

Abstract
NADH pyrophosphatase (NudC) catalyses the hydrolysis of NAD(H) to AMP and NMN(H) [nicotinamide mononucleotide (reduced form)]. NudC multiple sequence alignment reveals that homologues from most Mycobacterium tuberculosis isolates, but not other mycobacterial species, have a polymorphism at the highly conserved residue 237. To elucidate the functional significance of this polymorphism, comparative analyses were performed using representative NudC isoforms from M. tuberculosis H37Rv (NudCRv) and M. bovis BCG (NudCBCG). Biochemical analysis showed that the P237Q polymorphism prevents dimer formation, and results in a loss of enzymatic activity. Importantly, NudCBCG was found to degrade the active forms of isoniazid (INH), INH-NAD and ethionamide (ETH), ETH-NAD. Consequently, overexpression of NudCBCG in Mycobacterium smegmatis mc2155 and M. bovis BCG resulted in a high level of resistance to both INH and ETH. Further genetic studies showed that deletion of the nudC gene in M. smegmatis mc2155 and M. bovis BCG resulted in increased susceptibility to INH and ETH. Moreover, inactivation of NudC in both strains caused a defect in drug tolerance phenotype for both drugs in exposure assays. Taken together, these data suggest that mycobacterial NudC plays an important role in the inactivation of INH and ETH.
Introduction
World Health Organization estimates suggest that Mycobacterium tuberculosis kills over 1 million people each year and causes over 8 million incident cases of tuberculosis (TB). The ever increasing incidence of drug-resistant TB further compounds this global health crisis. By the year 2007, there were approximately 0.5 million reported cases of multidrug-resistant TB. Moreover, by November 2009, 57 countries and territories had reported at least one case of extensively drug-resistant TB (World Health Organization, 2009). To counter this increasing threat of drug resistance, it is critical to understand fundamental aspects of TB-related biology. Such studies will not only uncover cellular mediators of susceptibility and resistance to existing anti-TB drugs, but will also provide new drug targets for the design of novel therapeutic agents.
Isoniazid (INH) was first described in the early 1900s, and since 1952 has prevailed as one of the most potent and widely administered anti-TB drugs (Bernstein et al., 1952; Fox, 1952). Over the last few decades, several genetic and biochemical studies have delineated the series of events involved in INH action. Upon entry into the cell, the hydrazide group of INH is activated by catalase-peroxidase (KatG) to generate an isonicotinic acyl radical. Once formed, this radical spontaneously reacts with NAD(H) at the 4th position of the nicotinamide ring, yielding an INH-NAD adduct (Zhang et al., 1992; Johnsson and Schultz, 1994; Rozwarski et al., 1998; Wilming and Johnsson, 1999). This adduct acts as a slow-onset, tight-binding inhibitor of the NADH-dependent enoyl-ACP reductase (InhA) of the mycobacterial fatty acid synthase II (Banerjee et al., 1994; Rozwarski et al., 1998; Wilming and Johnsson, 1999; Rawat et al., 2003). Competitive inhibition of NADH binding by INH-NAD results in cessation of mycolic acid biosynthesis and cellular lysis (Takayama et al., 1972; Vilchèze et al., 2000).
INH-resistant mutants of M. tuberculosis were recognized within the first year of the clinical use of INH as an anti-tubercular drug (Middlebrook and Cohn, 1953). Several mechanisms of INH resistance have since been described. These include failure to form the INH-NAD adduct due to loss of function mutations in katG (Heym and Cole, 1992; Zhang et al., 1992; Heym et al., 1995); failure of the INH-NAD adduct to associate with the target due to active-site mutations in inhA (Banerjee et al., 1994; Vilchèze et al., 2006); out-competition of the INH-NAD adduct with excessive cytoplasmic levels of NADH due to mutations in ndh (Miesel et al., 1998; Lee et al., 2001; Vilchèze et al., 2005); and titration of the INH-NAD adduct by increased expression of InhA due to promoter-up mutations (Larsen et al., 2002; Vilchèze et al., 2006). Importantly, resistance mechanisms for up to 22% of the INH-resistant M. tuberculosis clinical isolates still remain unknown (Hazbon et al., 2006).
The action of INH has been shown to be linked with NAD metabolism in M. tuberculosis, early studies showed that the NAD content of M. tuberculosis decreased when cells were exposed to INH (Bekierkunst, 1966). However, the NAD metabolism of M. tuberculosis has not been thoroughly investigated in spite of efforts made during the last few years (Boshoff et al., 2008; Vilchèze et al., 2010), and the link between INH and NAD metabolism is still unclear.
Ethionamide (ETH) is an important component of second-line therapy for the treatment of multidrug-resistant TB, and shares a common target with INH. Like INH, ETH is a pro-drug that requires activation to form adducts with NAD that subsequently inhibit InhA (Banerjee et al., 1994; Baulard et al., 2000; DeBarber et al., 2000; Vannelli et al., 2002; Wang et al., 2007). Resistance to ETH has been reported to result from various mechanisms, including mutations altering EthA/EthR (Baulard et al., 2000; DeBarber et al., 2000; Morlock et al., 2003), InhA and its promoter (Banerjee et al., 1994; Larsen et al., 2002; Vilchèze et al., 2006), the NADH dehydrogenase encoded by ndh (Miesel et al., 1998; Vilchèze et al., 2005), and the MshA enzyme which is involved in mycothiol biosynthesis (Vilchèze et al., 2008). However, the mechanism of resistance to ETH of 19% of the ETH-resistant isolates remains unknown (Brossier et al., 2011).
Rv3199c of M. tuberculosis H37Rv and BCG_3224c of Mycobacterium bovis BCG Pasteur strain 1173P2 (hereafter referred to as M. bovis BCG) encode a NADH pyrophosphatase (NudC, EC 3.6.1.22), which belongs to the Nudix hydrolase superfamily of nucleotide pyrophosphatases. The Nudix hydrolases comprise a large family of proteins characterized by a highly conserved 23-amino-acid Nudix motif (Nudix box), GX5EX7REUXEEXGU, where U represents a bulky, hydrophobic, amino acid, usually Ile, Leu or Val (Bessman et al., 1996). The sequence SQPWPFPQS located 10 residues downstream of the Nudix box is found in many characterized NADH hydrolases and may confer pyridine nucleotide specificity (Dunn et al., 1999). The Nudix superfamily proteins (InterPro IPR000086; PfamPF00293) act on substrates with a general structure (nucleoside diphosphate linked to another moiety X, NDP-X), yielding NMP and P-X (Bessman et al., 1996). The hydrolysis of dinucleotide pyrophosphates requires divalent metal ions (Mg2+ or Mn2+) and yields two mononucleoside 5′-phosphates (Frick and Bessman, 1995). In addition to NADH and NAD+, structurally related compounds like NADPH, ADP-ribose and diadenosine polyphosphates are also substrates of this superfamily of proteins (Frick and Bessman, 1995).
Since NudC has a rather broad range of substrates as mentioned above, we speculated that mycobacterial NudC can hydrolyse the INH-NAD and ETH-NAD adducts, resulting in the inactivation of these inhibitors. Thus, overexpression of NudC should lead to INH or ETH resistance in mycobacteria. On the contrary, inactivation of NudC should increase susceptibility to INH and ETH. To verify this hypothesis, we first characterized NudC from M. tuberculosis H37Rv (NudCRv) and M. bovis BCG (NudCBCG) biochemically. Their abilities to hydrolyse INH-NAD or ETH-NAD were verified by mass-spectrometry and also enzymatic assays. The corresponding NudC-encoding genes were then overexpressed in Mycobacterium smegmatis mc2155 and M. bovis BCG to examine changes in drug susceptibility. Furthermore, nudC of M. smegmatis mc2155 and M. bovis BCG was deleted to look into the changes of drug susceptibilities.
Results
Alignment of NudC protein sequences from different origins
Mycobacterium tuberculosis H37Rv nudC gene (Rv3199c) encoding NudC consists of 942 base pairs corresponding to 313 amino acid residues. Multiple alignments of mycobacterial NudC show that M. tuberculosis NudC has a polymorphism (P237Q) not present in any other mycobacteria which lies in a very conserved region of NudC (SQPWPFPQS) and is predicted to confer pyridine nucleotide specificity (Dunn et al., 1999) (Fig. 1A). Subsequently, alignments of NudC between M. bovis BCG and M. tuberculosis clinical isolates (http://www.broadinstitute.org/annotation/genome/mycobacterium_tuberculosis_diversity/GenomesIndex.html) were also performed and multiple differences were noted (Fig. 1B). There were two amino acid differences (residues 254 and 268) between NudC from M. bovis BCG and M. tuberculosis K85 (Mostowy et al., 2004), and between M. tuberculosis T85 and M. tuberculosis w_148 (Hirsh et al., 2004; Gagneux et al., 2006) (residues 237 and 239). However, the sequence of NudC from the three M. tuberculosis clinical isolates M. tuberculosis T46, M. tuberculosis CPHL_A and M. tuberculosis EAS054 (Hirsh et al., 2004; Mostowy et al., 2004) was found to be identical to NudCBCG. nudC genes from a further 137 M. tuberculosis clinical isolates from China were sequenced and analysed (Table S1). Results showed that the P237Q mutation of NudC was present in all of these isolates. In addition, about 75% of these isolates harboured a second mutation (P239R) in the NudC conserved signature sequence, and a few isolates were found to harbour mutations at other sites.
