAM and MAL on this TLR4 dimer. Both adapters were predicted to bind at symmetry-related sites at the TLR4 dimer interface indicated in Fig. 9, D and F. These predicted docking sites are not particularly conserved, and mutations at these sites do not affect adapter binding in our MAPPIT assays nor any of the tested TLR4 signaling pathways in the study of Ronni et al. (25). We therefore favor a model with two adjacent binding sites I as binding sites for MAL and TRAM.
The MAPPIT technique allows detecting mutations that affect binding specificity for different interaction partners. Nevertheless, in this study, we did not find mutations that specifically affect adapter recruitment. Instead, we find that mutations in binding site I and II affect both the TLR4-TLR4 and TLR4-adapter interactions in MAPPIT and AlphaScreenTM assays. This indicates that the TLR4 TIR domain needs to dimerize to allow recruitment of MAL and TRAM. Conversely, we were able to show that overexpression of TRAM enhances dimerization of the intracellular TLR4 domain. This suggests that binding of MAL or TRAM can stabilize the TLR4 TIR dimerization in our MAPPIT assays. This cooperativity between TLR4 TIR dimerization and adapter recruitment can explain why we did not detect any mutation that affects adapter-receptor TIR-TIR interaction without affecting receptor-receptor TIR-TIR interaction. Interestingly, the chemical compound TAK-242 specifically targets Cys-747 in binding site II in TLR4 and inhibits the MyD88-dependent and -independent TLR4 signaling pathways (40). We suggest that this compound inhibits TLR4 TIR dimerization and therefore adapter recruitment.
All mutations that affect MAL binding also affect TRAM binding in our MAPPIT assays, and our data suggest that the binding sites for MAL and TRAM on TLR4 strongly overlap. In line with this, thorough alanine scanning analysis of the TLR4 TIR domain by Ronni et al. (25) did not find any mutations that affect specific signal transduction pathways. In our MAPPIT assays, MAL and TRAM seem to compete for this common or overlapping binding site, which in our model is formed by the adjacent binding sites I. It is unclear whether this competition plays a role in TLR4 signal transduction, as the MAL/MyD88 signaling pathway starts at the plasma membrane, whereas the TRAM/TRIF-dependent pathway requires endocytosis (6, 7).
The model presented here for TLR TIR domain dimerization and adapter recruitment may apply for most if not all TLR TIR domains. Analysis of conservation of a TLR2 and TLR10 dimer for example shows a similar conserved binding site I platform (data not shown). Interestingly, TLR1 acquires the ability to bind MyD88 after mutation of residue 672 in its box1 motif (41). This residue corresponds to residue Asp-711 in binding site I of TLR4. Mutation of Arg-748, Phe-749, Leu-752, and Arg-753 in the DD loop and αD helix of TLR2 decreases TLR2/TLR1 signaling (42). These residues are in or at the edge of the predicted binding site II, which may form the TLR-TLR TIR-TIR interface in TLR2-mediated signaling.
Although mutations in predicted binding site I and II in this work affect all tested aspects of TLR4 signal transduction, we find that mutations in binding site III only affect the TLR4 TIR-TLR4 TIR interaction in our MAPPIT assay. Most mutations in binding site III did not strongly affect TLR4-induced NF-κB signaling or MAL or TRAM recruitment. Interestingly, our data suggest that binding site III may be specifically involved in IRF-3 activation. This is probably not a consequence of defective TRAM binding, but it may be related to the effect of binding site III mutations on the TLR4-TLR4 interaction as observed via MAPPIT. The inhibitory effect of the site III mutations on TLR4ic-TLR4ic interaction could be rescued by overexpression of TRAM. This probably means that binding site III mutations only affect the weak interaction between isolated TLR4ic domains. The mutations lose their effect when this interaction is stabilized or enhanced by overexpression of TRAM or by additional interactions in the activated full-length TLR4 receptor complex. We cannot rule out a role for this area of the TIR domain for receptor oligomerization. The higher order MyD88-IRAK4 “myddosome” complexes suggest that TLR4 may indeed form oligomers upon activation (4).
