

Abstract
Hepatocyte-specific, albumin-Cre recombinase-mediated deletion of the entire mouse Stat5a-Stat5b locus was carried out to evaluate the role of STAT5a and STAT5b (STAT5ab) in the sex-dependent transcriptional actions of GH in the liver. The resultant hepatocyte STAT5ab-deficient mice were fertile, and unlike global STAT5b-deficient male mice, postnatal body weight gain was normal, despite a 50% decrease in serum IGF1. Whole liver STAT5ab RNA decreased by ∼65-85%, and residual STAT5 immunostaining was observed in a minority of the hepatocytes, indicating incomplete excision by Cre-recombinase. Quantitative PCR analysis of twenty sexually dimorphic, liver-expressed genes revealed significant down-regulation of 10 of 11 male-specific genes in livers of male hepatocyte STAT5ab-deficient mice. Class I female-specific liver genes (Holloway et al (2006) Molec Endocrinol 20: 647-660) were markedly up regulated (de-repressed), whereas the expression of class II female genes, belonging to the Cyp3a subfamily, was unaffected by the loss of hepatocyte STAT5ab. STAT5ab is thus required in the liver for positive regulation of male-specific genes and for negative regulation of a subset of female-specific genes. Continuous GH infusion strongly induced (>500-fold) the class II female gene Cyp3a16 in both wild type and hepatocyte STAT5ab-deficient male mice, indicating sex-specific transcriptional regulation by GH that is STAT5ab-independent. In contrast, hepatocyte STAT5ab deficiency abolished the strong suppression of the male-specific Cyp2d9 by continuous GH seen in control mouse liver. Analysis of global STAT5a-deficient mice indicated no essential requirement of STAT5a for expression of these sex-specific liver Cyp genes. Thus, the major loss of liver sexual dimorphism in hepatocyte STAT5ab-deficient mice can primarily be attributed to the loss of STAT5b.
Introduction
Pituitary GH secretion is sexually differentiated in many species including rats, mice and humans (1-3). In adult male rats, pituitary GH secretion is highly pulsatile, with little or no GH detected in plasma between pulses, while adult females are characterized by a more continuous GH secretory profile. These sexually dimorphic plasma GH profiles control the sex-dependent expression of a large number of hepatic genes, including cytochrome P450 (Cyps) and other enzymes involved in oxidative metabolism of lipophilic drugs and steroids (4-6). GH binding to its cell surface receptor induces Janus kinase 2-catalyzed tyrosine phosphorylation of GH receptor at multiple residues, creating docking sites for downstream cytoplasmic signaling proteins including signal transducer and activator of transcription 5b (STAT5b). STAT5b, in turn, is phosphorylated on tyrosine 699, and then dimerizes and translocates into the nucleus, where it binds specific DNA response elements and activates gene transcription (7, 8).
In adult male rats, there is a close temporal relationship between the plasma GH profile and hepatic STAT5 activity, with each successive plasma GH pulse directly leading to the activation of liver STAT5b. In contrast, the more continuous pituitary GH secretory profile of adult female rats generally maintains STAT5 activity at a low but persistent level (9-11). STAT5 displays a similar sex difference in mouse liver (12). The essential nature of STAT5b was established by the characterization of male mice with a targeted disruption of the Stat5b gene (global STAT5b-deficient mice), which display a loss of male-characteristic body growth rates and loss of the male pattern of liver gene expression (13, 14). Although STAT5b is essential for the sexual dimorphism of male mouse liver, it plays only a minor role in female mouse liver, as revealed by qPCR analysis of 15 sex-dependent liver genes (15). Male-specific liver genes down-regulated in global STAT5b KO male liver are designated class I male genes, whereas male-specific genes down-regulated in both sexes are designated class II genes (15). Female-specific liver genes up-regulated in global STAT5b KO male liver are designated class I female genes, while female-specific genes that are unaffected by the global loss of STAT5b are designated class II female genes. In a follow-up, large-scale gene expression study, 90% of 850 male-predominant genes identified were down-regulated in male mice with a global deficiency in STAT5b, while 61% of 753 female-predominant genes were up-regulated to near wild type female levels. In contrast, 90% of the sexually dimorphic liver genes examined were unaffected by the loss of STAT5b in females (16).
The widespread effects that global STAT5b deficiency has on sex-dependent liver gene expression can be explained by two distinct mechanisms: 1) The loss of STAT5b in the liver may directly impair GH signaling in hepatocytes leading to the observed loss of sex-dependent gene expression. Alternatively, 2) GH-activated STAT5b may contribute to the feedback inhibition of somatostatin neurons in the hypothalamus (17), such that the loss of hypothalamic STAT5b impairs the negative feedback inhibition of pituitary GH release and perturbs the plasma GH profile in a manner that feminizes liver gene expression and body growth rates. Indeed, plasma GH levels may be elevated in global STAT5b-deficient mice (13). In hypophysectomized mice, GH pulse replacement restores male-characteristic body growth and male liver gene expression in the case of wild type, but not global STAT5b deficient mice, evidencing the intrinsic GH pulse resistance of mice with a global deficiency in STAT5b (18). Nevertheless, these studies do not establish whether the loss of liver STAT5b per se is a major cause of the observed feminization of liver and body growth phenotypes.
Presently, we characterize mice with a hepatocyte-specific deletion of the entire Stat5a-Statb locus (Stat5ab) to assess the requirement of hepatic STAT5ab for the liver gene expression and body growth phenotypes previously associated with global STAT5b deficiency. Hepatocyte-specific STAT5b deficiency was introduced using the Cre-Lox system to delete the entire Stat5b gene in hepatocytes, in an effort to avoid any potential complications of hypomorphic alleles associated with residual STAT5 protein fragments present in some Stat5b-disrupted mouse models (14, 19). The deletion was extended to include the adjacent Stat5a gene, which codes for a protein >90% identical to STAT5b that exhibits many similar, but also some unique properties (20, 21). Using this mouse model, we compare the effects of STAT5ab loss in hepatocytes to that of global STAT5b deficiency. Our findings lead us to conclude that hepatocyte STAT5b is not required for normal postnatal growth but plays an essential role in the establishment and/or maintenance of sexually dimorphic gene expression in the liver. These findings are discussed in terms of the mechanisms through which hepatocyte STAT5b regulates liver sexual dimorphism.
Materials and Methods
Knockout mice
Hepatocyte-specific STAT5ab-deficient mice were generated by mating C57Bl/6 × 129J mice having a floxed Stat5a-Stat5b locus (22) with albumin promoter-regulated Cre transgenic mice (FVB/N) (23). Livers from 8-12 wk old hepatocyte STAT5ab-deficient males and females, and floxed controls, were excised, snap frozen in liquid nitrogen and stored at -80°C until use. Livers were excised from 8-9 wk global Stat5b gene-disrupted mice and corresponding wild-type controls (13). Stat5a gene-deleted livers (24, 25) were from 7-9 wk old mice, except for two control males and two STAT5a-deficient males, which were from 6 wk old mice. Hepatocyte-specific STAT5ab-deficient male mice, and male floxed controls, were given a continuous infusion of rat GH at 20 ng/g BW/h for 7 or 14 d using Alzet osmotic mini-pumps using methods described previously (15). Serum IGF1 was measured by direct radioimmunoassay (ALPCO Diagnostics, cat. # 22-IGF-R21) carried out by Oksana Gavrilova of the NIDDK metabolism core (NIH, Bethesda, MD). Serum GH levels were measured by radioimmunoassay carried out by Dr. A. F. Parlow (National Hormone and Pituitary Program, UCLA Medical Center, Torrance, CA).