Fig. 1.
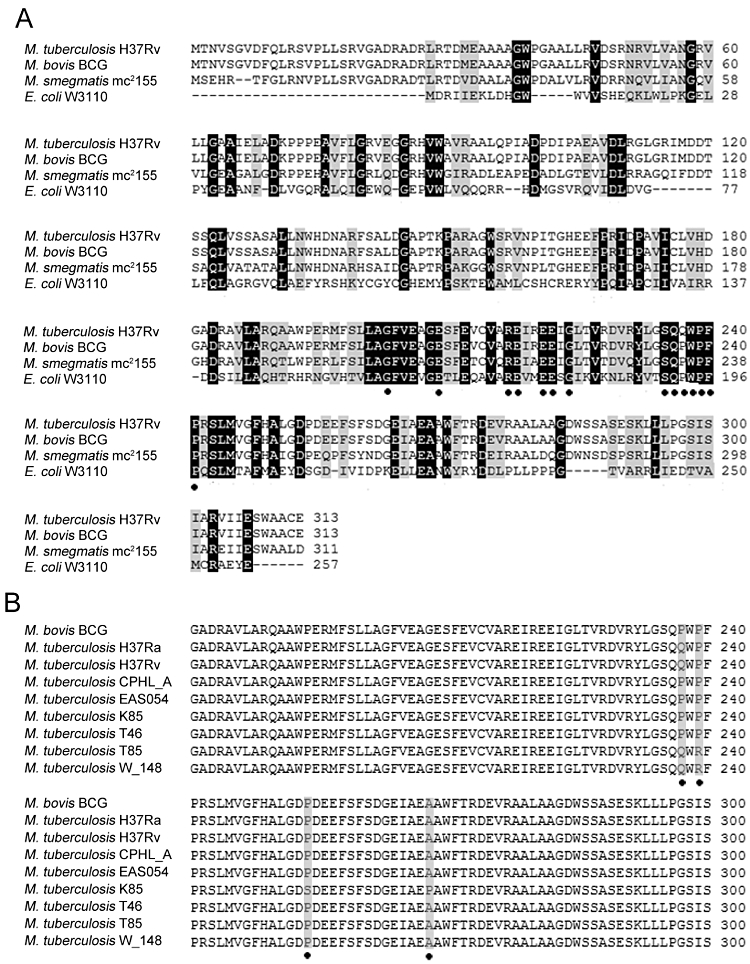
Multiple alignments of NudC from different origins. Sequences aligned using clustal w.
A. Alignments of NudC from E. coli W3110 (AP_003822), M. tuberculosis H37Rv (NP_217715), M. smegmatis mc2155 (YP_886312) and M. bovis BCG (YP_979308). Conserved residues (Nudix box and NudC characteristic sequences) in NudC are highlighted (•).
B. Alignments of NudC from different M. tuberculosis clinical isolates and M. bovis BCG. Only regions containing differences are highlighted (grey and •).
Physicochemical characterization of NudCRv and NudCBCG
NudCRv and NudCBCG were successfully expressed in Escherichia coli BL21 (DE3) by using the expression vector pET32a containing the thioredoxin protein (Trx) tag, which has been shown to be very effective in reducing inclusion body formation (LaVallie et al., 1993). SDS-PAGE analysis shows (Fig. 2A) that the molecular weight of purified recombinant NudC (fused with Trx tag) is about 51 kDa, which is consistent with its theoretical molecular weight.
Fig. 2.
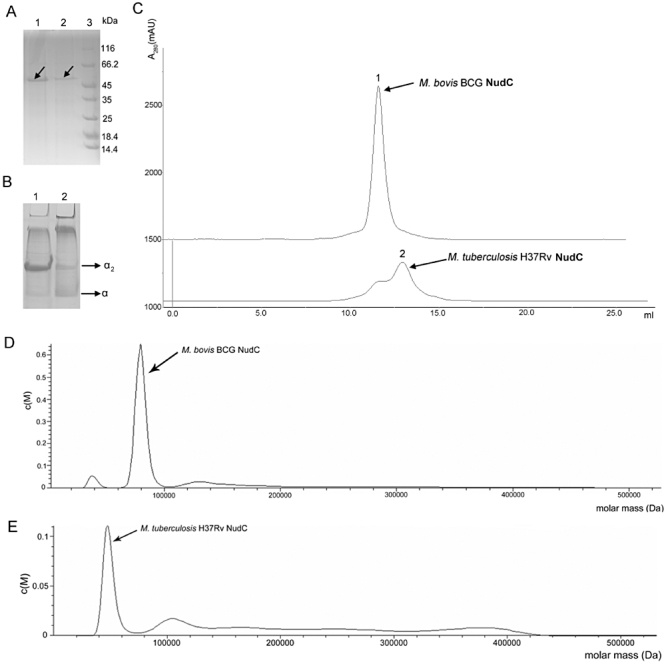
Analysis of NudC native structure.
A. Nickel affinity-purified NudCBCG (lane 1, arrow) and NudCRv (lane 2, arrow) were electrophoresed on 10% (v/v) SDS-PAGE; lane 3: protein molecular weight marker.
B. Native PAGE of purified NudCBCG (lane 1) and NudCRv (lane 2) on 9% (v/v) PAGE. Two NudC bands were detected and are indicated as α2 and α (arrows indicated).
C. Gel exclusion chromatography analysis of the native structure of purified NudCRv and NudCBCG. Nickel affinity-purified NudCBCG and NudCRv were concentrated then loaded onto a Superdex 200 10/300GL column and eluted.
D and E. Analytical ultracentrifugation analysis of the native structure of purified NudCBCG (D) and NudCRv (E). Expected products are indicated by arrows.
The native molecular weight of recombinant NudC was determined by gel exclusion chromatography. As shown in Fig. 2C, NudCRv has two adjacent elution peaks (peak 1 and 2 as indicated by arrows) whereas NudCBCG has only one peak (peak 1). Subsequent SDS-PAGE analysis revealed that both peaks 1 and 2 contained recombinant NudC proteins, NudCBCG being present mainly in elution peak 1 while NudCRv was present mainly in peak 2 (Fig. 2C). According to results from elution curve analysis, it was clear that NudCBCG was eluted faster than NudCRv, suggesting that NudCBCG and NudCRv may have two structural forms under native conditions. The native molecular weight of recombinant NudC was further analysed by analytical ultracentrifugation; recombinant NudCBCG was estimated to be about 80 kDa, although a small portion of protein was estimated to be about 40 kDa (Fig. 2D). The molecular weight of NudCRv was estimated to be about 50 kDa, although a small portion of protein was estimated to be about 100 kDa (Fig. 2E). The molecular weights of recombinant NudC estimated from gel exclusion chromatography analysis were thus consistent with results from analytical ultracentrifugation. To further analyse differences in protein native structure between NudCRv and NudCBCG, non-denaturing PAGE experiments were performed (Fig. 2B). Under non-denaturing conditions two bands were observed for both proteins after staining. NudCBCG was present mainly in the upper-band (possible dimeric form, indicated as α2), but NudCRv was present mainly in the lower band (possible monomeric form, indicated as α). These data, combined with data from gel exclusion chromatography and analytical ultracentrifugation, suggest that NudCRv is mainly present as a monomeric protein, while NudCBCG is mainly dimeric under native conditions.
NudC in most M. tuberculosis clinical isolates from China harboured double mutations (P237Q and P239R) (hereafter referred to as NudCQR) which were identical to those of M. tuberculosis clinical isolates T85 and W_148. This NudC double mutant protein was purified and then analysed by gel exclusion chromatography (Fig. S1A) and native PAGE (Fig. S1B) as described above. Results showed that NudCQR mainly forms a monomeric protein and small of dimeric protein under native conditions.
Taken together, our results indicate that residue 237 of NudCRv plays an important role in maintaining its dimeric structure. As a consequence, the mutation of this residue (P237Q) in M. tuberculosis NudC leads to the failure of protein dimer formation. In addition, residue 239 of NudC also affects dimer formation.
Enzymatic assays of NudCBCG and NudCRv
Results from the alignment of M. tuberculosis NudC with NudC from other species show that the Nudix box and NudC characteristic sequences are highly conserved. NADH has already been shown to be the substrate favoured over all other nucleoside diphosphate derivatives by NudC from other species (Frick and Bessman, 1995; Bessman et al., 1996). In addition, divalent metal ions have been shown to be essential for NudC enzymic activity, with the most efficient stimulators being Mg2+ and Mn2+, as previously reported (Frick and Bessman, 1995).