It is unclear whether equivalents of the binding sites defined in this work are present in the adapters MAL, MyD88, TRAM, and TRIF. The inhibitory effect of BB loop mutations on adapter functionality suggests that they may use similar type I- and II-binding sites for interaction via the “S interfaces” and “A interfaces” as defined above. Ohnishi et al. (16) reported three possible binding sites in their MyD88 structure, based on the effects of MyD88 TIR mutations on the inhibitory capacity of the MyD88 TIR domain in TLR4-mediated NF-κB activation. Binding sites II and III in this study were reported to mediate interaction with MAL. Superposition and alignment of the MyD88 and TLR4 structures and sequences show that these sites correspond to our binding sites I and III. Mutations of MAL at position Asp-198 inhibit TLR2 and TLR4 signaling and affect its interaction with TLR4 and MyD88 (43, 44). Asp-198 is found in a position that overlaps with the binding site III as defined in this study (43).
In conclusion, we demonstrated the importance of two binding sites in TLR4 adapter binding and signaling. Our data support a model of TLR4 TIR domain dimerization as found in the TLR10 TIR domain crystal structure. This dimerization is required for formation of a large conserved platform that contains the BB loop and box 1 motifs and that forms a potential binding site for the adapters MAL and TRAM. MAL and TRAM both bind to this platform, and adapter binding stabilizes the complex. It remains to be determined how MAL and TRAM interact with this binding site and how this leads to recruitment of MyD88. The MAPPIT method allows the detection of TLR4-MAL, TLR4-TRAM, MAL-MAL, and MAL-MyD88 interactions. A strategy that combines MAPPIT with mutagenesis of the adapter proteins as applied in this study can help to further define the interfaces in the TLR-adapter complexes.
Supplementary Material
Acknowledgments
We thank Dr. Stefan Constantinescu, Dr. Shinichiroh Saito, Dr. Alain Israel, Dr. Douglas Golenbock, Dr. Takashi Fujita, and Dr. Luke O'Neill for the kind gifts of different plasmids.
This work was supported by Ugent BOF Grant 01Z03306 and FWO “Krediet aan Navorsers” Grant 1.5.256.08N.

This article contains supplemental Fig. 1 and Tables 1 and 2.
- TLR
- Toll-like receptor
- TIR domain
- Toll/interleukin-1 receptor domain
- MAL
- MyD88 adapter-like
- Trif
- TIR-domain-containing adapter inducing interferon-β
- TRAM
- TRIF-related adapter molecule
- MAPPIT
- mammalian protein-protein interaction trap
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- SVT
- SV40 large T.
REFERENCES
- 1. Gay N. J., Gangloff M. (2007) Structure and function of Toll receptors and their ligands. Annu. Rev. Biochem. 76, 141–165 [DOI] [PubMed] [Google Scholar]
- 2. Hennessy E. J., Parker A. E., O'Neill L. A. (2010) Targeting Toll-like receptors. Emerging therapeutics? Nat. Rev. Drug Discov. 9, 293–307 [DOI] [PubMed] [Google Scholar]
- 3. Kawai T., Akira S. (2010) The role of pattern-recognition receptors in innate immunity. Update on Toll-like receptors. Nat. Immunol. 11, 373–384 [DOI] [PubMed] [Google Scholar]
- 4. Gay N. J., Gangloff M., O'Neill L. A. (2011) What the Myddosome structure tells us about the initiation of innate immunity. Trends Immunol. 32, 104–109 [DOI] [PubMed] [Google Scholar]
- 5. Jenkins K. A., Mansell A. (2010) TIR-containing adapters in Toll-like receptor signaling. Cytokine 49, 237–244 [DOI] [PubMed] [Google Scholar]
