

Abstract
LRRK2 (PARK8) is the most common genetic determinant of Parkinson's disease (PD), with dominant mutations in LRRK2 causing inherited PD and sequence variation at the LRRK2 locus associated with increased risk for sporadic PD. Although LRRK2 has been implicated in diverse cellular processes encompassing almost all cellular compartments, the precise functions of LRRK2 remain unclear. Here, we show that the Drosophila homolog of LRRK2 (Lrrk) localizes to the membranes of late endosomes and lysosomes, physically interacts with the crucial mediator of late endosomal transport Rab7 and negatively regulates rab7-dependent perinuclear localization of lysosomes. We also show that a mutant form of lrrk analogous to the pathogenic LRRK2G2019S allele behaves oppositely to wild-type lrrk in that it promotes rather than inhibits rab7-dependent perinuclear lysosome clustering, with these effects of mutant lrrk on lysosome position requiring both microtubules and dynein. These data suggest that LRRK2 normally functions in Rab7-dependent lysosomal positioning, and that this function is disrupted by the most common PD-causing LRRK2 mutation, linking endolysosomal dysfunction to the pathogenesis of LRRK2-mediated PD.
INTRODUCTION
Parkinson's disease (PD) is the second most common neurodegenerative disorder, and has no cure. Dominant mutations in Leucine-rich repeat kinase 2 (LRRK2) cause an inherited form of PD (1,2), and genome-wide association studies have identified variants at the LRRK2 locus as having increased risk for sporadic PD (3,4). In certain populations, LRRK2 mutations are found in up to 40% of PD patients (5), making LRRK2 the most common genetic determinant of PD identified to date (6,7). Studies of LRRK2 therefore offer tremendous potential to elucidate the mechanisms of PD, and to identify novel therapies for the disease.
LRRK2 encodes a large protein of the ROCO family (8), characterized by leucine-rich repeats (LRR), a Roc GTPase domain and a kinase domain (9). LRRK2 has been implicated in diverse cellular processes encompassing nearly all cellular compartments, including mitochondrial function (10), regulation of transcription (11) and translation (12,13), Golgi protein sorting (14), apoptosis (15) and dynamics of actin (16,17) and microtubules (18–20). There is equally little consensus regarding the subcellular localization of LRRK2, as it has been variably reported to localize to mitochondria (21–24), the endoplasmic reticulum (23,25), Golgi (22–24) and microtubule structures (18,23); however, most authors are in agreement that LRRK2 associates with intracellular membranes (22–24,26,27). Interestingly, LRRK2 has also been found to localize to vesicles in the endolysosomal pathway (22,24,26,28,29), and there is some evidence implicating LRRK2 in intracellular membrane transport and lysosomal function. Most notably, LRRK2 physically interacts with the early endosomal GTPase Rab5 (28), and knockdown of LRRK2 in cultured neurons alters synaptic vesicle endocytosis (28,30) and the kinetics and distribution of presynaptic vesicles (30). Expression of a fragment of human LRRK2 in yeast impairs trafficking to the yeast lysosome (31), and overexpression of a pathogenic mutant form of LRRK2 in cultured neurons causes defects in neurite morphology associated with the formation of lysosome inclusions (32). Moreover, LRRK2 knockout mice show accumulation of lipofuscin in the kidney, suggestive of lysosome dysfunction (33). While these data hint at a possible role for LRRK2 in regulating endolysosomal membrane transport, an in vivo cell biological analysis is needed to characterize the precise cellular functions of LRRK2.
Drosophila melanogaster is a valuable model system for the study of genes associated with neurodegenerative diseases, including PD (34–36). The Drosophila genome encodes a single homolog that shares the domain structure of human LRRK2 (12,37,38), and residues affected by PD-causing mutations in LRRK2 are conserved in Drosophila Lrrk (12). Overexpression of either human LRRK2 or Drosophila lrrk in flies causes similar phenotypes (13,39), including degeneration of dopaminergic neurons (12,40,41), suggesting that the two proteins are functionally conserved. While endogenous lrrk appears to be dispensable for the survival of dopaminergic neurons in Drosophila (12,37,38), lrrk null mutants show two major phenotypes at the organismal level: altered morphology of the neuromuscular junction (NMJ) (39), and impaired female fertility (12,37), the latter of which has not been explored. A role for lrrk in the regulation of protein translation (12,13) has also been described.