NADH/NAD+ hydrolysis by NudC in the presence of different metal ions was verified according to a standard protocol (Frick and Bessman, 1995). Parallel experiments were performed for NudCRv and NudCBCG. As shown in Table 1, NudCBCG and NudCRv differ in several respects. First, in the presence of Mg2+, NudCRv barely hydrolysed either NADH or NAD+, whereas hydrolysis by NudCBCG was significant, and showed a marked preference for NADH over NAD+. Second, in the presence of Mn2+, NudC from both species could hydrolyse NADH, but the specific activity of NudCRv was only approximately one-fifth that of NudCBCG. Third, even in the presence of Mn2+, NudCRv could barely hydrolyse NAD+. Other than Mg2+ and Mn2+, NudCBCG showed some activity on NADH in the presence of Fe2+ as well. However, neither enzyme had detectable activity in the presence of Ca2+, Cu2+, Zn2+ or Fe3+.
Table 1.
Comparison of specific activities of NudCRv and NudCBCG in the presence of different metal ionsa
| M. bovis BCG (U·mg−1) | M. tuberculosis H37Rv (U·mg−1) | |||
|---|---|---|---|---|
| NADH | NAD+ | NADH | NAD+ | |
| Mg2+ | 1.59 ± 0.2 | 0.03 ± 0.01 | 0.02 ± 0.01 | 0 |
| Mn2+ | 1.44 ± 0.1 | 0.58 ± 0.08 | 0.29 ± 0.1 | 0.05 ± 0.03 |
| Zn2+ | 0.06 ± 0.01 | 0.01 ± 0.01 | 0.05 ± 0.01 | 0.04 ± 0.01 |
| Ca2+ | 0 | 0 | 0 | 0 |
| Cu2+ | 0 | 0 | 0 | 0 |
| Fe3+ | 0.02 ± 0.01 | 0 | 0.03 ± 0.01 | 0 |
| Fe2+ | 0.39 ± 0.05 | 0.05 ± 0.02 | 0.03 ± 0.01 | 0 |
Measurements were performed in triplicate. Values are means ± standard deviations.
In addition to NADH and NAD+, several nucleotide pyrophosphate analogues (NADP+, NADPH, ADP-ribose) were also tested as substrates for NudCRv and NudCBCG, and their relative activities are shown in Table S2. As expected, NADH was favoured over all other substrates tested. Both enzymes preferred the reduced forms of the substrates, consistent with results for NudC from other sources (Xu et al., 2000). NudCBCG was found to have a broader range of substrates than NudCRv.
The enzymatic activity of NudCQR from a representative M. tuberculosis clinical isolate was also determined. The enzymatic activity of this double mutant could hardly be detected in the presence of any metal ion (Mg2+, Mn2+ or any other ion), no matter what substrate was used.
Taken together, these results indicate that residue 237 of M. tuberculosis NudC is important, not only for its dimerization, but also for its metal ion binding and enzymatic activity. Residue 239 may also play a role in its enzymatic activity.
Using NADH as the substrate, the optimal pH and temperature of NudCBCG were determined to be pH 8.0 and 40°C respectively (Fig. S2A and B). However, enzyme activity rapidly decreased below pH 7.5, or when the temperature was below 35°C or above 45°C.
NudCBCG hydrolyses the INH-NAD and ETH-NAD adducts
As described above, NudC from both M. bovis BCG and M. tuberculosis were verified to be NADH pyrophosphatases, and NudCBCG was shown to have a rather broad range of substrates. Since INH-NAD and ETH-NAD adducts are analogues of NAD, we wondered whether NudCBCG might be also able to hydrolyse it. To test this hypothesis, INH-NAD and ETH-NAD adducts were synthesized and purified according to published protocols (Lei et al., 2000; Nguyen et al., 2001; 2002; Wang et al., 2007). Synthesized INH-NAD (Fig. S3) and ETH-NAD (Fig. S4) adducts were analysed by HPLC-MS (Figs S3A and S4A) and UV/VIS spectrometry (Figs S3B and S4B), and results obtained were found to be consistent with previous reports (Lei et al., 2000; Nguyen et al., 2001; 2002; Wang et al., 2007).
After incubation of INH-NAD or ETH-NAD with NudC, the reaction mixture was filtered through a Microcon 3 centricon filter unit to remove all proteins. Filtrates collected were then analysed by HPLC-MS, and results indicated that NudCBCG could hydrolyse the INH-NAD and ETH-NAD adducts in the presence of Mg2+, yielding AMP [(M-H)-] (peak of m/z 346) and INH-NMNH [(M-H)-] (peak of m/z 440) (Fig. 3A) or AMP [(M-H)-] (peak of m/z 346) and ETH-NMNH [(M-H)-] (peak of m/z 468) (Fig. 3D). However, NudCRv could not hydrolyse the INH-NAD or ETH-NAD adducts, and only had a peak of m/z 769 [(M-H)-] representing the intact INH-NAD adduct (Fig. 3B) or a peak of m/z 797 [(M-H)-] representing the ETH-NAD adduct (Fig. 3C).
Fig. 3.

HPLC-MS analysis of the INH-NAD and ETH-NAD adducts hydrolysed by NudC. Purified NudC was incubated with INH-NAD (A and B) or ETH-NAD (C and D) adducts in the presence of Mg2+, reacted sufficiently, and the reaction mixture was then filtered to remove protein before analysing the filtrate by HPLC-MS. The data from mass spectral analysis of the NudCBCG hydrolysis products are shown in (A) and (D), and those of NudCRv are shown in (B) and (C). The product peaks are indicated by arrows and chemical structures showing the composition and structure of the molecules are shown. INH-NAD, calculated weight = 770 and found weight = 769 [(M-H)-]; ETH-NAD, calculated weight = 798 and found weight = 797 [(M-H)-]; AMP, calculated weight = 347 and found weight = 346 [(M-H)-]; INH-NMNH, calculated weight = 441 and found weight = 440 [(M-H)-]; ETH-NMNH, calculated weight = 469 and found weight = 468 [(M-H)-]. All MS data were acquired in the negative mode.
Results indicated that the NudCQR could not hydrolyse INH-NAD or ETH-NAD adduct, and only had a peak of m/z 769 [(M-H)-] representing the intact INH-NAD adduct (Fig. S5) or m/z 797 [(M-H)-] representing the ETH-NAD adduct (Fig. S6).
Overexpression of NudC in mycobacteria
The nudC gene from M. bovis BCG and M. tuberculosis H37Rv were both cloned into pMV261. The resulting plasmids pMV261-NudCBCG, pMV261-NudCRv and empty vector pMV261 were electroporated into M. smegmatis mc2155, yielding mc2155 pMV261::nudCBCG, mc2155 pMV261::nudCRv and mc2155 pMV261 respectively. To confirm the overexpression of NudCRv and NudCBCGin vivo, polyclonal anti-NudCRv and anti-NudCBCG antibodies were raised. Western blotting showed that NudC from both species was expressed successfully in M. smegmatis mc2155 (Fig. S7).
As mentioned above, since NudCBCG is able to hydrolyse the INH-NAD and ETH-NAD adducts and thus inactivate them, overexpression of this gene should result in INH and ETH resistance. Subsequently, tests for drug susceptibility to INH, ETH, Rifampicin (RIF) and Ethambutol (EMB) were performed on the NudCBCG overexpressing M. smegmatis mc2155 strain (mc2155 pMV261::nudCBCG). As shown in Table 2, mc2155 pMV261::nudCBCG was co-resistant to INH and ETH, with 20- to 40-fold increases in MIC (Minimum inhibitory concentration) for each compound, but its susceptibility to the other two drugs was not changed. The susceptibility of mc2155 pMV261::nudCRv and mc2155 pMV261 to these four drugs was not changed. These results suggest that the co-resistance of mc2155 pMV261::nudCBCG to INH and ETH was caused by overexpression of NudCBCG, and not from other causes. Similarly, overexpression of M. smegmatis mc2155 NudC (NudCSm) in M. smegmatis mc2155 (mc2155 pMV261::nudCSm) also resulted in a high level of resistance to both INH and ETH and susceptibility to the other two drugs (RIF and EMB) was not changed (Table 2).
Table 2.