- 6. Kagan J. C., Medzhitov R. (2006) Phosphoinositide-mediated adapter recruitment controls Toll-like receptor signaling. Cell 125, 943–955 [DOI] [PubMed] [Google Scholar]
- 7. Kagan J. C., Su T., Horng T., Chow A., Akira S., Medzhitov R. (2008) TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-β. Nat. Immunol. 9, 361–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. O'Neill L. A., Bowie A. G. (2007) The family of five. TIR-domain-containing adapters in Toll-like receptor signaling. Nat. Rev. Immunol. 7, 353–364 [DOI] [PubMed] [Google Scholar]
- 9. Lu Y. C., Yeh W. C., Ohashi P. S. (2008) LPS/TLR4 signal transduction pathway. Cytokine 42, 145–151 [DOI] [PubMed] [Google Scholar]
- 10. Fitzgerald K. A., Palsson-McDermott E. M., Bowie A. G., Jefferies C. A., Mansell A. S., Brady G., Brint E., Dunne A., Gray P., Harte M. T., McMurray D., Smith D. E., Sims J. E., Bird T. A., O'Neill L. A. (2001) Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature 413, 78–83 [DOI] [PubMed] [Google Scholar]
- 11. Fitzgerald K. A., Rowe D. C., Barnes B. J., Caffrey D. R., Visintin A., Latz E., Monks B., Pitha P. M., Golenbock D. T. (2003) LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J. Exp. Med. 198, 1043–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu Y., Tao X., Shen B., Horng T., Medzhitov R., Manley J. L., Tong L. (2000) Structural basis for signal transduction by the Toll/interleukin-1 receptor domains. Nature 408, 111–115 [DOI] [PubMed] [Google Scholar]
- 13. Tao X., Xu Y., Zheng Y., Beg A. A., Tong L. (2002) An extensively associated dimer in the structure of the C713S mutant of the TIR domain of human TLR2. Biochem. Biophys. Res. Commun. 299, 216–221 [DOI] [PubMed] [Google Scholar]
- 14. Khan J. A., Brint E. K., O'Neill L. A., Tong L. (2004) Crystal structure of the Toll/interleukin-1 receptor domain of human IL-1RAPL. J. Biol. Chem. 279, 31664–31670 [DOI] [PubMed] [Google Scholar]
- 15. Nyman T., Stenmark P., Flodin S., Johansson I., Hammarström M., Nordlund P. (2008) The crystal structure of the human toll-like receptor 10 cytoplasmic domain reveals a putative signaling dimer. J. Biol. Chem. 283, 11861–11865 [DOI] [PubMed] [Google Scholar]
- 16. Ohnishi H., Tochio H., Kato Z., Orii K. E., Li A., Kimura T., Hiroaki H., Kondo N., Shirakawa M. (2009) Structural basis for the multiple interactions of the MyD88 TIR domain in TLR4 signaling. Proc. Natl. Acad. Sci. U.S.A. 106, 10260–10265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Valkov E., Stamp A., Dimaio F., Baker D., Verstak B., Roversi P., Kellie S., Sweet M. J., Mansell A., Gay N. J., Martin J. L., Kobe B. (2011) Crystal structure of Toll-like receptor adapter MAL/TIRAP reveals the molecular basis for signal transduction and disease protection. Proc. Natl. Acad. Sci. U.S.A. 108, 14879–14884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jiang Z., Georgel P., Li C., Choe J., Crozat K., Rutschmann S., Du X., Bigby T., Mudd S., Sovath S., Wilson I. A., Olson A., Beutler B. (2006) Details of Toll-like receptor:adapter interaction revealed by germ-line mutagenesis. Proc. Natl. Acad. Sci. U.S.A. 103, 10961–10966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Poltorak A., He X., Smirnova I., Liu M. Y., Van Huffel C., Du X., Birdwell D., Alejos E., Silva M., Galanos C., Freudenberg M., Ricciardi-Castagnoli P., Layton B., Beutler B. (1998) Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice. Mutations in Tlr4 gene. Science 282, 2085–2088 [DOI] [PubMed] [Google Scholar]