Drosophila oogenesis is an ideal cell biological system. The large size and ease of visualization of cells in the Drosophila ovary allows for the examination of subcellular structures with a resolution difficult to accomplish in neurons. Thus, we exploited the role of lrrk in female fertility as a model system in which to explore the cell biological functions of Lrrk in vivo by aDDRessing three questions: what is the localization of Lrrk in vivo? Does the subcellular localization of the protein define the compartment in which Lrrk functions? Does a pathogenic mutation alter the functions of the protein at this site? Using this approach, we have identified a role for lrrk at late endosomes and lysosomes as a regulator of rab7-dependent lysosome transport and positioning, with this function intrinsically altered by introducing the most common PD-associated mutation into lrrk. These data point to differential roles for endogenous and pathogenic forms of lrrk in rab7-dependent lysosomal positioning.
RESULTS
lrrk is required in follicle cells to ensure proper female fertility
lrrke03680 is a null allele resulting from a PiggyBac transposable element insertion in the lrrk coding region (Fig. 1A), and produces no detectable transcript (Fig. 1B) (12). Flies homozygous for lrrke03680 or trans-heterozygous for lrrke03680 over a chromosomal deficiency removing the lrrk genomic region are viable, but show a dramatic reduction in female fertility (Fig. 1C and D) (12,37). We generated a genomic rescue transgene expressing a Myc-tagged version of lrrk under the endogenous promoter. Anti-Myc western blot from flies carrying this transgene revealed a single band at the expected size (data not shown), and a single copy of this transgene was able to significantly rescue the female fertility defect of lrrke03680 flies (Fig. 1C).
Figure 1.

Lrrk is required in follicle cells for proper female fertility. (A) Schematic depicting domain organization of Drosophila Lrrk and location of PiggyBac insertion in the lrrke03680 allele and G-to-S substitution in lrrkGS. Above is an alignment of the amino acid sequences of human LRRK2 (top) and Drosophila Lrrk (bottom) in the region of the GS mutation (marked with an asterisk). LRR, leucine-rich repeats; Roc, GTPase domain; Cor, C-terminal of Roc domain. (B) lrrke03680 homozygous flies produce no detectable lrrk transcript by RT–PCR. (C) Fertility is reduced in lrrke03680 homozygous mutants, and the phenotype is partially rescued by a Myc-tagged genomic rescue transgene (p[lrrk-myc]). (D) Fertility is fully restored to lrrke03680/Df null mutant flies by follicle cell-specific expression of either wild-type lrrk (fc>lrrkWT) or lrrkGS (fc>lrrkGS).
Developing Drosophila egg chambers consist of the germline oocyte, its clonally related germline nurse cells and the somatic follicle cells, which form an epithelial monolayer surrounding the oocyte (42). Flies carrying a single copy of the lrrk-myc genomic rescue transgene displayed striking Lrrk-myc signal in follicle cells but not in germline cells, and this was restricted to stages 11–13 of the 14 stages of egg chamber development (Fig. 2A). To confirm that lrrk is autonomously required in follicle cells to ensure fertility, we generated UAS-lrrk (hereafter lrrkWT) and expressed it under the control of a follicle cell-specific Gal4 driver in lrrk null mutants. Indeed, targeted expression of lrrkWT specifically in follicle cells fully restored fertility to lrrke03680/Df flies (Fig. 1D).
Figure 2.

Lrrk localizes to late endosomes and lysosomes in follicle cells. (A) Anti-Myc staining of egg chambers from flies carrying a single copy of the C-terminally Myc-tagged lrrk genomic rescue transgene. Lrrk-Myc was not detected in egg chambers prior to stage 11 (A), and persisted through stages 12 (A″) and 13 (A″′). Lrrk-Myc staining reveals a patchy distribution, with expression in subsets of follicle cells (including main body and stretched follicle cells), but not in germline cells. (B) Schematic depicting the endolysosomal pathway and proteins labeling specific compartments. (C–J) Images of single main body follicle cells with Lrrk-Myc staining in red and costaining for various cellular compartments in green. Nuclei are outlined by dashed gray lines, and insets depict enlargements of the boxed regions. Lrrk localizes to discrete cytosolic puncta that often appear as halos surrounding a non-stained central region, indicative of an association with vesicle membranes. These large Lrrk-positive halos colocalize with the late endosomal markers Rab7 (C) and Rab9:GFP (D). The large Lrrk-positive structures also contain Lamp1:GFP (E). Note that while Rab7 and Rab9:GFP label late endosome membranes, Lamp1:GFP accumulates in the lumena of late endosomes and lysosomes. Small Lrrk-positive puncta occasionally colocalize with the early endosomal marker Hrs (F). In contrast, there is little colocalization between Lrrk and Rab5 (G) or Rab11 (H), although these markers often appear adjacent to the Lrrk-positive vesicles. There is no significant colocalization between Lrrk and the mitochondrial marker mitoGFP (I) or the Golgi marker Grasp65:GFP (J). Scale bars represent 5 μm.