Primer design and qPCR analysis
qPCR primers specific to each gene were designed using Primer Express software (Applied Biosystems) and are shown in Table 1 or as reported earlier (15, 26). qPCR primers selected for Mup 1/2/6/8 were unable to distinguish between Mup genes 1, 2, 6 and 8, as the percent nucleotide identities of these genes ranged up to 97%. Similarly, the Gstπ primers do not distinguish Gstπ1 and Gstπ2 RNAs (∼98% identity) (27). Expression profiles obtained using the Mup and Gstπ primers therefore reflect a composite expression pattern based on the most abundant RNAs in each liver sample. For convenience, the Mup and Gstπ genes are referred to as single genes in the text. Methods for liver RNA isolation, cDNA synthesis, and real time qPCR using SYBR Green I chemistry to quantify relative levels of each RNA were described previously (15). Amplification of a single, specific product during qPCR cycling was verified by examination of dissociation curves of each amplicon. Data are graphed after normalization to the 18S rRNA content of each sample. Statistical analysis was carried as indicated in each figure using GraphPad Prism software version 4 (San Diego, CA). P-values less than 0.05 were considered significant. Each of the 20 sex-specific liver-expressed genes examined showed a very similar pattern of sex-dependent expression in all three mouse strains used in this study, minimizing the impact of any background strain differences between the mouse models.
Table 1. Mouse qPCR primer sets and GenBank accession numbers.
qPCR primer pairs specific to each gene were designed as described in “Materials and Methods.” The position of the resultant PCR amplicon is indicated by nucleotide numbering based on the indicated GenBank accession numbers. Primer sequences and GeneBank accession numbers for other genes characterized in this study are listed elsewhere (15, 26).
| Gene | Oligo Numbers | GenBank | Amplicon (Nucleotides) | Forward Primer | Reverse Primer |
|---|---|---|---|---|---|
| Cyp3a16a | 1738/1739 | NM_007820 | 851-949 | AGCACCGCGTGGACTTTATT | GGGCTGTGATCTCGATTTCAG |
| Cyp39a1 | 1410/1411 | NM_018887 | 918-968 | TTCTGGAACCCTCTTGCAGG | CGTGTTTCCGTCTCCACCAC |
| Elovl3 | 1650/1651 | NM_007703 | 1421-1471 | GGACAGAGGCACACACAAACA | GCGCCTACCAGGCCTAGAAT |
| Hsd3b5 | 1632/1633 | NM_008295 | 1380 - 1432 | AGTCCTAAGCACTTGCCCAGTAAT | CACAGCAGCTGAGTCACAACAG |
| Moxd1 | 1646/1647 | NM_021509 | 1120 – 1170 | GGGTGAGCCTCTTCCACACA | CAGAATGGAACTCGGGCATC |
| Nnmt | 1642/1643 | NM_010924 | 549-599 | GAAGGGACCTGAGAAGGAGGA | AGTACCTGCTTGATTGCACGC |
| Slco1a1 | 1628/1629 | NM_013797 | 2400- 2451 | TTCATTTTCACATGGCATTTTCTC | AACACAACTCCCCTTGATTGAGTTA |
| Sutl1e1 | 1640/1641 | NM_023135 | 174 – 227 | TGGACAAACGGTTCACCAAA | GCCTTGCCAAGAACATTTCAA |
| Stat5a | 1511/1512 | NM_011488 | 44 - 165 | TGCGCCAGATGCAAGTGTT | CAAGTCAATAGCATCCCACGG |
Antibodies
Rabbit polyclonal anti-STAT5b (sc-835) and anti-GAPDH (sc-25778) antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Mouse monoclonal anti-β-catenin antibody was obtained from BD Transduction Laboratories (BD Biosciences, San Jose, CA).
Western blotting
Whole liver extracts (15 μg) were electrophoresed through standard 7.5% Laemmli SDS polyacrylamide gels and transferred to nitrocellulose membranes. The membranes were blocked for 1 h at 25°C in 5% dry milk in 10 mM Tris HCl pH 7.5, 0.1% Tween 20, 0.1M NaCl followed by incubation at 25°C for 1 h with anti-STAT5b antibody (1:3000 dilution) or anti-GAPDH antibody (1:400 dilution). Washing of the blots, probing with secondary antibody and detection using enhanced chemiluminescence (ECL kit, Amersham Pharmacia Biotech) used standard methods.
Immunostaining
Male STAT5ab-deficient and floxed (control) mice were injected with 2 μg GH/g body weight and euthanized 15 min later. Livers were excised, fixed in 4% paraformaldehyde overnight at 4°C, embedded in paraffin and sectioned at 5 μm. Sections were cleared in xylene and rehydrated. Digital Decloaking Chamber (Biocare Medical; Walnut Creek, CA) was utilized for antigen retrieval. After blocking for 30 min in PBS containing 0.1% Tween 20 and 3% goat serum, sections were incubated with antibodies against STAT5b (1:100). β-Catenin antibody (1:200) was used for counterstaining. The primary antibodies were allowed to bind overnight at 4°C in the presence of blocking buffer. Fluorescent ligand-conjugated secondary antibodies (1:400, Alexa Fluor 488 and 594, Molecular Probes) were applied to sections for 30-60 min in the dark at room temperature and mounted with VectaShield containing DAPI (Vector Laboratories; Burlingame, CA). Sections were viewed under an epifluorescence equipped Olympus BX51 microscope. Images were captured with a Q Imaging Retiga Exi digital camera (Image Systems, Inc.; Columbia, MD) and Image-Pro Plus 5.1 software. The percentage of STAT5-positive cells was estimated from the ratio of red stained to DAPI stained nuclei based on 3 Flox control livers and 2 STAT5ab-deficient livers.
Results
Hepatocyte STAT5ab-deficient mice
Mice deficient in STAT5ab expression in the liver were generated by mating mice with a floxed Stat5a-Stat5b locus (22) with albumin-Cre transgenic mice (23). The latter mouse model expresses the Cre recombinase under the control of the albumin promoter and can be used to effect hepatocyte-specific deletion in postnatal mice. Analysis of STAT5a and STAT5b expression by quantitative PCR (qPCR) and Western blotting showed that the extent of knock out ranged from 65% to 85% at the RNA level (Fig. 1A, 1B), with a somewhat greater decrease seen at the protein level (Fig. 1C). Immunofluorescence analysis of liver sections was performed on mice killed 15 min after GH pulse treatment, which concentrates STAT5ab in the nucleus. Strong nuclear STAT5ab staining was observed in > 90% of the hepatocytes from GH-treated floxed (control) mice, whereas only up to ∼ 20% of hepatocytes from the GH-treated hepatocyte STAT5ab-deficient mice displayed nuclear STAT5ab staining, which was generally weaker than in the control mice (Fig. 1D). The residual STAT5ab staining seen in individual hepatocytes indicates incomplete excision of the floxed STAT5ab-locus by the albumin-Cre-recombinase. In contrast, no residual STAT5a and STAT5b protein was detected in livers of mice with a global deficiency in STAT5a and STAT5b, respectively (13, 25) (also see Fig. 1C, lanes 2 and 3).