Drug susceptibility tests of mycobacterial strains
| MIC (µg ml−1) | |||||
|---|---|---|---|---|---|
| Strain | Description | INH | ETH | EMB | RIF |
| mc2155 | M. smegmatis mc2155 | 5 | 10 | 0.5 | 10 |
| mc2155 pMV261 | mc2155 transformed with pMV261 | 5 | 10 | 0.5 | 10 |
| mc2155 pMV261::nudCRv | mc2155 transformed with pMV261-NudCRv | 5 | 10 | 0.5 | 10 |
| mc2155 pMV261::nudCBCG | mc2155 transformed with pMV261-NudCBCG | > 200 | 200 | 0.5 | 10 |
| mc2155 ΔnudC | mc2155 knockout nudC | 1 | 0.5 | 0.5 | 10 |
| mc2155 pMV261::nudCSm | mc2155 transformed with pMV261-NudCSm | 200 | 50 | 0.5 | 10 |
| BCG | M. bovis BCG Pasteur1173P2 | 0.1 | 5 | 2.5 | 0.1 |
| BCG pMV261 | BCG transformed with pMV261 | 0.1 | 5 | 2.5 | 0.1 |
| BCG pMV261::nudCRv | BCG transformed with pMV261-NudCRv | 0.1 | 5 | 2.5 | 0.1 |
| BCG pMV261::nudCBCG | BCG transformed with pMV261-NudCBCG | > 2 | > 50 | 2.5 | 0.1 |
| BCG ΔnudC | BCG knockout nudC | 0.05 | 1 | 2.5 | 0.1 |
| H37Ra | M. tuberculosis H37Ra | 0.1 | 1 | 2.5 | 0.01 |
| H37Ra ΔnudC | H37Ra knockout nudC | 0.1 | 1 | 2.5 | 0.01 |
| mc2155 ΔnudC pMV261::nudCSm | mc2155 ΔnudC transformed with pMV261-NudCSm | > 200 | 200 | 0.5 | 10 |
| BCG ΔnudC pMV261::nudCBCG | BCG ΔnudC transformed with pMV261-NudCBCG | > 2 | > 50 | 2.5 | 0.1 |
Recombinant plasmids pMV261-NudCBCG, pMV261-NudCRv and empty vector pMV261 were electroporated into M. bovis BCG yielding BCG pMV261::nudCBCG, BCG pMV261::nudCRv and BCG pMV261 respectively. We found that the efficiency of transformation of pMV261-NudCBCG was extremely low in comparison with that of pMV261-NudCRv and the blank vector pMV261 (less than 20 transformants per µg plasmid versus thousands of transformants per µg plasmid). To verify the transformants obtained, specific sequences from the overexpression vector containing nudC were amplified and then sequenced. Interestingly, most sequences amplified from pMV261-NudCBCG transformants contained deletions and mutations of the nudC gene, but there were no mutations in the chromosomal copy of nudC gene (data not shown), and only a very few transformants (about one in 30) had an intact coding sequence of NudC, indicating that toleration of the overexpression of NudC may require secondary mutations. A very similar phenomenon was also observed in M. tuberculosis H37Ra, suggesting that unlike M. smegmatis mc2155, overexpression of functional NudC in slow-growing mycobacteria is not tolerated. This may reflect differences in NAD metabolism between fast and slow growing mycobacteria.
Subsequent drug susceptibility tests showed that, BCG pMV261::nudCBCG was also co-resistant to INH and ETH, whereas the susceptibility of BCG pMV261::nudCRv and BCG pMV261 to these two drugs was unchanged (Table 2). Susceptibility to RIF and EMB in these strains also remained unchanged (Table 2).
Inactivation of nudC resulted in increased susceptibility to both INH and ETH
To further investigate the physiological role of mycobacterial nudC on cellular metabolism of INH and ETH, the nudC gene was deleted from M. smegmatis mc2155, M. bovis BCG and M. tuberculosis H37Ra and verified by PCR (Fig. S8). As expected, the M. smegmatis mc2155, M. tuberculosis H37Ra and M. bovis BCG strains in which nudC was knocked out, mc2155 ΔnudC and BCG ΔnudC, were more sensitive to both INH and ETH in MIC testing; however, the susceptibility of the M. tuberculosis H37Ra nudC knock out strain (H37Ra ΔnudC) to these two drugs was unchanged (Table 2). Complemented strains mc2155 ΔnudC pMV261::nudCSm and BCG ΔnudC pMV261::nudCBCG also showed resistance to both INH and ETH (Table 2), consistent with results obtained in our nudC overexpression experiments. NudC was also overexpressed in the complemented strains and the level of NudC expressed was much higher than that in the wild-type strain, also conferring resistance to INH and ETH on these complemented strains. Susceptibilities to RIF and EMB remained unchanged in these strains (Table 2).
Drug exposure experiments were performed and results showed that both mc2155 ΔnudC and BCG ΔnudC had a defect in the INH tolerance phenotype (Fig. 4A and B). The INH killing curve of mc2155 ΔnudC differed from that of the wild-type strain. Deletion of nudC resulted in greater killing in the mc2155 ΔnudC mutant strain throughout the course of treatment (96 h) than in the wild-type strain at the same concentration of INH. In the wild-type strain, the killing effect of INH peaked after 24 h of treatment, and then bacteria started to grow again. In the mc2155 ΔnudC mutant, the killing effect of INH was plateaued between 24 and 60 h, but increased thereafter as INH continued to inhibit the growth of the bacteria (Fig. 4A).
Fig. 4.
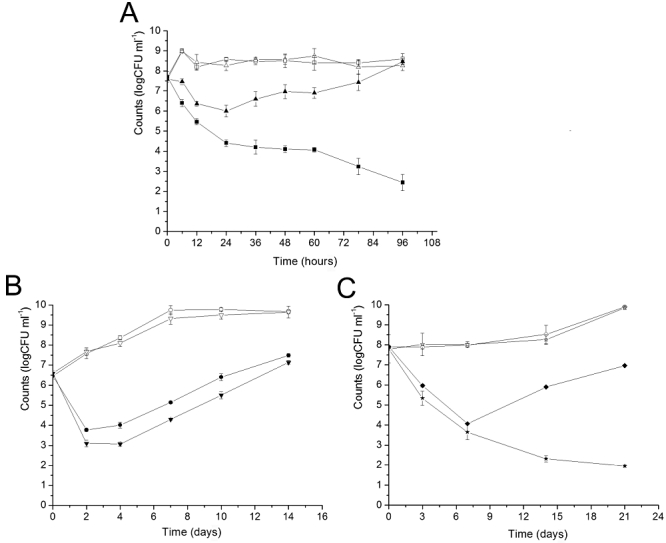
INH and ETH mycobacterial killing curves. Mycobacterial cells were grown to mid-log phase (OD600 of 0.5–1.0) and diluted to about 106–107 cfu ml−1 in fresh medium. INH or ETH was then added and aliquots were removed at regular intervals. Serial dilutions were performed before plating.
A. M. smegmatis mc2155 (▵) and mc2155 ΔnudC (□) with no drug added; M. smegmatis mc2155 (▴) and mc2155 ΔnudC ( ) with 50 µg ml−1 INH added.
) with 50 µg ml−1 INH added.
B. M. bovis BCG (○) and BCG ΔnudC (▿) with no drug added; M. bovis BCG (•) and BCG ΔnudC (▾) with 5 µg ml−1 INH added.
C. M. bovis BCG (◊) and BCG ΔnudC ( ) with no drug added; M. bovis BCG (⋄) and BCG ΔnudC (★) with 50 µg ml−1 ETH added. Experiments were performed in triplicate. Standard deviations are indicated by error bars.
) with no drug added; M. bovis BCG (⋄) and BCG ΔnudC (★) with 50 µg ml−1 ETH added. Experiments were performed in triplicate. Standard deviations are indicated by error bars.
Although deletion of M. bovis BCG nudC only resulted in an about 1 log-fold increase in killing by INH compared with the wild-type strain (Fig. 4B), it led to continuous killing by ETH (Fig. 4C). In the wild-type strain, the killing effect of ETH peaked after 7 days of treatment, and then bacteria began to grow again. However, in the BCG ΔnudC mutant, the killing effect continued to 21 days of treatment.
These results provide further strong support showing that, although previously unrecognized, mycobacterial NudC plays a role in the cellular metabolism of INH and ETH which is very important for the action and also the tolerance of these two drugs.
Mycolic acid analysis of mycobacteria following INH or ETH treatment
As mentioned above, overexpression of NudCBCG in M. smegmatis mc2155 and M. bovis BCG resulted in resistance to INH and ETH, while deletion of the nudC gene in M. smegmatis mc2155 and M. bovis BCG caused increased susceptibility to both drugs. Since treatment with INH or ETH leads to inhibition of mycobacterial mycolic acid biosynthesis, we expect that changes in susceptibility to these drugs resulting from overexpression of NudC or deletion of the nudC gene may alter their effect on mycobacterial mycolic acid biosynthesis. Mycobacterial strains (M. smegmatis mc2155, mc2155 ΔnudC, mc2155 pMV261::nudCBCG, M. bovis BCG, BCG ΔnudC and BCG pMV261::nudCBCG) were grown in the presence INH and ETH at various concentrations, after which cultures were labelled with 1, 2-[14C] acetate. Combined fatty acid methyl esters (FAMEs) and mycolic acid methyl esters (MAMEs) were extracted, resolved and fractionated on TLC plates. Treatment of mc2155 ΔnudC with INH (from 0 to 10 µg ml−1) (Fig. 5A) or ETH (from 0 to 500 µg ml−1) (Fig. 5B) resulted in very strong inhibition of mycolic acid biosynthesis as shown by the absences of MAMEs at a relatively low concentration of each drug (1 µg ml−1 INH, 25 µg ml−1 ETH) compared with the wild-type strain. On the contrary, INH (from 0 to 250 µg ml−1) and ETH (from 0 to 500 µg ml−1) showed very weak or nearly no inhibition of mycolic acid biosynthesis in NudCBCG overexpressed M. smegmatis mc2155 strain (mc2155 pMV261::nudCBCG) (Fig. 5A and B). Similarly, INH and ETH inhibition of mycolic acid biosynthesis in BCG ΔnudC was markedly stronger compared with that in the wild-type strain, while it was drastically diminished by overexpression of NudCBCG (Fig. 5C and D). INH and ETH at relatively low concentrations (0.1 µg ml−1 INH, 10 µg ml−1 ETH) completely abolished MAMEs synthesis in BCG ΔnudC, while higher concentrations of the drugs (0.5 µg ml−1 INH, 50 µg ml−1 ETH) were required to achieve the same effect in the wild-type strain. On the other hand, treatment of the NudCBCG overexpressing strain BCG pMV261::nudCBCG with INH (from 0 to 1 µg ml−1) or ETH (from 0 to 50 µg ml−1) had a rather weak or nearly no effect on the synthesis of MAMEs and FAMEs.