- 20. Toshchakov V. U., Basu S., Fenton M. J., Vogel S. N. (2005) Differential involvement of BB loops of toll-IL-1 resistance (TIR) domain-containing adapter proteins in TLR4- versus TLR2-mediated signal transduction. J. Immunol. 175, 494–500 [DOI] [PubMed] [Google Scholar]
- 21. Toshchakov V. Y., Fenton M. J., Vogel S. N. (2007) Cutting Edge. Differential inhibition of TLR signaling pathways by cell-permeable peptides representing BB loops of TLRs. J. Immunol. 178, 2655–2660 [DOI] [PubMed] [Google Scholar]
- 22. Bartfai T., Behrens M. M., Gaidarova S., Pemberton J., Shivanyuk A., Rebek J., Jr. (2003) A low molecular weight mimic of the Toll/IL-1 receptor/resistance domain inhibits IL-1 receptor-mediated responses. Proc. Natl. Acad. Sci. U.S.A. 100, 7971–7976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Loiarro M., Sette C., Gallo G., Ciacci A., Fantò N., Mastroianni D., Carminati P., Ruggiero V. (2005) Peptide-mediated interference of TIR domain dimerization in MyD88 inhibits interleukin-1-dependent activation of NF-κB. J. Biol. Chem. 280, 15809–15814 [DOI] [PubMed] [Google Scholar]
- 24. Loiarro M., Capolunghi F., Fantò N., Gallo G., Campo S., Arseni B., Carsetti R., Carminati P., De Santis R., Ruggiero V., Sette C. (2007) Pivotal Advance. Inhibition of MyD88 dimerization and recruitment of IRAK1 and IRAK4 by a novel peptidomimetic compound. J. Leukocyte Biol. 82, 801–810 [DOI] [PubMed] [Google Scholar]
- 25. Ronni T., Agarwal V., Haykinson M., Haberland M. E., Cheng G., Smale S. T. (2003) Common interaction surfaces of the toll-like receptor 4 cytoplasmic domain stimulate multiple nuclear targets. Mol. Cell. Biol. 23, 2543–2555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ulrichts P., Peelman F., Beyaert R., Tavernier J. (2007) MAPPIT analysis of TLR adapter complexes. FEBS Lett. 581, 629–636 [DOI] [PubMed] [Google Scholar]
- 27. Piessevaux J., Lavens D., Montoye T., Wauman J., Catteeuw D., Vandekerckhove J., Belsham D., Peelman F., Tavernier J. (2006) Functional cross-modulation between SOCS proteins can stimulate cytokine signaling. J. Biol. Chem. 281, 32953–32966 [DOI] [PubMed] [Google Scholar]
- 28. Pattyn E., Lavens D., Van der Heyden J., Verhee A., Lievens S., Lemmens I., Hallenberger S., Jochmans D., Tavernier J. (2008) MAPPIT (MAmmalian Protein-Protein Interaction Trap) as a tool to study HIV reverse transcriptase dimerization in intact human cells. J. Virol. Methods 153, 7–15 [DOI] [PubMed] [Google Scholar]
- 29. Yan J., Li Q., Lievens S., Tavernier J., You J. (2010) Abrogation of the Brd4-positive transcription elongation factor B complex by papillomavirus E2 protein contributes to viral oncogene repression. J. Virol. 84, 76–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Eyckerman S., Verhee A., der Heyden J. V., Lemmens I., Ostade X. V., Vandekerckhove J., Tavernier J. (2001) Design and application of a cytokine-receptor-based interaction trap. Nat. Cell Biol. 3, 1114–1119 [DOI] [PubMed] [Google Scholar]
- 31. Kobayashi M., Saitoh S., Tanimura N., Takahashi K., Kawasaki K., Nishijima M., Fujimoto Y., Fukase K., Akashi-Takamura S., Miyake K. (2006) Regulatory roles for MD-2 and TLR4 in ligand-induced receptor clustering. J. Immunol. 176, 6211–6218 [DOI] [PubMed] [Google Scholar]
- 32. Yoneyama M., Suhara W., Fukuhara Y., Fukuda M., Nishida E., Fujita T. (1998) Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J. 17, 1087–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Peelman F., Van Beneden K., Zabeau L., Iserentant H., Ulrichts P., Defeau D., Verhee A., Catteeuw D., Elewaut D., Tavernier J. (2004) Mapping of the leptin-binding sites and design of a leptin antagonist. J. Biol. Chem. 279, 41038–41046 [DOI] [PubMed] [Google Scholar]