Figure 1. Knock down of hepatocyte STAT5ab by Cre-loxP recombination.
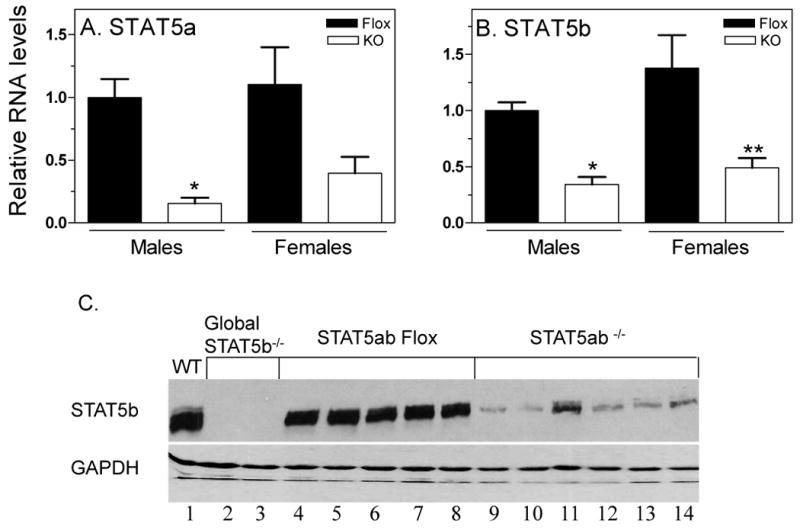
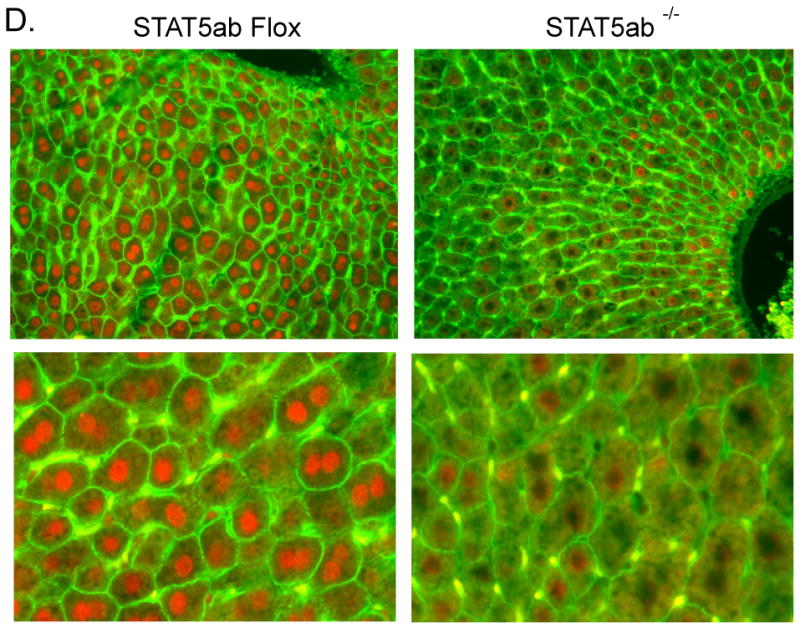
Control (STAT5ab-flox; ‘Flox’) and hepatocyte STAT5ab-deficient (‘KO’) male and female mouse livers were analyzed for the expression of STAT5a RNA (A), STAT5b RNA (B) and STAT5b protein (C) as described under “Materials and Methods.” Panels A and B -RNA levels are graphed as mean +/- SE values (n=8 livers/group), normalized to the 18S rRNA content of each individual liver. Mean male STAT5ab-flox RNA levels were set to 1. Statistical analysis (one-way ANOVA with Bonferroni post hoc test) was reported as follows: ** and *, p<0.01 and p<0.05, respectively, for control male vs. hepatocyte STAT5ab-deficient male, or for control female vs. hepatocyte STAT5ab-deficient female. Panel C - Whole liver lysates from STAT5ab-flox and hepatocyte STAT5ab-deficient female livers were analyzed on Western blots probed with antibody to STAT5 and antibody to GAPDH (loading control), as indicated. Partial knockdown of STAT5b protein was evident in the STAT5ab-deficient livers (lanes 9-14 vs. lanes 4-8), whereas no residual STAT5b protein was detected in global STAT5b-deficient livers (lanes 2, 3). Lane 1, wild type male liver. Similar results were obtained with male livers (data not shown). Panel D - Deletion of STAT5b was assessed by immunofluorescence analysis of control (left) and STAT5ab-deficient mice (right). Most hepatocytes in GH-treated STAT5ab-deficient mice were devoid of STAT5b, as revealed by staining with anti-STAT5b (red) and anti-β-catenin (green) antibodies. Top, 200× magnification; bottom, enlarged fields from an independent pair of liver sections.
The hepatocyte STAT5ab-deficient mice were viable and fertile. In contrast to global STAT5b-deficient mice (13), no significant difference in growth rate was noted between floxed (control) male mice and hepatocyte STAT5ab-deficient male mice (e.g., body weight = 20.2 ± 3.5 g and 20.7 ± 3.0 g, respectively, at 6 wk; 26.9 ± 4.2 g and 21.7 ± 3.2 g, respectively, at 9 wk; and 27.0 ± 2.4 g and 28.7 ± 3.4 g, respectively, at 17 wk). Serum IGF1 levels were decreased by 50% at 8 wk in liver STAT5ab-deficient males compared to controls (160 ± 53 ng/ml and 325 ± 56 ng/ml, respectively; n=4-5, p<0.005), somewhat greater than the 30% decrease seen in global STAT5b-deficient mice (13), but less extensive than the 75% serum IGF1 decrease described in liver IGF1-deficient mice (28). Analysis of serum GH indicated an increase in pituitary GH secretion, reflecting a decrease in feedback inhibition by IGF1 (28). Thus, 4 of 15 hepatocyte STAT5ab-deficient males had serum GH levels <10 ng/ml at the time of killing, while 8 of 14 floxed control mice had GH levels <10 ng/ml; serum GH levels were >100 ng/ml in 6 of 15 hepatocyte STAT5ab-deficient and 0 of 14 floxed control males (p=0.01, Student t-test).
Dependence of male-specific genes on hepatocyte STAT5ab
Class I male-specific genes are down-regulated in global STAT5b-deficient male but not female livers (15). qPCR analysis of five class I male genes revealed significant decreases in expression in livers of hepatocyte STAT5ab-deficient male mice, with little or no effect seen in the corresponding females (Fig. 2). These results are similar to those seen in global STAT5b-deficient mice (15, 16), although the down-regulation of three of the genes (Gstπ, Slp and Moxd1) was more complete in the global STAT5b-deficient male mice, most likely reflecting the incomplete loss of STAT5ab in the hepatocyte-specific STAT5ab mouse livers. Thus, hepatocyte STAT5ab is required for the high level, male-specific expression of all five genes.