Lrrk localizes specifically to the membranes of late endosomes and lysosomes
We decided to exploit the attributes of follicle cells as a cell biological system to explore the in vivo functions of Lrrk, starting by determining its in vivo subcellular localization. Using flies carrying the lrrk-myc genomic rescue transgene, we found that Lrrk localized predominantly to discrete halo-shaped cytoplasmic structures (Fig. 2C). Double labeling with markers of membrane-bound compartments revealed striking colocalization between Lrrk and the late endosomal and lysosomal markers Rab7 (43) (Fig. 2C), Rab9 (44) (Fig. 2D) and Lamp1 (45,46) (Fig. 2E). Lrrk rarely colocalized with the early/sorting endosomal marker Hrs (43,47) (Fig. 2F), and did not colocalize with the early endsomal marker Rab5 (48–50) (Fig. 2G). These early endosomal markers, however, were sometimes seen adjacent to the larger Lrrk-positive structures (Fig. 2F and G). Lrrk also failed to colocalize with markers of recycling endosomes (51,52) (Fig. 2H), mitochondria (53) (Fig. 2I) or the Golgi (54) (Fig. 2J). Thus, in vivo, Lrrk localizes specifically to the membranes of late endosomes and lysosomes.
Abnormal Rab7-positive compartment in lrrk null mutants
The localization of Lrrk to endolysosomal membranes suggested a role in membrane transport. Thus, we examined a battery of endolysosomal markers in wild-type and lrrk null mutant follicle cells to identify potential morphology defects in specific endosomal compartments that might reflect such a role. The distribution of the early endosomal markers Rab5 (Fig. 3A versus B) and Hrs (Fig. 3C versus D) were indistinguishable between wild-type and lrrk null mutants, as was that of the lysosomal markers Lysotracker (Fig. 3G versus H) and Lamp1:GFP (Fig. 3I versus J). In contrast, striking defects were observed in the Rab7-positive late endosomal compartment. Consistently, ∼15% of lrrk mutant follicle cells displayed dramatically enlarged Rab7-positive structures (Fig. 3F), which were never observed in stage-matched wild-type cells (Fig. 3E). That no similarly enlarged structures are observed in lrrk null mutants when stained with the acidophilic dye Lysotracker (Fig. 3H) suggests that these aberrant Rab7-positive structures are not acidified. This could be due to a direct failure of acidification, or an indirect consequence of a block in the maturation of these late endosomal structures before acidification has begun.
Figure 3.

Altered Rab7-positive late endosomal compartments in lrrk loss-of-function mutants. (A–J) Examination of a battery of endolysosomal markers in wild-type (A, C, E, G, I) and lrrk null mutant (B, D, F, H, J) follicle cells reveals no distinguishable difference between the early endosomal markers Rab5 (A versus B) or Hrs (C versus D), or the lysosomal markers Lysotracker (G versus H) or Lamp1:GFP (I versus J). A small portion of lrrk mutant follicle cells, however, did display dramatically expanded Rab7-positive late endosomes (marked with arrowheads in F) that were never observed in stage-matched wild-type follicle cells (E). (K and L) After a 30 min chase, fluorescently labeled dextran was taken up into wild-type follicle cells and trafficked predominantly to Rab7-positive late endosomes (K). In lrrk mutant follicle cells (L), some enlarged Rab7-positive structures accumulated massive amounts of dextran, while others were devoid of the tracer. Follicle cell nuclei are outlined with dashed gray lines in (A–J). Scale bars represent 5 μm.