- 34. Eyckerman S., Waelput W., Verhee A., Broekaert D., Vandekerckhove J., Tavernier J. (1999) Analysis of Tyr to Phe and fa/fa leptin receptor mutations in the PC12 cell line. Eur. Cytokine Netw. 10, 549–556 [PubMed] [Google Scholar]
- 35. Eswar N., Eramian D., Webb B., Shen M. Y., Sali A. (2008) Protein structure modeling with MODELLER. Methods Mol. Biol. 426, 145–159 [DOI] [PubMed] [Google Scholar]
- 36. Katoh K., Kuma K., Toh H., Miyata T. (2005) MAFFT version 5. Improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33, 511–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera. A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 38. Núñez Miguel R., Wong J., Westoll J. F., Brooks H. J., O'Neill L. A., Gay N. J., Bryant C. E., Monie T. P. (2007) A dimer of the Toll-like receptor 4 cytoplasmic domain provides a specific scaffold for the recruitment of signaling adapter proteins. PLoS One 2, e788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Monie T. P., Moncrieffe M. C., Gay N. J. (2009) Structure and regulation of cytoplasmic adapter proteins involved in innate immune signaling. Immunol. Rev. 227, 161–175 [DOI] [PubMed] [Google Scholar]
- 40. Takashima K., Matsunaga N., Yoshimatsu M., Hazeki K., Kaisho T., Uekata M., Hazeki O., Akira S., Iizawa Y., Ii M. (2009) Analysis of binding site for the novel small molecule TLR4 signal transduction inhibitor TAK-242 and its therapeutic effect on mouse sepsis model. Br. J. Pharmacol. 157, 1250–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Brown V., Brown R. A., Ozinsky A., Hesselberth J. R., Fields S. (2006) Binding specificity of Toll-like receptor cytoplasmic domains. Eur. J. Immunol. 36, 742–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gautam J. K., Ashish, Comeau L. D., Krueger J. K., Smith M. F., Jr. (2006) Structural and functional evidence for the role of the TLR2 DD loop in TLR1/TLR2 heterodimerization and signaling. J. Biol. Chem. 281, 30132–30142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ulrichts P., Bovijn C., Lievens S., Beyaert R., Tavernier J., Peelman F. (2010) Caspase-1 targets the TLR adapter Mal at a crucial TIR-domain interaction site. J. Cell Sci. 123, 256–265 [DOI] [PubMed] [Google Scholar]
- 44. Miggin S. M., Pålsson-McDermott E., Dunne A., Jefferies C., Pinteaux E., Banahan K., Murphy C., Moynagh P., Yamamoto M., Akira S., Rothwell N., Golenbock D., Fitzgerald K. A., O'Neill L. A. (2007) NF-κB activation by the Toll-IL-1 receptor domain protein MyD88 adapter-like is regulated by caspase-1. Proc. Natl. Acad. Sci. U.S.A. 104, 3372–3377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Thompson J. D., Gibson T. J., Plewniak F., Jeanmougin F., Higgins D. G. (1997) The CLUSTAL_X windows interface. Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Slack J. L., Schooley K., Bonnert T. P., Mitcham J. L., Qwarnstrom E. E., Sims J. E., Dower S. K. (2000) Identification of two major sites in the type I interleukin-1 receptor cytoplasmic region responsible for coupling to pro-inflammatory signaling pathways. J. Biol. Chem. 275, 4670–4678 [DOI] [PubMed] [Google Scholar]
- 47. Hubbard S. J., Thornton J. M. (1993), NACCESS Computer Program, University College London [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.