Figure 2. Hepatocyte STAT5ab positively regulates class I male-specific liver genes.

Liver RNA isolated from control (Flox) and hepatocyte STAT5ab-deficient (KO) mouse livers was analyzed for expression of the five indicated genes by qPCR as described under “Materials and Methods”. Data are graphed as relative RNA levels, mean +/- SE (n=8 livers/group), normalized to the 18S rRNA content of each sample, with the mean control male RNA levels set to 1. Statistical analyses (one-way ANOVA followed by Bonferroni post hoc test) were reported as follows: ** and *, p<0.01 and p<0.05, respectively, for control (Flox) male vs. hepatocyte STAT5ab-deficient male, or for control (Flox) female vs. hepatocyte STAT5ab-deficient female; ++ and +, p<0.01 and p<0.05, respectively, for control male vs. control female, or for hepatocyte STAT5ab-deficient male vs. hepatocyte STAT5ab-deficient female. Data for Moxd1 in males are based on n=7 individuals/group; an 8th individual in each male group showed extraordinarily high expression levels, corresponding to values of 17.4 and 5.5 relative to the male Flox control, for the male Flox control and the male hepatocyte STAT5ab-deficient groups, respectively.
Class II male genes differ from class I male genes insofar as they are down-regulated in both male and female liver with partial retention of male specificity in global STAT5b-deficient mice (15). qPCR analysis of six class II male genes revealed that the loss of hepatocyte STAT5ab significantly decreased expression of five of the genes (Fig. 3, panels A-D and F). These decreases were observed in both males and females for four of the genes (panels A-D). For three of the five genes (Mup3, Hsd3b5 and Mup1/2/6/8), the decreases were somewhat less dramatic than those seen in global STAT5b-deficient mice (15, 16). The sixth class II male gene, Cyp2d9, did not show any significant or consistent decrease in expression in hepatocyte STAT5ab-deficient liver, either in males or in females (Fig. 3E). Indeed, Cyp2d9 RNA levels were increased in female liver in the absence of STAT5ab. Thus, STAT5ab expression in hepatocytes is required for high level, male-specific expression of a majority of the class II male genes investigated.
Figure 3. Response of class II male genes to loss of hepatocyte STAT5ab.

qPCR analysis of liver RNA samples, data presentation and statistical analysis were the same as in Fig. 2, with mean control male levels set to 1. Significant down-regulation of five of the six class II male genes was observed in response to the loss of hepatocyte STAT5ab (panels A-D, F). Cyp2d9 did not show a consistent decrease in expression in either male or female livers.
Impact of hepatocyte-specific loss of STAT5ab on female-specific gene expression
Whole body loss of STAT5b results in strong up-regulation of class I female genes in male liver, indicating that these genes are repressed in male mice, either directly or indirectly, by a mechanism that requires STAT5b (15). Presently, we assayed the effect of hepatocyte STAT5ab deletion on six class I female genes, three of which were previously characterized by qPCR in the global STAT5b-deficient mouse model (Cyp2b9, Cyp2b13, Cyp2a4), and three of which were shown by microarray and qPCR analysis to have the same pattern of dependence on STAT5b (Cyp39a1, Nnmt and Sult1el) ((16) and data not shown). All six genes exhibited the expected female-specificity, which reached >1000-fold in the case of Cyp2b13 in this mouse strain. Moreover, the loss of STAT5ab in male hepatocytes led to increased expression (de-repression) of all six genes, albeit to different extents (Fig. 4), indicating a requirement for hepatocyte STAT5ab for negative regulation of these genes in males.
Figure 4. De-repression of class I female genes in STAT5ab-deficient male livers.

qPCR analysis of liver RNA samples, data presentation and statistical analysis were as described in Fig. 2, with mean control male levels set to 1. All six class I female genes were up-regulated to varying extents upon loss of STAT5ab in male mouse liver. In the case of Cyp39a1, Cyp2a4 and Cyp2b13, up regulation was seen in 5, 6 and 8 of the 8 hepatocyte-specific STAT5ab deficient male livers examined, respectively. Mean RNA levels were significantly different from floxed male controls for these three genes as judged by t-test but did not reach significance by the more stringent ANOVA with Bonferroni post hoc test. Data for Cyp2b13 in STAT5ab-deficient males are based on n=7 liver samples; the 8th liver displayed an RNA level 8-fold higher than the average of the 7 other male samples.
Next, we characterized the impact of hepatocyte STAT5ab deficiency on the expression of three class II (i.e., STAT5b-independent) female genes, Cyp3a16, Cyp3a41 and Cyp3a44. The loss of STAT5ab in hepatocytes had no effect on the expression of these genes (Fig. 5), as was previously observed in global STAT5b-deficient mice (15).
Figure 5. Class II female gene expression is independent of hepatocyte STAT5ab.

STAT5ab-flox and hepatocyte STAT5ab-deficient livers were analyzed for the expression of three Cyp3a genes by qPCR. No changes in expression were observed in either males or females. RNA samples, data presentation and statistical analysis were the same as in Fig. 2, with mean control male levels set to 1.
Responsiveness of hepatocyte STAT5ab-deficient mice to continuous GH infusion
The above studies indicate that hepatocyte STAT5ab plays a minor role in sex-specific gene expression in female liver. We therefore investigated whether hepatocyte STAT5ab is required for the feminizing effect of continuous GH infusion, which mimics the GH profile of females. This feminization is readily evident in wild-type male mice implanted with osmotic mini-pumps that release GH in a continuous manner for 7-14 d, which overrides the endogenous male plasma GH pulses and down-regulates male-specific liver genes while markedly inducing the expression of female-specific genes (15). The feminizing effect of continuous GH was evaluated in hepatocyte STAT5ab-deficient male mice by analyzing two sex-specific liver genes whose expression is not already feminized in the untreated mice, namely, Cyp3a16 (Fig. 5A) and Cyp2d9 (Fig. 3E). As shown in Fig. 6A, continuous GH treatment induced Cyp3a16 expression in both control and hepatocyte STAT5ab-deficient male mouse liver (≥500-fold increase). In contrast, GH suppressed Cyp2d9 expression to female-like levels in control males but not in hepatocyte STAT5ab-deficient males (Fig. 6B). Thus, the absence of hepatocyte STAT5ab abolishes the suppressive effects of continuous GH with respect to Cyp2d9 but does not block the inductive effects of continuous GH with respect to Cyp3a16.
Figure 6. Response of Cyp3a16 and Cyp2d9 to continuous GH infusion.