To confirm that these enlarged Rab7-positive structures indeed represented late endosomes, we performed endocytic tracer uptake experiments. Live egg chambers were pulse incubated with Texas Red-dextran, which is internalized into cells via endocytosis, and then examined at various time points as the dextran was trafficked through endocytic compartments. After a 30 min chase, dextran was primarily found within Rab7-positive endosomes in wild-type cells (Fig. 3K). The enlarged Rab7-positive structures in lrrk mutant cells, however, displayed marked heterogeneity with some of these structures accumulating massive amounts of dextran while others were devoid of the tracer (Fig. 3L). Collectively, these results suggest that lrrk loss-of-function alters the morphology of the Rab7 compartment and affects trafficking to these structures. It is unclear whether these aberrant structures are the cause of the reduced fertility in lrrk mutant females given that they are seen in a minority of cells. However, it is important to note that even during stage 12, when Lrrk expression in follicle cells is at its highest, it is expressed in only a subset of cells and there is marked cell–cell variation in the level of Lrrk expression (Fig. 2A). This may indicate that Lrrk expression is activated only in follicle cells that have achieved a particular developmental state. In any case, the fact that all follicle cells do not show abnormalities in the Rab7 compartment is consistent with the finding that all follicle cells do not appear to be expressing lrrk simultaneously.
lrrk genetically and physically interacts with rab7
The striking colocalization of Lrrk with Rab7 combined with the late endosome defects observed in lrrk mutants suggested a role for lrrk in rab7-dependent processes. A crucial function of rab7 is to promote the perinuclear localization of lysosomes (55) by driving their transport along microtubules (56–58). This perinuclear localization of lysosomes is vital for multiple cellular functions, including the regulation of autophagy (59). In mammalian cells, expression of a constitutively active form of Rab7 results in the formation of large perinuclear lysosome clusters, while dominant-negative Rab7 disperses lysosomes throughout the cytosol (55). Whether this occurs in Drosophila has not been reported. In wild-type follicle cells, ∼40% of the total lysosome bulk localized to clusters, defined as having a cross-sectional area >1 μm (Fig. 4A and G). Expression of constitutively active rab7 (rab7CA) caused a 35% increase in lysosome clustering, with these clusters primarily perinuclear (Fig. 4D versus A and G). In contrast, expression of dominant-negative rab7 (rab7DN) caused a striking 46% decrease in the fraction of clustered lysosomes, and resulted in more even dispersal of lysosomes throughout the cytoplasm (Fig. 6C versus A and G).
Figure 4.

lrrk and rab7 genetically interact. (A–F) Lysotracker staining of follicle cells from stage 12 egg chambers. In wild-type follicle cells (A), most Lysotracker-positive vesicles are individual and distributed throughout the cytosol, although small clusters are often seen. Neither lrrk loss-of-function (B), nor overexpression of wild-type lrrk (C) significantly alters lysosome distribution. Expression of constitutively active rab7Q67L (D) results in an increase in the formation of Lysotracker-positive clusters, and these tend to localize to the peri-nuclear region. lrrk loss-of-function significantly enhances lysosome clustering in follicle cells expressing rab7Q67L (E), while overexpression of wild-type lrrk significantly reduces rab7Q67L-induced lysosome clustering (F). Follicle cell nuclei are outlined with dashed gray lines in all Lysotracker images. (G) Quantification of lysosome clustering in the genotypes depicted in (A)–(F). Scale bars represent 5 μm.
Figure 6.

The GS mutation abrogates the ability of lrrk to inhibit rab7-induced lysosome clustering. (A–F) Lysotracker staining of follicle cells from stage 12 egg chambers of the indicated genotypes. Expression of constitutively active Rab7 (rab7Q67L, B) causes perinuclear clustering of Lysotracker-positive structures similar to that seen with expression of lrrkGS (D), while expression of rab7Q67L and lrrkGS together (E) causes a phenotype that is not significantly different from expression of lrrkGS alone. Expression of dominant-negative Rab7 (rab7T22N, C) causes lysosome dispersal. Expression of rab7T22N along with lrrkGS significantly reduces the lrrkGS-induced lysosome clustering. (G) Quantification of lysosome clustering in the indicated genotypes. Follicle cell nuclei are outlined with dashed gray lines in all Lysotracker images. Scale bars represent 5 μm.
Using this assay of perinuclear lysosome localization, we tested for genetic interactions between lrrk and rab7. As noted above, lrrk loss-of-function alone had no effect on the morphology or distribution of lysosomes (Fig. 4B and G), nor did overexpression of lrrkWT alone (Fig. 4C and G). Interestingly, however, expression of rab7CA in the lrrk null mutant background resulted in a 36% increase in perinuclear lysosome clustering compared with expression of rab7CA in the wild-type background (Fig. 4E versus D and G). Conversely, overexpression of rab7CA along with lrrkWT resulted in a significant 56% reduction in lysosome clustering compared with rab7CA alone (Fig. 4F versus D and G). These results are consistent with a role of lrrk as a negative regulator of rab7-mediated perinuclear clustering and localization of lysosomes.