Fig. 5.

Inhibition of mycobacterial mycolic acid biosynthesis by INH and ETH. M. smegmatis mc2155, mc2155 ΔnudC and mc2155 pMV261::nudCBCG were treated with serial concentrations of INH (A) or ETH (B) for 4 h, and then labelled with 1, 2-[14C] acetate for another 4 h. FAMEs and MAMEs were then extracted and separated by TLC. 14C-labelled FAMEs and MAMEs were detected by autoradiography after overnight exposure to a Kodak BioMax MR film. Similarly, M. bovis BCG, BCG ΔnudC and BCG pMV261::nudCBCG were treated with serial concentrations of INH (C) or ETH (D) for 24 h. Then 1, 2-[14C] acetate was added and cultures were further incubated for another 24 h. FAMEs and MAMEs were then extracted and separated by TLC. 14C-labelled FAMEs and MAMEs were detected by autoradiography after overnight exposure to a Kodak BioMax MR film.
Discussion
In this study, we report a unique character of M. tuberculosis NAD metabolism, and propose a novel mechanism of INH and ETH inactivation in mycobacteria.
NudC, the NADH pyrophosphatase, is a component of NAD salvage pathway in many bacteria species including mycobacteria. Protein sequence alignment showed that NudC from M. tuberculosis H37Rv harbours a polymorphism at residue 237 that is not present in other mycobacteria. Although this residue is conserved among NudC from different species and is also predicted to be one of the active site of the enzyme, so far no experimental data have been presented for this specific residue. In this study, through comparative biochemical analysis of purified NudCBCG and NudCRv, we found that the mutation at residue 237 results in disruption of protein dimer formation, changes in metal ion binding and loss of enzymatic activity. In addition, sequence analysis of nudC genes amplified from 137 M. tuberculosis clinical isolates from China revealed that nudC from most isolates harboured another mutation (residue 239), and a few isolates even had multiple mutations. The standard laboratory strain M. tuberculosis H37Rv was isolated in the year 1905. Since it was not possible to analyse the sequence of nudC from different clinical isolates at that time, it is now unclear whether the additional mutations of nudC identified in clinical isolates here were already present then or whether they have been acquired subsequently. Biochemical analysis of the purified double mutant NudCQR showed that the mutations at residues 237 and 239 lead to complete loss of enzymatic activity. Alignment of mycobacterial NudC and E. coli NudC revealed that residues P237 and P239 are both located in a very conserved region (SQPWPFPQS) predicted to confer pyridine nucleotide specificity (Dunn et al., 1999) (Fig. 1B), suggesting that the mutation of these two residues may affect the binding of substrates. According to the results of amino acid sequence alignment, residue 237 of NudCRv corresponds to residue 193 of E. coli NudC. The crystal structure of E. coli NudC (PDB code 1vk6 and PDB code 2gb5) has been resolved and it was found to be a dimeric protein. Residue 193 of E. coli NudC is located on the surface of the interface between subunits (Fig. S9A and B). It may thus be the case that the mutation of residue 237 in NudCRv affects the interaction between subunits, causing the failure in protein dimerization observed here. In addition, residue 239 of NudCRv corresponds to residue 195 in E. coli NudC. Residue 195 is also known to be close to the surface of the protein interface between subunits (Fig. S9A and B). Thus it is not surprising to find that double mutation of residues 237 and 239 in NudCQR also affect protein dimer formation. However, it is still remains unclear whether protein dimerization is necessary for the enzymatic activity of NudC.
From the above data, it is very clear that, in contrast to M. bovis BCG and other mycobacteria, M. tuberculosis H37Rv and most M. tuberculosis clinical isolates have acquired mutations in the nudC gene, leading to the loss of NADH pyrophosphatase activity. As mentioned above, both residues 237 and 239 are located in a conserved region (SQPWPFPQS) of NudC that is predicted to confer pyridine nucleotide specificity. It is possible that NudCRv may have gained additional as yet unknown functions, which remain to be identified in further studies.
One unique property of M. tuberculosis NAD metabolism is that it secretes niacin into the growth medium under in vitro growth conditions, and previous research revealed that, niacin secretion of M. tuberculosis was mainly due to its high NAD glycohydrolase activity (Kasărov and Moat, 1972). Here we report another unique property of M. tuberculosis NAD metabolism. Most M. tuberculosis clinical isolates have an inactive NADH pyrophosphatase, although the reason remains unknown.
Previous studies have shown that NudC has a rather broad range of substrates, including NADH/NAD+ and nucleoside diphosphate derivatives (Frick and Bessman, 1995). This raises the possibility that, as homologues of NAD, INH-NAD and ETH-NAD adducts could also be substrates of NudC. We validated this hypothesis here by biochemical assays including HPLC-MS. As expected, NudCBCG was able to hydrolyse both adducts whereas NudCRv was not. Since NudCBCG was capable of hydrolysing INH-NAD and ETH-NAD, we reasoned that its overexpression should lead to co-resistance to both drugs, and that deletion of the nudC gene should result in increased sensitivity to both drugs. This speculation was verified by in vivo experiments. Overexpression of NudCBCG in M. smegmatis mc2155 and M. bovis BCG resulted in co-resistance to INH and ETH, but overexpression of NudCRv had no effect on susceptibility to both drugs. Meanwhile, overexpression of NudCSm in M. smegmatis mc2155 also resulted in co-resistance to INH and ETH, suggesting that NudCSm is also functional and able to hydrolyse INH-NAD and ETH-NAD adducts. On the other hand, deletion of the nudC gene in M. smegmatis mc2155 and M. bovis BCG resulted in increased susceptibility to INH and ETH in MIC tests. More interestingly, inactivation of the nudC gene also caused a defect in the INH or ETH tolerance phenotype (Fig. 4). Furthermore, data from the analysis of mycolic acids showed that overexpression of a functional NudC effectively alleviated the inhibition of mycolic acid biosynthesis caused by INH and ETH, and deletion of nudC elevated the effects of both drugs on mycolic acid biosynthesis (Fig. 5).
Based on the above results, we propose a novel mechanism of INH and ETH inactivation in mycobacteria, as shown in Fig. 6: INH and ETH are activated by KatG or EthA, then form INH-NAD and ETH-NAD adducts with NAD which inhibit the activity of InhA and result in inhibition of mycolic acid biosynthesis. Functional mycobacterial NudC, however, can hydrolyse INH-NAD and ETH-NAD adducts, blocking the effects of these drugs.
Fig. 6.

Diagram of a novel mechanism of INH an ETH inactivation. The INH prodrug requires activation by catalase-peroxidase KatG or Mn3+. Similarly, the ETH prodrug is activated by the flavin monooxygenase EthA. Activated intermediate products then react with NAD+ and yield an INH-NAD or ETH-NAD adduct. These adducts inhibit target protein InhA, the NADH-dependent enoyl-ACP reductase of the fatty acid synthase type II system, resulting in inhibition of mycolic acid biosynthesis and cell lysis. However, functional NudC hydrolyses the INH-NAD and ETH-NAD adducts, resulting in inactivation of these adducts and INH or ETH inactivation.
Inactivation of ETH and INH by mycobacterial NudC was first discovered in this manuscript; however, inactivation of INH by another mycobacterial enzyme Arylamine N-acetyltransferase (NAT) was observed by other researchers previously (Upton et al., 2001). The new finding that mycobacterial NudC can also inactivate INH adds to the complexity of metabolism of this drug in mycobacteria.
Except for M. bovis BCG, which was verified to be able to inactivate INH and ETH through the new mechanism, M. smegmatis mc2155 may also be able to inactivate both drugs by the same mechanism according to our results. Therefore it is important to investigate whether this new mechanism exists in other mycobacteria, especially other members of the M. tuberculosis complex. When comparing the NudC sequence of M. bovis BCG with those from clinical isolates of M. tuberculosis with known genome sequences (http://www.broadinstitute.org/annotation/genome/mycobacterium_tuberculosis_diversity/GenomesIndex.html), identical NudC sequences were found in the three M. tuberculosis clinical isolates T46, CPHL_A and EAS054 (Hirsh et al., 2004; Mostowy et al., 2004). This indicates that, although the NudC from M. tuberculosis H37Rv and most M. tuberculosis clinical isolates is inactive, intact and functional NudC exists in a small portion of M. tuberculosis clinical isolates, implying the new mechanism of INH and ETH inactivation may also exist in these isolates. This needs to be verified by further studies.