Expression of Cyp3a16 (panel A) and Cyp2d9 (panel B) RNAs was assayed by qPCR in individual livers of flox controls and hepatocyte STAT5ab-deficient (KO) male (M) and female (F) mice, without or with a continuous infusion of GH for 7 or 14 d, as indicated (+GH). Male KO mouse controls were implanted with vehicle-filled mini-pumps and showed no significant difference with untreated KO mouse controls regarding the expression of either Cyp gene. Data shown are based on the following number of individual mice per group: n=4 (GH-treated KO male groups) and n=3 (all other groups). RNA levels (mean ± SE) were normalized to the 18S rRNA content of each liver and are presented relative to the average expression level in the untreated flox male (panel A) or flox female (panel B) group, which was set to 1. GH-treated male samples were compared by two-tailed, unpaired student t-test to the corresponding untreated controls, with * and ** indicating significance at p<0.05 and p<0.01, respectively. The large error bar for Cyp3a16 in the flox male + GH 14 d group reflects the variability of Cyp3a16 induction in this group, which ranged from 45-fold to >2000-fold in individual mice. The induction of Cyp3a16 in the corresponding KO male + GH 14 d group was also substantial but variable in individual livers, ranging from 57-fold to 783-fold compared to the sham-treated male-KO controls.
Impact of Stat5a gene disruption on sexually dimorphic liver gene expression
The general consistency of the sex-specific liver gene expression profiles between hepatocyte STAT5ab-deficient mice (above) and global STAT5b-deficient mice (15) supports the hypothesis that STAT5b, rather than STAT5a, is the key required factor for liver sexual dimorphism. This hypothesis was tested by analyzing the impact of global Stat5a disruption on sexually dimorphic liver gene expression. Class I and class II male liver genes showed no substantial changes in their expression, either in males or in females deficient in STAT5a (Table 2). STAT5a is thus dispensable in liver, and in other tissues, for male-specific liver gene expression. Similarly, global loss of STAT5a had no significant effect on the expression of either class I or class II female genes (Table 2), supporting the conclusion that hepatic STAT5b, rather than hepatic STAT5a, is essential for the sex-specificity of liver gene expression.
Table 2. Sex-specific liver gene expression in global STAT5a-deficient male and female mice.
qPCR analysis was carried out using cDNA prepared from total liver RNA isolated from STAT5a wild-type male (n=6), STAT5a-deficient male (n=8), STAT5a-wild type female (n=4) and STAT5a-deficient female mice (n = 4). Data shown represent relative RNA levels, mean ± S.E. normalized to the 18S rRNA content of each sample and set to 1.0 for wild type male or female liver, as indicated. Statistical differences, determined by one-way ANOVA with Bonferroni post hoc test, represent the following comparisons: STAT5a male vs. STAT5a female (+ = p<0.05 and ++ = p<0.01). None of the genes displayed a significant difference in expression at p<0.05 between STAT5a-wild type and STAT5a-deficient male or female liver. Sex-specificity and class of each gene is represented as male (M) or female (F) and I or II.
| Gene | Sex specificity and Class | STAT5a +/+ Male | STAT5a -/- Male | STAT5a +/+ Female | STAT5a -/- Female |
|---|---|---|---|---|---|
| 18S rRNA | 1.00 ± 0.04 | 0.94 ± 0.02 | 0.95_± 0.02 | 1.00_± 0.06 | |
| Cyp4a12 | MI | 1.00 ± 0.27 | 1.20 ± 0.34 | ≤0.01 + | ≤0.01 + |
| Gst π | MI | 1.00 ± 0.41 | 1.10 ± 0.43 | 0.09 ± 0.06 | 0.20 ± 0.08 |
| Slp | MI | 1.00 ± 0.21 | 0.55 ± 0.11 | 0.02_± 0.01 | 0.01 ± 0.00 |
| Elovl3 | MI | 1.00 ± 0.34 | 0.56 ± 0.17 | ≤0.01 + | 0.01 ± 0.00 |
| Moxd1 | MI | 1.00 ± 0.66 | 0.85 ±0.57 | 0.01_± 0.00 | 0.01 ± 0.00 |
| Mup3 | MII | 1.00 ± 0.20 | 0.83 ± 0.17 | 0.08 ± 0.03 ++ | 0.24 ± 0.04 + |
| Cyp7b1 | MII | 1.00 ± 0.23 | 0.53 ± 014 | 0.07 ± 0.02 + | 0.08 ± 0.01 + |
| Hsd3b5 | MII | 1.00 ± 0.15 | 1.38 ± 0.37 | ≤0.01 ++ | ≤0.01 + |
| Slco1a1 | MII | 1.00 ± 0.25 | 0.57 ± 0.11 | 0.10 ± 0.01 + | 0.16 ± 0.07 + |
| Cyp2d9 | MII | 1.00 ± 0.41 | 1.50 ± 0.37 | 0.05 ± 0.03 ++ | 0.02 ± 0.00 + |
| Mup1/2/6/8 | MII | 1.00 ± 0.17 | 0.97 ± 0.20 | 0.15 ± 0.09 ++ | 0.45 ± 0.38 |
| Cyp2b9 | FI | ≤0.01 | ≤0.01 | 1.00 ± 0.29 ++ | 0.37± 0.11 ++ |
| Cyp2b13 | FI | ≤0.01 | ≤0.01 | 1.00 ± 0.54 + | 0.41 ± 0.25 + |
| Cyp2a4 | FI | 0.21 ± 0.07 | 0.18 ± 0.04 | 1.00 ± 0.20 ++ | 0.87 ± 0.23 ++ |
| Cyp39a1 | FI | 0.31 ± 0.09 | 0.55 ± 0.27 | 1.00 ± 0.24 + | 2.13 ± 0.36 |
| Nnmt | FI | 0.37 ± 0.16 | 0.38 ± 0.10 | 1.00 ± 0.42 | 1.85 ± 0.41 ++ |
| Sult1e1 | FI | 0.02 ± 0.01 | 0.04 ± 0.02 | 1.00 ± 0.18 ++ | 1.92 ± 1.42 |
| Cyp3a16 | FII | ≤0.01 | ≤0.01 | 1.00 ± 0.19 ++ | 0.54 ± 0.13 ++ |
| Cyp3a41 | FII | ≤0.01 | ≤0.01 | 1.00 ± 0.17 ++ | 0.78 ± 0.23 ++ |
| Cyp3a44 | FII | ≤0.01 | ≤0.01 | 1.00 ± 0.34 ++ | 0.78 ± 0.14 ++ |
Discussion
STAT5b is proposed to be a key mediator of the sex-dependent actions of GH in male liver. Global disruption of Stat5b is associated with GH pulse insensitivity, loss of male-characteristic body growth rates (13, 18), and feminization of male liver gene expression, as evidenced by the down-regulation of ∼90% of male-predominant liver genes and by the up-regulation (de-repression) of ∼61% of female-dominant genes (16). It is unclear, however, whether this dramatic feminization of the male liver reflects the loss of liver STAT5b per se, or alternatively, whether it is an indirect response to the loss of STAT5b in other tissues, e.g., the hypothalamus, which may disrupt the feedback inhibition of pituitary GH secretion and effectively feminize plasma GH profiles, thereby feminizing liver gene expression. Similarly, the reduced pubertal growth rate that is seen in global STAT5b-deficient male mice (13, 14) could either result from impaired liver STAT5b signaling, e.g., impacting production of the growth promoting factor IGF1 (13, 29), which is a direct target of STAT5b (30, 31), or may result from a perturbation of pituitary GH secretory profiles secondary to the global loss of STAT5b.