We next sought to determine whether Lrrk might interact physically with Rab7 by performing co-immunoprecipitation experiments on lysates from transiently transfected insect cells. Mammalian LRRK2 has previously been reported to bind to the early endosomal GTPase Rab5 (28), so we included Rab5 in our experiments as a positive control. We found that Drosophila Lrrk immunoprecipitated with both Rab5 and Rab7, but not GFP alone (Fig. 7A), suggesting that Lrrk physically interacts with both Rab5 and Rab7.
Figure 7.

The GS mutation alters the characteristics of physical binding between Lrrk and Rab7 forms. (A) In lysates from cultured Drosophila S2 cells transfected with the depicted constructs, LrrkWT immunoprecipitates with both Rab7-GFP and Rab5-GFP, but not with GFP alone. The GS mutation reduces the physical interaction between Lrrk and Rab7, but does not affect binding to Rab5. (B) Whereas LrrkWT preferentially immunoprecipitates with the GTP-binding deficient dominant-negative version of Rab7, LrrkGS binds to Rab7DN and Rab7CA equally.
Expression of lrrkGS results in perinuclear lysosome clustering similar to activated rab7
Next, we sought to determine whether PD-causing mutations in lrrk affect rab7-dependent lysosome positioning. We chose the G2019S mutation, as it is the most common pathogenic LRRK2 mutation (60,61). This mutation is located in the activation loop of the kinase domain and causes an increase in LRRK2 kinase activity (16,21,32,62,63). We generated flies expressing a form of Lrrk bearing the equivalent mutation (LrrkG1914S, hereafter LrrkGS) (Fig. 1A). Expression of lrrkGS in follicle cells restored fertility to lrrk null mutants as effectively as expression of lrrkWT (Fig. 1D), indicating that LrrkGS retains at least some of the functions of the wild-type protein.
Next we examined lysosome positioning in flies overexpressing lrrkGS in follicle cells. Strikingly, we found that, as with constitutively active rab7 (Fig. 4D), lrrkGS expression resulted in a dramatic 65% increase in perinuclear lysosome clustering compared with the wild-type (Fig. 5B versus A and D). Similar to expression of rab7CA, lrrkGS expression did not appear to alter the total number of lysosomes per cell, as at higher magnification lysosome clusters were clearly composed of numerous individual lysosomes (data not shown). Lysosome clustering was also apparent in lrrkGS-expressing cells using the marker Lamp1:GFP (Fig. 5E′ versus F′), which colocalized with Lysotracker in lrrkGS-expressing cells (Fig. 5F″). It is important to note that Lamp1:GFP is normally rapidly degraded upon reaching the lysosome (46), which can be appreciated by the low degree of colocalization between Lamp1:GFP and Lysotracker in wild-type cells (Fig. 5E″). In contrast, Lamp1:GFP almost exclusively colocalized with Lysotracker in lrrkGS-expressing cells (Fig. 5F″), in which it also accumulated to a much higher degree (Fig. 5F′) relative to the wild-type (Fig. 5E′). This suggests that lrrkGS expression may also result in impairment in the degradative activities of the lysosome. Notably, the effects of lrrkGS on vesicle positioning were specific to lysosomes as there was no difference between wild-type and lrrkGS-expressing cells with regard to the distribution of Hrs (Fig. 5G versus H), which labels early endosomes, nor with Rab7 itself (Fig. 5I versus J), which labels late endosomes. Moreover, as shown above, no changes in lysosome clustering were observed with equivalent expression of wild-type lrrk (Fig. 5C and D). Taken together, these results suggest that LrrkGS, but not LrrkWT, specifically affects lysosome position by driving their perinuclear transport, and may also impair the functional activities of the lysosome.
Figure 5.