Experimental procedures
Bacterial strains, plasmids and media
Strains used are listed in Table S3. E. coli HB101 and E. coli TOP10 were used for constructing mycobacterial mutant strains. E. coli BL21 (DE3) was used as a host strain for recombinant protein expression. M. smegmatis mc2155, M. bovis BCG and M. tuberculosis H37Ra were kindly provided by Dr William R. Jacobs Jr (Howard Hughes Medical Institute, Albert Einstein College of Medicine). Plasmids pET32a, pET28a (Novagen) and pMV261 (kindly provided by Dr Guofeng Zhu) were used for construction of expression plasmids. M. smegmatis mc2155 was grown in Middlebrook 7H9 Broth or 7H10 agar medium (Difco) supplemented with 0.5% (v/v) glycerol and 0.05% (v/v) Tween-80. M. bovis BCG and M. tuberculosis H37Ra were grown in Middlebrook 7H9 Broth or 7H10 agar medium (Difco) supplemented with 10% (v/v) OADC (Difco), 0.5% (v/v) glycerol and 0.05% (v/v) Tween-80. Antibiotics (Sigma) were added at the following concentrations: kanamycin, 25 µg ml−1 for mycobacteria and 50 µg ml−1 for E. coli; hygromycin, 75 µg ml−1 for mycobacteria and 150 µg ml−1 for E. coli, and ampicillin, 100 µg ml−1 for E. coli.
Molecular cloning
To generate expression constructs for purification of NudC, the nudC gene was amplified from M. bovis BCG and M. tuberculosis H37Rv genomic DNA using primers NudCNcoIFP and NudCXhoIRP (Table S3), digested with NcoI and XhoI, and then ligated to the expression vector pET32a, yielding pET32a-NudCBCG and pET32a-NudCRv. To generate in vivo overexpression constructs, nudC from M. tuberculosis H37Rv and M. bovis BCG were amplified using primers 261NudCBamHIFP and 261NudCHindIIIRP (Table S3), digested with BamHI and HindIII, and then cloned into pMV261, yielding pMV261-NudCBCG and pMV261-NudCRv. nudC from M. smegmatis mc2155 was amplified using primers 261NudCBamHIFPSm and 261NudCHindIIIRPSm (Table S3), digested with BamHI and HindIII, and then cloned into pMV261, yielding pMV261-NudCSm. The inhA and EthA genes of M. tuberculosis H37Rv were amplified using primers InhANcoIFP/InhAHindIIIRP and EthABamHIFP/EthAHindIIIRP (Table S3), inserted into the NcoI/HindIII sites of pET28a and BamHI/HindIII sites of pET32a, creating pET28a-InhA and pET32a-EthA respectively.
Protein expression and purification
Recombinant strains E. coli BL21 (DE3)/pET32a-NudCBCG, E. coli BL21 (DE3)/pET32a-NudCRv and E. coli BL21 (DE3)/pET28a-InhA were all induced with 0.5 mM isopropyl β-d-thiogalactopyranoside (IPTG) at 25°C for 10 h. Bacterial cells were harvested, suspended in binding buffer (50 mM Tris·HCl, 0.5 M NaCl, 25 mM imidazole, pH 8.0) and disrupted by sonication. All proteins were purified by nickel affinity chromatography. NudC protein used for analytical ultracentrifugation was then purified by gel filtration with a Superdex 200 10/300GL column (Pharmacia BioTech) which had previously been equilibrated with 50 mM Tris-HCl buffer (pH 8.0). The enzyme was eluted with 50 mM Tris-HCl buffer (pH 8.0), and peak fractions containing the enzyme were pooled.
Analytical ultracentrifugation
The molecular weight of recombinant NudC was analysed using an XL-I analytical ultracentrifuge (Beckman Coulter, Fullerton, CA, USA) equipped with a four-cell An-60 Tirotor. Purified NudC (0.8 mg ml−1 in 50 mM Tris·HCl, pH 8.0) was centrifuged at 4°C under 40000 rpm for 4 h, using 50 mM Tris-HCl buffer (pH 8.0) as a control. After ultracentrifugation, data were analysed using SEDFIT (Schuck, 2000; http://www.analyticalultracentrifugation.com/download.htm).
NudC activity assays
NudC activity was quantified by measuring the conversion of the phosphatase-insensitive substrate NADH to phosphatase-sensitive products AMP and NMNH (Xu et al., 2000). The standard reaction mixture (50 µl) contained: 50 mM Tris-HCl, pH 8.0; 5 mM MgCl2; 2.5 mM NADH; 4 units of calf intestine alkaline phosphatase (EC 3.1.3.1) and 0.2–2 milliunits of enzyme. After incubation at 37°C for 15 min, the reaction was terminated by addition of 250 µl of 4 mM EDTA. Inorganic orthophosphate was measured as previously reported (Ames and Dubin, 1960). A unit of enzyme catalysed the hydrolysis of 1 µmol of NADH per min under these conditions.
Synthesis and isolation of INH-NAD and ETH-NAD adducts
First, the [MnIII(H2P2O7)3]Na3 complex which is necessary for synthesis of the INH-NAD adduct was synthesized as previously described (Nguyen et al., 2001). In brief, an aqueous solution of sodium pyrophosphate (200 mM) containing MnIII(OAc)3 (5.5 mM) was acidified to pH 4.5 (or pH 6.5) with pyrophosphoric acid, then stirred at RT for 24 h. The INH-NAD adduct was then synthesized as reported (Nguyen et al., 2002). The reaction mixture containing 100 mM phosphate buffer (pH 7.5), 2 mM INH, 2 mM NAD and 4 mM Mn(III) pyrophosphate (introduced in 10 consecutive additions of 400 µM each, every 2 min) was stirred at RT for 20 min after addition of the last ingredient. To isolate the INH-NAD adduct, purified InhA (400 µM) was added into the above reaction mixture and stirred at RT for 2 h. The sample was then concentrated and the buffer was exchanged with 50 mM Tris-HCl by using a Millipore Amicon Ultra 10 K centrifugal device. The InhA-adduct complex was denatured by boiling for 40 s and immediately cooling on ice and then transferred into a Microcon 3 centricon filter unit and spun to recover the released adduct in the filtrate (Lei et al., 2000). The ETH-NAD adduct was synthesized as previously described (Wang et al., 2007). Briefly, the plasmids pET28a-InhA and pET32a-EthA were co-transformed into E. coli BL21 (DE3). The strain containing these two plasmids was cultured in LB media containing 50 µg ml−1 kanamycin and 100 µg ml−1 ampicillin at 37°C until OD600 reached 0.8, then induced at 16°C for 20 h by addition of 0.5 mM IPTG. ETH of 100 µg ml−1 was also added to the culture during induction. Recombinant InhA was purified according to the above method. The InhA purified from the strains containing both pET28a-InhA and pET32a-EthA plasmids was concentrated and heated for 40 s at 100°C. After heat treatment, ETH-NAD was separated from the denatured InhA by filtration using a Microcon 3 centricon filter unit.
HPLC-MS analysis of INH-NAD and ETH-NAD adducts and their NudC hydrolysis products
The INH-NAD adduct and its NudC hydrolysis products were analysed by HPLC-ESI/MS (Shimadzu LCMS-2010EV, Tokyo, Japan). HPLC separations were performed according to Rawat (Rawat et al., 2003). A Shim-pack VP-ODS column (Shimadzu, 250 × 2.0 mm i.d., 5 µm) fitted with a C18 guard column (Shimadzu) was used. ESI/MS working parameters were as follows: capillary voltage was 4.5 kV, curved desolvation line (CDL) was 250, and heat block temperatures for the analysis was set at 200°C. Nitrogen drying and nebulizer gases were set at 1.5 l min−1 with a pressure of 0.02 MPa. The detector voltage was set at 1.4 eV. The ETH-NAD adduct and its NudC hydrolysis products were analysed on a HPLC-MicrOTOF_Q mass spectrometer (Agilent 1200 HPLC and Bruker Daltonic MicrOTOF_Q mass spectrometer). HPLC separations were carried out under the same conditions as those for INH-NAD. The ESI/MS working parameters were as follows: the ESI source was operated with a nebulizer pressure of 0.8 bar while the drying gas was delivered at a flow rate of 8 l min−1 and the capillary voltage was 4000 V. All MS data were acquired in a scan range between 200 and 1000 m/z under the negative ionization mode.