These questions were investigated in the present study, where the entire 110 kb Stat5a-Stat5b locus was specifically deleted in hepatocytes using Cre recombinase under the control of the albumin promoter. Quantification of whole liver STAT5a and STAT5b RNA revealed a 65-85% decrease in expression compared to floxed controls. This decrease is substantially less than the ≥98% decreases observed in the case of liver-specific genes, such as albumin (23) and Hnf4 (26) using the same albumin-Cre knockout strategy. This difference could reflect the presence of STAT5ab in non-parenchymal cells in the liver (32), where albumin is not expressed (33), or it could be the result of incomplete Cre excision of the Stat5ab locus in hepatocytes. Immunofluorescence analysis using GH-treated male mice revealed the presence of residual STAT5ab protein in up to ∼20% of the hepatocytes (Fig. 1D), supporting the latter hypothesis. These findings are consistent with the incomplete excision of the Stat5ab locus, perhaps due to the large size (110 kb) of the floxed gene sequences.
In contrast to the whole body growth retardation phenotype seen in global Stat5b-deleted mice (13, 14) and in STAT5b mutated humans (34, 35), mice with hepatocyte-specific STAT5ab-deficiency showed no major changes in body growth rate compared to floxed controls, despite a 50% decrease in circulating IGF1. These observations establish that hepatocyte STAT5ab makes an important contribution to circulating IGF1, and furthermore, demonstrate that hepatocyte STAT5ab is not essential for postnatal growth, which apparently requires the presence of STAT5ab in one or more extrahepatic tissues, such as bone and skeletal muscle (36). As a note of caution, albumin-Cre-mediated gene deletion is not manifest until after birth, and in the case of hepatocyte STAT5ab is still not 100% complete at puberty and in early adulthood, leaving open the possibility that the residual hepatocyte STAT5ab fulfills an essential role in growth. The apparent hepatocyte STAT5ab-independence of postnatal growth reported here is, however, consistent with the lack of a growth phenotype in mice with a liver-specific deletion of the Igf1 gene (23) and contrasts with the striking requirement of hepatocyte STAT5ab for liver sexual dimorphism discussed below. These two sex-dependent phenotypes are also distinguished by their dependencies on the frequency of exogenous GH pulse administration in a hypophysectomized rat model (37).
Ten of the eleven male-specific genes presently examined were substantially down regulated in livers of male hepatocyte STAT5ab-deficient mice. While we cannot rule out the possibility that the elevated plasma GH levels seen in some individual hepatocyte STAT5ab-deficient males might contribute to the down-regulation of these genes, or to the observed up-regulation of class I female genes, two findings suggest that the GH profiles in these mice are not feminized, making this possibility less likely. First, body growth rates were not feminized, and second, certain female-specific genes, e.g., Cyp3a16, were not induced to female-like levels in the hepatocyte STAT5ab-deficient male mice until the plasma GH profiles were feminized by continuous infusion of exogenous GH. Nevertheless, it is still possible that the plasma GH profile requirements for suppression of male genes, or for induction of class I female genes, differ from the requirements that govern body growth rates and the induction of class II female genes. Indeed, the distinct plasma GH concentration requirements for regulation of individual rat liver CYP genes (38) are consistent with the latter possibility.
One of the male-specific genes that is down-regulated in global STAT5b-deficient male liver, Cyp2d9 (15), was not significantly suppressed in hepatocyte STAT5ab-deficient livers. Thus, this gene did not show the strong dependence on hepatocyte STAT5ab that was seen with other male-specific genes. Nevertheless, STAT5ab was required for the suppression of Cyp2d9 by continuous GH, a finding that may help explain the up-regulation of this gene in females with hepatocyte-specific STAT5ab deficiency. The difference between mouse models in the response of Cyp2d9 to the loss of STAT5b is not likely to reflect feminization by the circulating GH profiles in the global STAT5b-deficient male mice, given the intrinsic unresponsiveness of Cyp2d9 to suppression by continuous GH presently seen in hepatocyte STAT5ab-deficient livers.
Class I female genes were strongly up regulated in male but not female mouse liver in the absence of hepatocyte STAT5ab, demonstrating that hepatocyte STAT5ab plays an essential role in the silencing of these genes that occurs in wild type male liver. STAT5ab could effect this negative regulation by a direct mechanism, e.g., by binding to putative negative regulatory elements associated with these female-specific genes. Alternatively, the inhibitory effect of STAT5ab could be indirect, e.g., mediated by epigenetic mechanisms or by male-specific transcriptional repressors whose expression is induced by STAT5ab (6), consistent with the delayed induction of class I female genes seen in livers of male mice infused with GH continuously (15). The increased secretion of GH secondary to the loss of hepatocyte STAT5ab, presently seen in individual hepatocyte STAT5ab-deficient male mice, could also contribute to the up regulation of class I female genes, as noted above. However, class II female genes, belonging to the Cyp3a gene family, did not respond to the loss of STAT5b in either model of STAT5b deficiency, indicating that their expression is STAT5ab-independent. Our finding that continuous GH treatment induces Cyp3a16 even in males with hepatocyte STAT5ab deficiency strengthens this conclusion. In contrast, continuous GH treatment down-regulated the male-specific Cyp2d9 in control male mice but not in hepatocyte STAT5ab-deficient male mice, highlighting the requirement of STAT5ab for some, but not all, of the sex-specific hepatic effects of continuous plasma GH stimulation.
Comparison of the effects of hepatic STAT5ab deficiency to the response to global deletion of either Stat5a or Stat5b indicated that the major effects of hepatocyte STAT5ab deficiency described here can largely be ascribed to the loss of STAT5b. Thus, ablation of STAT5a, whose coding sequence is >90% identical to that of STAT5b and whose protein and mRNA abundance is 90-95% lower than that of STAT5b (25), had little effect on the sex-dependent liver genes examined. This finding does not, of course, rule out a role for STAT5a in the regulation of other sex-specific hepatic genes. Previously, we had observed a loss of expression of certain liver Cyp proteins and liver Cyp-catalyzed testosterone hydroxylase activities in global STAT5a-deficient female liver, suggesting a requirement of STAT5a for expression of the corresponding gene products (25). The present qPCR analysis of specific, individual Cyp genes revealed no major effect of global STAT5a deficiency on sex-dependent liver gene expression. Decreases in two Cyp2b RNAs were, however, seen in STAT5a-deficient female livers (Table 2), in agreement with the decrease in Cyp2b protein(s) previously seen in the same mouse model (25), although the present RNA decreases did not reach statistical significance due to individual variation in expression levels.
In summary, the present study provides strong support for the conclusion that the loss of sex-specific liver gene expression in global STAT5b-deficient mice can primarily be ascribed to the loss of STAT5b in hepatocytes. STAT5b is not required for all of the sex-dependent, liver transcriptional regulatory effects of GH, however, as demonstrated by the striking induction of the female-specific Cyp3a16 gene by continuous GH treatment in male mice with hepatocyte-specific STAT5ab deficiency. Hepatocyte STAT5ab-deficient mice did not display the reduced male pubertal growth rate seen earlier in global STAT5b-deficient mice, supporting the conclusion that male GH pulse-stimulated postnatal growth requires STAT5b in one or more extrahepatic tissues. Finally, the lack of major changes in sex-dependent gene expression in global STAT5a-deficient mouse liver indicates that this quantitatively minor liver STAT5 form does not make a global contribution to sex-dependent liver gene expression. Future studies will focus on the molecular mechanisms, both positive and negative, through which hepatocyte STAT5b regulates sexually dimorphic liver gene expression.