Expression of lrrkGS drives perinuclear positioning and clustering of lysosomes. (A–C) Lysotracker staining of follicle cells from stage 12 egg chambers. In wild-type follicle cells (A), most Lysotracker-positive vesicles are individual and distributed throughout the cytosol, although small clusters are often seen. Expression of lrrkGS (B) in follicle cells causes almost all Lysotracker-positive structures to collapse into one to four compact perinuclear clusters, while equivalent overexpression of lrrkWT (C) results in no significant changes in the distribution of Lysotracker-positive structures relative to the wild-type. (D) Quantification of lysosome clustering from the genotypes depicted in (A)–(C). (E and F) Lysotracker staining of wild-type (E) and lrrkGS-expressing (F) follicle cells also expressing the lysosomal marker Lamp1:GFP. Lamp1:GFP-positive vesicles are clustered in lrrkGS-expressing cells (F′), colocalizing with Lysotracker (F″). Note that Lamp1:GFP accumulates to a much higher degree in lrrkGS-expressing cells (F′) versus wild-type (E′) in images taken with equivalent microscope settings, suggesting that expression of lrrkGS stabilizes Lamp1:GFP. (G–J) Labeling of early endosomes with the marker Hrs (G and H) and late endosomes with the marker Rab7 (I and J) reveals no significant difference between wild-type (G and I) and lrrkGS-expressing cells (H and J), demonstrating that the clustering effect of lrrkGS is specific to lysosomes. Follicle cell nuclei are outlined with dashed gray lines in all images. Scale bar in (A) represents 5 μm in (A)–(F); scale bar in (G) represents 5 μm in (G)–(J).
The GS mutation abrogates the ability of lrrk to inhibit rab7-induced lysosome clustering
Next, we probed whether the altered function of lrrkGS extends to its genetic relationship with rab7. As reported above, lrrk negatively regulates rab7 such that expression of wild-type lrrk suppresses the perinuclear lysosome clustering caused by rab7CA. In contrast, we found that expressing lrrkGS along with rab7CA results in a degree of lysosome clustering that is identical to that caused by expression of lrrkGS alone (Fig. 6E versus D and G). This is consistent with the hypothesis that the GS mutation might abrogate the ability of Lrrk to negatively regulate Rab7, although it does not rule out the possibility that LrrkGS might simultaneously inhibit Rab7 activity while promoting lysosome clustering via a distinct mechanism. To distinguish between these possibilities, we asked whether expression of rab7DN was able to suppress lrrkGS-induced lysosome clustering. Indeed, we found that lrrkGS-induced clustering of lysosomes was significantly reduced by 41% when rab7DN was also expressed (Fig. 6F versus D and G). Thus, the clustering of lysosomes driven by lrrkGS is at least partially rab7-dependent. Collectively, these data indicate that the GS mutation alters the genetic relationship between lrrk and rab7. Whereas wild-type lrrk negatively regulates rab7, lrrkGS promotes rab7-dependent effects on lysosome position.
The GS mutation alters the physical interaction between Lrrk and Rab7
Given that the GS mutation strikingly alters the genetic relationship between Lrrk and Rab7, we next wished to probe whether the GS mutation also alters the physical interaction between the two proteins. Interestingly, we found that LrrkGS shows reduced binding to Rab7 when compared with LrrkWT (Fig. 7A). This was specific to the interaction between Lrrk and Rab7, as LrrkWT and LrrkGS show equivalent binding to Rab5 (Fig. 7A). To gain further insight into this differential interaction, we explored the physical binding of LrrkWT and LrrkGS with mutant forms of Rab7. Strikingly, we found that LrrkWT shows a markedly stronger binding with the GTP-binding deficient dominant-negative form of Rab7 compared with the constitutively active form of Rab7 that is deficient in GTP hydrolysis (Fig. 7B). In contrast, LrrkGS appears to bind equally well with both mutant forms of Rab7 (Fig. 7B). Thus, the GS mutation alters both the genetic and physical interactions between Lrrk and Rab7, suggesting that differential binding with Rab7 may be the basis for the differences in the functions of LrrkWT and LrrkGS with respect to Rab7.
lrrkGS-driven perinuclear clustering of lysosomes is dependent on dynein-dependent microtubule transport
Through multiple binding partners, Rab7CA drives perinuclear clustering of lysosomes by recruiting the dynein–dynactin motor complex to lysosome membranes, thereby promoting microtubule minus-end directed motility (56–58). Interestingly, LRRK2 has been shown to bind to and phosphorylate tubulin (18,19), and to modulate microtubule dynamics both in vitro and in vivo (20,39,41). Thus, we hypothesized that the effects of lrrkGS on lysosome position might also be microtubule- and dynein-dependent. Consistent with this notion, we found that treatment of lrrkGS-expressing follicle cells with the microtubule destabilizing agent nocodazole resulted in a dramatic shift in the localization of lysosome clusters from the perinuclear region to the cell periphery (Fig. 8D versus C). That cluster formation itself is not affected by nocodazole suggests that either cluster formation is independent of perinuclear localization, or that once formed, the maintenance of lysosome clusters no longer requires microtubules due to membrane tethering events. In support of the latter hypothesis, we found that the loss of a single copy of the gene encoding Dynein heavy chain significantly reduced cluster formation in lrrkGS-expressing cells by 20% (Fig. 8F versus C and G), and also reduced the size of those clusters that were present (Fig. 8F versus C and H). Thus, both clustering and perinuclear localization of lysosomes induced by lrrkGS-expression are dependent on dynein-dependent microtubule transport.