Construction of mycobacterial mutant strains
Temperature-sensitive specialized transducing phasmids (Bardarov et al., 2002) were constructed and used for allelic exchange-mediated deletion of the nudC gene in different mycobacteria (MSMEG_1946, Rv3199c/MRA_3236 and BCG_3224c). DNA segments of approximately 1 kb flanking the gene of interest were amplified by PCR, digested with Van91I and ligated to compatible fragments of the counter-selectable vector p0004S (a kind gift from Dr William R. Jacobs Jr). Allelic exchange plasmids were selected and propagated following transformation of E. coli TOP10 to hygromycin resistance. Sequences of inserted DNA fragments were verified. Allelic exchange plasmids were digested with PacI and ligated to PacI-digested phAE159 (Lee et al., 2004). Ligation products were packaged using MaxPlax™ Lambda Packaging Extracts (EPICENTRE Biotechnologies, USA) and transduced into E. coli HB101 for propagation as cosmids. Cosmid DNA was purified and electroporated into M. smegmatis mc2155 for phage propagation at the permissive temperature (30°C). Subsequently, M. smegmatis mc2155 strains were transduced at the non-permissive temperature (37°C) and recombinant strains with integration of the allelic exchange marker were selected on LB agar plates containing hygromycin. Integration of the allelic exchange marker at the locus of interest was verified by PCR using one primer that anneals within the exchange marker and one primer that anneals distal to the genomic region that was involved in allelic exchange.
Drug susceptibility tests
Mycobacterial cells were grown to mid-log phase (OD600 of 0.5–1.0) and diluted to 106 cfu ml−1 (cfu, colony-forming unit) in fresh 7H9 medium. Then 10-fold serial dilutions were plated onto 7H10 agar solid plates containing various concentrations of different drugs: for M. smegmatis mc2155 strains, INH (0, 0.1, 0.25, 0.5, 1.25, 2.5, 5, 10, 25, 50, 100, 150, 200 µg ml−1), ETH (0, 0.1, 0.25, 0.5, 1.25, 2.5, 5, 10, 25, 50, 100, 150, 200 µg ml−1), EMB (0, 0.1, 0.25, 0.5, 1, 2.5, 5 µg ml−1) and RIF (0, 5, 10, 25, 50, 75, 100 µg ml−1); for M. bovis BCG and M. tuberculosis H37Ra strains, INH (0, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.8, 1, 2 µg ml−1), ETH (0, 2.5, 5, 10, 15, 20, 50 µg ml−1), EMB (0, 0.1, 0.25, 0.5, 1, 2.5, 5, 10 µg ml−1) and RIF (0, 0.005, 0.01, 0.05, 0.1, 0.25, 0.5 µg ml−1). Cultures were incubated for 3 days (for M. smegmatis mc2155) or 21 days (for M. bovis BCG and M. tuberculosis H37Ra) at 37°C. MIC was defined as the lowest concentration of compound required to inhibit 99% of bacterial cfu.
Drug exposure experiments
Mycobacterial cells were grown to mid-log phase (OD600 of 0.5–1.0) and diluted to about 106–107 cfu ml−1 in fresh 7H9 medium with or without 10% OADC. For INH treatment of M. smegmatis mc2155, 50 µg ml−1 INH were added to each sample, for that of M. bovis BCG, 5 µg ml−1 INH was added to each sample. For ETH treatment of M. bovis BCG, 50 µg ml−1 ETH was added to each sample. Aliquots of samples were taken at regular intervals and serial dilutions were performed before plating.
Analysis of mycobacterial mycolic acids following ETH and INH treatment
Extraction of cell wall mycolic acids from mycobacterial cells was carried out as previously described (Baulard et al., 2000). M. smegmatis mc2155 or M. bovis BCG cultures (5 ml) were grown to mid-log phase (OD600 ≍ 0.3), then ETH or INH was added at various concentrations followed by further incubation for 4 h (for M. smegmatis mc2155) or 24 h (for M. bovis BCG). At this point, 1, 2-[14C] acetate (1 µCi ml−1) was added and the cultures were further incubated with gentle agitation at 37°C for 4 h (for M. smegmatis mc2155) or 24 h (for M. bovis BCG). After centrifugation, cell pellets were washed once with distilled water and then resuspended in 3 ml of 15% tetrabutylammonium hydroxide (TBAH). The cell suspension was heated at 100°C overnight. After cooling, water (2 ml), dichloromethane (1 ml) and iodomethane (250 µl) were added, and the entire reaction mixture was agitated for 30 min. After centrifugation, the upper aqueous phase was discarded and the lower organic phase was washed twice with water, dried in a sand bath, and the residue was resuspended in methylene chloride. The radiolabelled extracts were separated by thin-layer chromatography (TLC) on a silica gel 60 F254 plate and developed once in petroleum ether: acetone (95:5, v/v). Subsequent autoradiography revealed 14C-labelled FAMEs and MAMEs after overnight exposure of the TLC plates to Kodak BioMax MR film.
Acknowledgments
We thank William R. Jacobs Jr for providing strains M. smegmatis mc2155, M. bovis BCG, M. tuberculosis H37Ra and plasmids p0004S and phAE159; Guofeng Zhu for providing plasmid pMV261 and analysing the nudC gene of M. tuberculosis clinical isolates; and Anthony Baughn and Feng Wang for helpful discussions and critical reading of the manuscript. This project was supported by State Key Development Program for Basic Research of China (2009CB522605), National Basic Research Program of China (2011CB933600) and CAS (KSCX2-YW-R-164).
Supporting information
Additional supporting information may be found in the online version of this article.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Ames BN, Dubin DT. The role of polyamines in the neutralization of bacteriophage deoxyribonucleic acid. J Biol Chem. 1960;235:769–775. [PubMed] [Google Scholar]
- Banerjee A, Dubnau E, Quemard A, Balasubramanian V, Um KS, Wilson T, et al. inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science. 1994;263:227–230. doi: 10.1126/science.8284673. [DOI] [PubMed] [Google Scholar]
- Bardarov S, Bardarov S, Jr, Pavelka MS, Jr, Sambandamurthy V, Larsen M, Tufariello J, et al. Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology. 2002;148:3007–3017. doi: 10.1099/00221287-148-10-3007. [DOI] [PubMed] [Google Scholar]
- Baulard AR, Betts JC, Engohang-Ndong J, Quan S, McAdam RA, Brennan PJ, et al. Activation of the pro-drug ethionamide is regulated in mycobacteria. J Biol Chem. 2000;275:28326–28331. doi: 10.1074/jbc.M003744200. [DOI] [PubMed] [Google Scholar]
- Bekierkunst A. Nicotinamide-adenine dinucleotide in tubercle bacilli exposed to isoniazid. Science. 1966;152:525–526. doi: 10.1126/science.152.3721.525. [DOI] [PubMed] [Google Scholar]
- Bernstein JW, Lott A, Steinberg BA, Yale HL. Chemotherapy of experimental tuberculosis. Am Rev Tuberc. 1952;65:357–374. doi: 10.1164/art.1952.65.4.357. [DOI] [PubMed] [Google Scholar]
- Bessman MJ, Frick DN, O'Handley SF. The MutT proteins or ‘nudix’ hydrolases, a family of versatile, widely distributed, ‘housecleaning’ enzymes. J Biol Chem. 1996;271:25059–25062. doi: 10.1074/jbc.271.41.25059. [DOI] [PubMed] [Google Scholar]
- Boshoff HI, Xu X, Tahlan K, Dowd CS, Pethe K, Camacho LR, et al. Biosynthesis and recycling of nicotinamide cofactors in Mycobacterium tuberculosis. An essential role for NAD innonreplicating bacilli. J Biol Chem. 2008;283:19329–19341. doi: 10.1074/jbc.M800694200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brossier F, Veziris N, Truffot-Pernot C, Jarlier V, Sougakoff W. Molecular investigation of resistance to the antituberculous drug ethionamide in multidrug-resistant clinical isolates of Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2011;55:355–360. doi: 10.1128/AAC.01030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBarber AE, Mdluli K, Bosman M, Bekker LG, Barry CE., 3rd Ethionamide activation and sensitivity in multidrug-resistant Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2000;97:9677–9682. doi: 10.1073/pnas.97.17.9677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn CA, O'Handley SF, Frick DN, Bessman MJ. Studies on the ADP-ribose pyrophosphatase subfamily of the Nudix hydrolases and tentative identification of trgB, a gene associated with tellurite resistance. J Biol Chem. 1999;274:32318–32324. doi: 10.1074/jbc.274.45.32318. [DOI] [PubMed] [Google Scholar]
- Fox HH. The chemical approach to the control of tuberculosis. Science. 1952;116:129–134. doi: 10.1126/science.116.3006.129. [DOI] [PubMed] [Google Scholar]
- Frick DN, Bessman MJ. Cloning, purification, and properties of a novel NADH pyrophosphatase-evidence for a nucleotide pyrophosphatase catalytic domain in MutT-like enzymes. J Biol Chem. 1995;270:1529–1534. doi: 10.1074/jbc.270.4.1529. [DOI] [PubMed] [Google Scholar]