Acknowledgments
Supported in part by NIH Grant DK33765 (to D.J.W.) and the intramural program of NIDDK/NIH (to L.H.).
Abbreviations
- GH
growth hormone
- CYP
cytochrome P450
- STAT
signal transducer and activator of transcription
- STAT5ab
STAT5a and STAT5b
- Mup
major urinary protein
- qPCR
quantitative, real-time polymerase chain reaction
Footnotes
Authors' Disclosure Statement: The authors have nothing to disclose
This is an un-copyedited author manuscript copyrighted by The Endocrine Society. This may not be duplicated or reproduced, other than for personal use or within the rule of “Fair Use of Copyrighted Materials” (section 107, Title 17, U.S. Code) without permission of the copyright owner, The Endocrine Society. From the time of acceptance following peer review, the full text of this manuscript is made freely available by The Endocrine Society at http://www.endojournals.org/. The final copy edited article can be found at http://www.endojournals.org/. The Endocrine Society disclaims any responsibility or liability for errors or omissions in this version of the manuscript or in any version derived from it by the National Institutes of Health or other parties. The citation of this article must include the following information: author(s), article title, journal title, year of publication and DOI.
References
- 1.Jansson JO, Eden S, Isaksson O. Sexual dimorphism in the control of growth hormone secretion. Endocr Rev. 1985;6:128–150. doi: 10.1210/edrv-6-2-128. [DOI] [PubMed] [Google Scholar]
- 2.MacLeod JN, Pampori NA, Shapiro BH. Sex differences in the ultradian pattern of plasma growth hormone concentrations in mice. J Endocrinol. 1991;131:395–399. doi: 10.1677/joe.0.1310395. [DOI] [PubMed] [Google Scholar]
- 3.Veldhuis JD, et al. Neurophysiological regulation and target-tissue impact of the pulsatile mode of growth hormone secretion in the human. Growth Hormone IGF Res. 2001;11:S25–S37. doi: 10.1016/s1096-6374(01)80005-8. [DOI] [PubMed] [Google Scholar]
- 4.Shapiro BH, Agrawal AK, Pampori NA. Gender differences in drug metabolism regulated by growth hormone. Int J Biochem Cell Biol. 1995;27:9–20. doi: 10.1016/1357-2725(94)00056-5. [DOI] [PubMed] [Google Scholar]
- 5.Mode A, Ahlgren R, Lahuna O, Gustafsson JA. Gender differences in rat hepatic CYP2C gene expression--regulation by growth hormone. Growth Horm IGF Res. 1998;8 B:61–67. doi: 10.1016/s1096-6374(98)80025-7. [DOI] [PubMed] [Google Scholar]
- 6.Waxman DJ, O'Connor C. Growth Hormone Regulation of Sex-dependent Liver Gene Expression. Mol Endocrinol. 2006;20:2613–2629. doi: 10.1210/me.2006-0007. [DOI] [PubMed] [Google Scholar]
- 7.Herrington J, Carter-Su C. Signaling pathways activated by the growth hormone receptor. Trends Endocrinol Metab. 2001;12:252–257. doi: 10.1016/s1043-2760(01)00423-4. [DOI] [PubMed] [Google Scholar]
- 8.Darnell JE., Jr STATs and gene regulation. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 9.Waxman DJ, Ram PA, Park SH, Choi HK. Intermittent plasma growth hormone triggers tyrosine phosphorylation and nuclear translocation of a liver-expressed, Stat 5-related DNA binding protein. Proposed role as an intracellular regulator of male-specific liver gene transcription. J Biol Chem. 1995;270:13262–13270. doi: 10.1074/jbc.270.22.13262. [DOI] [PubMed] [Google Scholar]
- 10.Choi HK, Waxman DJ. Plasma growth hormone pulse activation of hepatic JAK-STAT5 signaling: developmental regulation and role in male-specific liver gene expression. Endocrinology. 2000;141:3245–3255. doi: 10.1210/endo.141.9.7638. [DOI] [PubMed] [Google Scholar]
- 11.Choi HK, Waxman DJ. Growth hormone, but not prolactin, maintains, low-level activation of STAT5a and STAT5b in female rat liver. Endocrinology. 1999;140:5126–5135. doi: 10.1210/endo.140.11.7106. [DOI] [PubMed] [Google Scholar]
- 12.Sueyoshi T, Yokomori N, Korach KS, Negishi M. Developmental action of estrogen receptor-alpha feminizes the growth hormone-Stat5b pathway and expression of Cyp2a4 and Cyp2d9 genes in mouse liver. Mol Pharmacol. 1999;56:473–477. doi: 10.1124/mol.56.3.473. [DOI] [PubMed] [Google Scholar]
- 13.Udy GB, Towers RP, Snell RG, Wilkins RJ, Park SH, Ram PA, Waxman DJ, Davey HW. Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. PNAS. 1997;94:7239–7244. doi: 10.1073/pnas.94.14.7239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Teglund S, McKay C, Schuetz E, van Deursen JM, Stravopodis D, Wang D, Brown M, Bodner S, Grosveld G, Ihle JN. Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell. 1998;93:841–850. doi: 10.1016/s0092-8674(00)81444-0. [DOI] [PubMed] [Google Scholar]
- 15.Holloway MG, Laz EV, Waxman DJ. Codependence of growth hormone-responsive, sexually dimorphic hepatic gene expression on signal transducer and activator of transcription 5b and hepatic nuclear factor 4alpha. Mol Endocrinol. 2006;20:647–660. doi: 10.1210/me.2005-0328. [DOI] [PubMed] [Google Scholar]
- 16.Clodfelter KH, Holloway MG, Hodor P, Park SH, Ray WJ, Waxman DJ. Sex-dependent liver gene expression is extensive and largely dependent upon signal transducer and activator of transcription 5b (STAT5b): STAT5b-dependent activation of male genes and repression of female genes revealed by microarray analysis. Mol Endocrinol. 2006;20:1333–1351. doi: 10.1210/me.2005-0489. [DOI] [PubMed] [Google Scholar]
- 17.Bennett E, McGuinness L, Gevers EF, Thomas GB, Robinson IC, Davey HW, Luckman SM. Hypothalamic STAT proteins: regulation of somatostatin neurones by growth hormone via STAT5b. J Neuroendocrinol. 2005;17:186–194. doi: 10.1111/j.1365-2826.2005.01296.x. [DOI] [PubMed] [Google Scholar]
- 18.Davey HW, Park SH, Grattan DR, McLachlan MJ, Waxman DJ. STAT5b-deficient mice are growth hormone pulse-resistant. Role of STAT5b in sex-specific liver p450 expression. J Biol Chem. 1999;274:35331–35336. doi: 10.1074/jbc.274.50.35331. [DOI] [PubMed] [Google Scholar]