Figure 8.

Perinuclear clustering of Lysotracker-positive structures in lrrkGS-expressing follicle cells is microtubule- and dynein-dependent. (A–F) Lysotracker staining of follicle cells. Treatment with the microtubule destabilizing agent nocodazole results in clearing of Lysotracker-positive structures from the perinuclear area in both wild-type (B versus A) and lrrkGS-expressing cells (D versus C), indicating that the localization of lysosomes to the perinuclear region under normal conditions, and the increased transport of these structures to the perinuclear region upon lrrkGS expression, both require microtubules. Single copy dynein heavy-chain loss-of-function (Dhc64C6-10/+, E) does not significantly alter lysosome position on its own, but partially blocks the increased perinuclear localization and clustering of lysosomes seen with lrrkGS expression (F). (G) Quantification of Lysotracker-positive clusters in the indicated genotypes (A, C, E, F) reveals a significant reduction in clustering in lrrkGS-expressing follicle cells with single copy dynein heavy-chain loss-of-function. (H) Those clusters that are present in lrrkGS-expressing follicle cells with single copy dynein heavy-chain loss-of-function are significantly smaller than those in lrrkGS-expressing cells alone. The dashed gray line in (H) indicates the size threshold above which a particle is categorized as a cluster. Follicle cell nuclei are outlined with dashed gray lines in all Lysotracker images. Scale bars represent 5 μm.
DISCUSSION
Here, we use Drosophila as an in vivo system to dissect the cell biological functions of lrrk by exploring the previously uncharacterized role of lrrk in oogenesis. Our studies point to a crucial role for lrrk in regulating rab7-dependent lysosomal positioning, and identify alterations in rab7-dependent lysosomal positioning and lysosome function as potential pathogenic mechanisms in LRRK2-mediated PD. Interestingly, lysosome dysfunction has been demonstrated in PD patient brains (64,65), and two strong genetic risk factors for PD, mutations in β-glucocerebrosidase (66,67) and ATP13A2 (68), are associated with lysosomal dysfunction, suggesting an important role for lysosome dysfunction in the pathogenesis of PD.
Mammalian LRRK2 has previously been found to localize to Rab5-positive early endosomes and to physically interact with Rab5, with knockdown of LRRK2 causing impairments in Rab5-dependent synaptic vesicle endocytosis (28). However, an interaction between LRRK2 and Rab7 had not been previously reported. We likewise detect a physical interaction between Drosophila Lrrk and Rab5, however we do not see localization of Lrrk to early endosomes, nor do we see evidence of early endosomal defects with genetic manipulation of lrrk. Rather, we have found that Lrrk binds to Rab7, and localizes predominately to Rab7-positive late endosomes and lysosomes. Furthermore, we see alterations in the morphology and distribution of late endosomal and lysosomal compartments associated with either loss of lrrk function or with expression of lrrkGS. While we cannot rule out the possibility that Lrrk also has early endosomal functions, our data clearly point to Rab7-positive late endosomes as a major site of Lrrk function in Drosophila. It is important to point out that lysosome dysfunction has been reported with genetic manipulation of LRRK2 in mammalian systems, as overexpression of mutant forms of LRRK2 in cultured neurons results in neurite morphology defects associated with the formation of lysosome inclusions (32), and LRRK2 knockout mice accumulate lipofuscin in the kidney, suggestive of lysosome dysfunction (33). Thus, the role of lrrk in regulating lysosomal processes is likely conserved in mammals. Further work is required to clarify whether LRRK2 has distinct functions at early endosomes and lysosomes.
What is the relationship between Lrrk and Rab7? We report that lrrk loss-of-function enhances while lrrk overexpression suppresses the increased perinuclear positioning of lysosomes due to rab7CA expression, consistent with the role for lrrk as a negative regulator of rab7 (Fig. 9A). However, we see no significant effect on lysosome position with either lrrk loss- or gain-of-function alone. Thus, uncovering the role of lrrk as a negative regulator of rab7 activity in this assay requires a sensitized genetic background in which rab7 activity is augmented. This suggests that the significance of lrrk's role as a negative regulator of rab7 may vary with rab7 activity, such as during periods of high flux through late endosomes. Interestingly, we have found that wild-type Lrrk preferentially binds to dominant-negative Rab7. While more work is required to dissect the molecular basis of the Lrrk/Rab7 interaction, it is tempting to speculate based on these data that Lrrk might exert its negative regulation of Rab7 by preferentially stabilizing its inactive GDP-bound form.