- Gagneux S, DeRiemer K, Van T, Kato-Maeda M, de Jong BC, Narayanan S, et al. Variable host–pathogen compatibility in Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2006;103:2869–2873. doi: 10.1073/pnas.0511240103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazbon MH, Brimacombe M, Bobadilla del Valle M, Cavatore M, Guerrero MI, Varma-Basil M, et al. Population genetics study of isoniazid resistance mutations and evolution of multidrug-resistant Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2006;50:2640–2649. doi: 10.1128/AAC.00112-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heym B, Cole ST. Isolation and characterization of isoniazid-resistant mutants of Mycobacterium smegmatis and M. aurum. Res Microbiol. 1992;143:721–730. doi: 10.1016/0923-2508(92)90067-x. [DOI] [PubMed] [Google Scholar]
- Heym B, Alzari PM, Honore N, Cole ST. Missense mutations in the catalase-peroxidase gene, katG, are associated with isoniazid resistance in Mycobacterium tuberculosis. Mol Microbiol. 1995;15:235–245. doi: 10.1111/j.1365-2958.1995.tb02238.x. [DOI] [PubMed] [Google Scholar]
- Hirsh AE, Tsolaki AG, DeRiemer K, Feldman MW, Small PM. Stable association between strains of Mycobacterium tuberculosis and their human host populations. Proc Natl Acad Sci USA. 2004;101:4871–4876. doi: 10.1073/pnas.0305627101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnsson K, Schultz PG. Mechanistic studies of the oxidation of isoniazid by the catalase peroxidase from Mycobacterium tuberculosis. J Am Chem Soc. 1994;116:7425–7426. [Google Scholar]
- Kasărov LB, Moat AG. Metabolism of nicotinamide adenine dinucleotide in human and bovine strains of Mycobacterium tuberculosis. J Bacteriol. 1972;110:600–603. doi: 10.1128/jb.110.2.600-603.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen MH, Vilchèze C, Kremer L, Besra GS, Parsons L, Salfinger M, et al. Overexpression of inhA, but not kasA, confers resistance to isoniazid and ethionamide in Mycobacterium smegmatis, M. bovis BCG and M. tuberculosis. Mol Microbiol. 2002;46:453–466. doi: 10.1046/j.1365-2958.2002.03162.x. [DOI] [PubMed] [Google Scholar]
- LaVallie ER, DiBlasio EA, Kovacic S, Grant KL, Schendel PF, McCoy JM. A thioredoxin gene fusion expression system that circumvents inclusion body formation in the E. coli cytoplasm. Bio/Technology. 1993;11:187–193. doi: 10.1038/nbt0293-187. [DOI] [PubMed] [Google Scholar]
- Lee AS, Teo AS, Wong SY. Novel mutations in ndh inisoniazid-resistant Mycobacterium tuberculosis isolates. Antimicrob Agents Chemother. 2001;45:2157–2159. doi: 10.1128/AAC.45.7.2157-2159.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Kriakov J, Vilcheze C, Dai Z, Hatfull GF, Jacobs WR., Jr Bxz1, a new generalized transducing phage for mycobacteria. FEMS Microbiol Lett. 2004;241:271–276. doi: 10.1016/j.femsle.2004.10.032. [DOI] [PubMed] [Google Scholar]
- Lei B, Wei CJ, Tu SC. Action mechanism of antitubercular isoniazid. Activation by Mycobacterium tuberculosis KatG, isolation, and characterization of InhA inhibitor. J Biol Chem. 2000;275:2520–2526. doi: 10.1074/jbc.275.4.2520. [DOI] [PubMed] [Google Scholar]
- Middlebrook G, Cohn ML. Some observations on the pathogenicity of isoniazid-resistant variants of tubercle bacilli. Science. 1953;118:297–299. doi: 10.1126/science.118.3063.297. [DOI] [PubMed] [Google Scholar]
- Miesel L, Weisbrod TR, Marcinkeviciene JA, Bittman R, Jacobs WR., Jr NADH dehydrogenase defects confer isoniazid resistance and conditional lethality in Mycobacterium smegmatis. J Bacteriol. 1998;180:2459–2467. doi: 10.1128/jb.180.9.2459-2467.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morlock GP, Metchock B, Sikes D, Crawford JT, Cooksey RC. ethA, inhA, and katG loci of ethionamide-resistant clinical Mycobacterium tuberculosis isolates. Antimicrob Agents Chemother. 2003;47:3799–3805. doi: 10.1128/AAC.47.12.3799-3805.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostowy S, Onipede A, Gagneux S, Niemann S, Kremer K, Desmond EP, et al. Genomic analysis distinguishes Mycobacterium africanum. J Clin Microbiol. 2004;42:3594–3599. doi: 10.1128/JCM.42.8.3594-3599.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen M, Claparols C, Bernadou J, Meunier B. A fast and efficient metal-mediated oxidation of isoniazid and identification of isoniazid-NAD(H) adducts. Chembiochem. 2001;2:877–883. doi: 10.1002/1439-7633(20011203)2:12<877::AID-CBIC877>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Nguyen M, Quémard A, Broussy S, Bernadou J, Meunier B. Mn(III) pyrophosphate as an efficient tool for studying the mode of action of isoniazid on the InhA protein of Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2002;46:2137–2144. doi: 10.1128/AAC.46.7.2137-2144.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawat R, Whitty A, Tonge PJ. The isoniazid-NAD adduct is a slow, tight-binding inhibitor of InhA, the Mycobacterium tuberculosis enoyl reductase: adduct affinity and drug resistance. Proc Natl Acad Sci USA. 2003;100:13881–13886. doi: 10.1073/pnas.2235848100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozwarski DA, Grant GA, Barton DH, Jacobs WR, Jr, Sacchettini JC. Modification of the NADH of the isoniazid target (InhA) from Mycobacterium tuberculosis. Science. 1998;279:98–102. doi: 10.1126/science.279.5347.98. [DOI] [PubMed] [Google Scholar]
- Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama K, Wang L, David HL. Effect of isoniazid on the in vivo mycolic acid synthesis, cell growth, and viability of Mycobacterium tuberculosis. Antimicrob Agents Chemother. 1972;2:29–35. doi: 10.1128/aac.2.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton AM, Mushtaq A, Victor TC, Sampson SL, Sandy J, Smith DM, et al. Arylamine N-acetyltransferase of Mycobacterium tuberculosis is a polymorphic enzyme and a site of isoniazid metabolism. Mol Microbiol. 2001;42:309–317. doi: 10.1046/j.1365-2958.2001.02648.x. [DOI] [PubMed] [Google Scholar]
- Vannelli TA, Dykman A, OrtizdeMontellano PR. The antituberculosis drug ethionamide is activated by a flavoprotein monooxygenase. J Biol Chem. 2002;277:12824–12829. doi: 10.1074/jbc.M110751200. [DOI] [PubMed] [Google Scholar]
- Vilchèze C, Morbidoni HR, Weisbrod TR, Iwamoto H, Kuo M, Sacchettini JC, Jacobs WR., Jr Inactivation of the inhA-encoded fatty acid synthase II (FASII) enoyl-acyl carrier protein reductase induces accumulation of the FASI end products and cell lysis of Mycobacterium smegmatis. J Bacteriol. 2000;182:4059–4067. doi: 10.1128/jb.182.14.4059-4067.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilchèze C, Weisbrod TR, Chen B, Kremer L, Hazbón MH, Wang F, et al. Altered NADH/NAD+ ratio mediates coresistance to isoniazid and ethionamide in mycobacteria. Antimicrob Agents Chemother. 2005;49:708–720. doi: 10.1128/AAC.49.2.708-720.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilchèze C, Wang F, Arai M, Hazbon MH, Colangeli R, Kremer L, et al. Transfer of a point mutation in Mycobacterium tuberculosis inhA resolves the target of isoniazid. Nat Med. 2006;12:1027–1029. doi: 10.1038/nm1466. [DOI] [PubMed] [Google Scholar]
- Vilchèze C, Av-Gay Y, Attarian R, Liu Z, Hazbón MH, Colangeli R, et al. Mycothiol biosynthesis is essential for ethionamide susceptibility in Mycobacterium tuberculosis. Mol Microbiol. 2008;69:1316–1329. doi: 10.1111/j.1365-2958.2008.06365.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilchèze C, Weinrick B, Wong KW, Chen B, Jacobs WR., Jr NAD+ auxotrophy is bacteriocidal for the tubercle bacilli. Mol Microbiol. 2010;76:365–377. doi: 10.1111/j.1365-2958.2010.07099.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Langley R, Gulten G, Dover LG, Besra GS, Jacobs WR, Jr, Sacchettini JC. Mechanism of thioamide drug action against tuberculosis and leprosy. J Exp Med. 2007;204:73–78. doi: 10.1084/jem.20062100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilming M, Johnsson K. Spontaneous formation of the bioactive form of the tuberculosis drug isoniazid. Angew Chem Int Ed Engl. 1999;38:2588–2590. doi: 10.1002/(sici)1521-3773(19990903)38:17<2588::aid-anie2588>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- World Health Organization. 2009. Global Tuberculosis Control. A short update to the 2009 report.
- Xu W, Dunn CA, Bessman MJ. Cloning and characterization of the NADH pyrophosphatases from Caenorhabditis elegans and Saccharomyces cerevisiae, members of a Nudix hydrolase subfamily. Biochem Biophys Res Commun. 2000;273:753–758. doi: 10.1006/bbrc.2000.2999. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Heym B, Allen B, Young D, Cole S. The catalase-peroxidase gene and isoniazid resistance of Mycobacterium tuberculosis. Nature. 1992;358:591–593. doi: 10.1038/358591a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.