- 19.Hoelbl A, Kovacic B, Kerenyi MA, Simma O, Warsch W, Cui Y, Beug H, Hennighausen L, Moriggl R, Sexl V. Clarifying the role of Stat5 in lymphoid development and Abelson-induced transformation. Blood. 2006;107:4898–4906. doi: 10.1182/blood-2005-09-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Soldaini E, John S, Moro S, Bollenbacher J, Schindler U, Leonard WJ. DNA binding site selection of dimeric and tetrameric Stat5 proteins reveals a large repertoire of divergent tetrameric Stat5a binding sites. Mol Cell Biol. 2000;20:389–401. doi: 10.1128/mcb.20.1.389-401.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Verdier F, Rabionet R, Gouilleux F, Beisenherz-Huss C, Varlet P, Muller O, Mayeux P, Lacombe C, Gisselbrecht S, Chretien S. A sequence of the CIS gene promoter interacts preferentially with two associated STAT5A dimers: a distinct biochemical difference between STAT5A and STAT5B. Mol Cell Biol. 1998;18:5852–5860. doi: 10.1128/mcb.18.10.5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cui Y, Riedlinger G, Miyoshi K, Tang W, Li C, Deng CX, Robinson GW, Hennighausen L. Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival, and differentiation. Mol Cell Biol. 2004;24:8037–8047. doi: 10.1128/MCB.24.18.8037-8047.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu JL, Yakar S, LeRoith D. Conditional knockout of mouse insulin-like growth factor-1 gene using the Cre/loxP system. Proc Soc Exp Biol Med. 2000;223:344–351. doi: 10.1046/j.1525-1373.2000.22349.x. [DOI] [PubMed] [Google Scholar]
- 24.Liu X, Robinson GW, Wagner KU, Garrett L, Wynshaw-Boris A, Hennighausen L. Stat5a is mandatory for adult mammary gland development and lactogenesis. Genes Dev. 1997;11:179–186. doi: 10.1101/gad.11.2.179. [DOI] [PubMed] [Google Scholar]
- 25.Park SH, Liu X, Hennighausen L, Davey H, Waxman DJ. Distinctive roles of STAT5a and STAT5b in sexual dimorphism of hepatic P450 gene expression. Journal of Biological Chemistry. 1999;274:7421–7430. doi: 10.1074/jbc.274.11.7421. [DOI] [PubMed] [Google Scholar]
- 26.Wiwi CA, Gupte M, Waxman DJ. Sexually dimorphic P450 gene expression in liver-specific hepatocyte nuclear factor 4alpha-deficient mice. Mol Endocrinol. 2004;18:1975–1987. doi: 10.1210/me.2004-0129. [DOI] [PubMed] [Google Scholar]
- 27.Bammler TK, Smith CA, Wolf CR. Isolation and characterization of two mouse Pi-class glutathione S-transferase genes. Biochem J. 1994;298(Pt 2):385–390. doi: 10.1042/bj2980385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sjogren K, Liu JL, Blad K, Skrtic S, Vidal O, Wallenius V, LeRoith D, Tornell J, Isaksson OG, Jansson JO, Ohlsson C. Liver-derived insulin-like growth factor I (IGF-I) is the principal source of IGF-I in blood but is not required for postnatal body growth in mice. Proc Natl Acad Sci U S A. 1999;96:7088–7092. doi: 10.1073/pnas.96.12.7088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davey HW, Xie T, McLachlan MJ, Wilkins RJ, Waxman DJ, Grattan DR. STAT5b is required for GH-induced liver IGF-I gene expression. Endocrinology. 2001;142:3836–3841. doi: 10.1210/endo.142.9.8400. [DOI] [PubMed] [Google Scholar]
- 30.Chia DJ, Ono M, Woelfle J, Schlesinger-Massart M, Jiang H, Rotwein P. Characterization of distinct Stat5b binding sites that mediate growth hormone-stimulated IGF-I gene transcription. J Biol Chem. 2006;281:3190–3197. doi: 10.1074/jbc.M510204200. [DOI] [PubMed] [Google Scholar]
- 31.Wang Y, Jiang H. Identification of a distal STAT5-binding DNA region that may mediate growth hormone regulation of insulin-like growth factor-I gene expression. J Biol Chem. 2005;280:10955–10963. doi: 10.1074/jbc.M412808200. [DOI] [PubMed] [Google Scholar]
- 32.Cao Q, Mak KM, Ren C, Lieber CS. Leptin stimulates tissue inhibitor of metalloproteinase-1 in human hepatic stellate cells: respective roles of the JAK/STAT and JAK-mediated H2O2-dependant MAPK pathways. J Biol Chem. 2004;279:4292–4304. doi: 10.1074/jbc.M308351200. [DOI] [PubMed] [Google Scholar]
- 33.Dudas J, Papoutsi M, Hecht M, Elmaouhoub A, Saile B, Christ B, Tomarev SI, von Kaisenberg CS, Schweigerer L, Ramadori G, Wilting J. The homeobox transcription factor Prox1 is highly conserved in embryonic hepatoblasts and in adult and transformed hepatocytes, but is absent from bile duct epithelium. Anat Embryol (Berl) 2004;208:359–366. doi: 10.1007/s00429-004-0403-4. [DOI] [PubMed] [Google Scholar]
- 34.Hwa V, Little B, Adiyaman P, Kofoed EM, Pratt KL, Ocal G, Berberoglu M, Rosenfeld RG. Severe growth hormone insensitivity resulting from total absence of signal transducer and activator of transcription 5b. J Clin Endocrinol Metab. 2005;90:4260–4266. doi: 10.1210/jc.2005-0515. [DOI] [PubMed] [Google Scholar]
- 35.Vidarsdottir S, Walenkamp MJ, Pereira AM, Karperien M, van Doorn J, van Duyvenvoorde HA, White S, Breuning MH, Roelfsema F, Kruithof MF, van Dissel J, Janssen R, Wit JM, Romijn JA. Clinical and biochemical characteristics of a male patient with a novel homozygous STAT5b mutation. J Clin Endocrinol Metab. 2006;91:3482–3485. doi: 10.1210/jc.2006-0368. [DOI] [PubMed] [Google Scholar]
- 36.Klover P, Hennighausen L. Postnatal body growth is dependent on the transcription factors Stat5a/b in muscle: a role for autocrine / paracrine IGF-1. Endocrinology. 2007 doi: 10.1210/en.2006-1431. in press. [DOI] [PubMed] [Google Scholar]
- 37.Waxman DJ, Pampori NA, Ram PA, Agrawal AK, Shapiro BH. Interpulse interval in circulating growth hormone patterns regulates sexually dimorphic expression of hepatic cytochrome P450. Proc Natl Acad Sci U S A. 1991;88:6868–6872. doi: 10.1073/pnas.88.15.6868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pampori NA, Shapiro BH. Feminization of hepatic cytochrome P450s by nominal levels of growth hormone in the feminine plasma profile. Mol Pharmacol. 1996;50:1148–1156. [PubMed] [Google Scholar]