Figure 9.
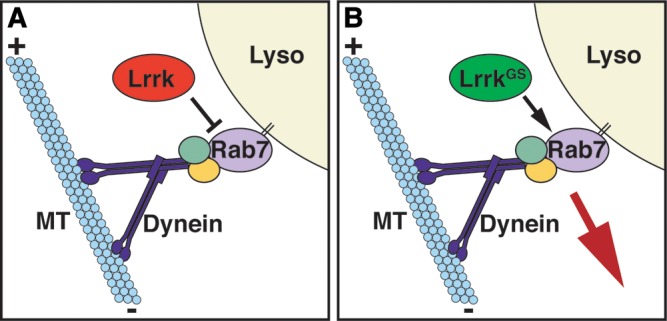
Schematic depicting the effects of wild-type lrrk and lrrkGS on rab7-dependent lysosome positioning. (A) Via multiple binding partners, Rab7 recruits the dynein–dynactin complex to lysosome membranes, thereby promoting their microtubule minus-end-directed motility. The net result is increased localization and clustering of lysosomes in the perinuclear region. Wild-type lrrk negatively regulates this function of rab7. (B) In contrast, lrrkGS actually promotes rab7-dependent perinuclear lysosome positioning in a microtubule- and dynein-dependent manner, resulting in increased localization and clustering of lysosomes in the perinuclear region.
Activated Rab7 induces perinuclear lysosome clustering by recruiting its binding partner Rab7-interacting lysosomal protein (RILP) to the vesicle membrane (57), which in turn recruits the dynein motor via an interaction between RILP and the dynactin/p150glued subunit (56), with the help of an additional Rab7-binding partner oxysterol binding protein-related protein 1 (58). The net result is increased localization of the dynein motor to lysosome membranes, and therefore increased transport of lysosomes toward microtubule minus ends in the perinuclear region. We have shown that the perinuclear clustering of lysosomes mediated by LrrkGS also requires dynein- and microtubule-based transport, suggesting a similar mechanism. Moreover, LrrkGS-induced lysosome clustering is inhibited by expression of dominant-negative Rab7, suggesting that LrrkGS may in fact induce lysosome clustering by acting through Rab7.
In addition to its effects on lysosome positioning, Rab7 plays a crucial role in regulating the maturation of late endosomes to lysosomes. This point is underscored by the fact that the dispersed lysosomes in the context of dominant-negative Rab7 expression are inaccessible to endocytosed substrates (55). Among other Rab7 functions that are crucial for late endosome to lysosome maturation, Rab7 mediates membrane tethering and fusion events between late endosomes and lysosomes via its interaction with the homotypic fusion and vacuole protein sorting complex (69). Our experiments suggest that in addition to promoting microtubule-based perinuclear transport, LrrkGS also promotes lysosome membrane tethering, as lysosomes remain clustered in the context of LrrkGS expression even when microtubules are destabilized. These data suggest that LrrkGS may also promote other Rab7 functions in addition to perinuclear positioning. Whether endogenous Lrrk likewise acts as a general regulator of Rab7 activity, or rather plays a specific role in regulating Rab7-dependent lysosome positioning, remains to be seen. However, it is interesting to note that lrrk null mutants accumulate enlarged Rab7-positive late endosomes that aberrantly accumulate an endocytic tracer, suggesting that lrrk loss-of-function may also disrupt aspects of Rab7-dependent late endosome to lysosome maturation.
lrrk and its mouse homolog play important roles in neuronal process morphology (32,39,70), and the Caenorhabditis elegans lrrk mutant causes defects in axonal-dendritic polarity (14). In Drosophila, lrrk mutants show defects in the NMJ, which are due in part to defects in microtubule dynamics (39). Moreover, expression of human LRRK2GS in Drosophila dopaminergic neurons causes dendrite degeneration associated with fragmentation of the microtubule network and mislocalization of the microtubule-associated protein Tau (41). LRRK2 has been shown to bind to tubulin in vitro (18,19), and our data demonstrate that microtubules are required for the effects of lrrkGS on lysosome positioning. Taken together, our findings raise the intriguing possibility that lrrk/LRRK2 might regulate neurite morphology and/or polarity through effects on microtubule-based transport of lysosomes and/or other vesicular compartments in the endolysosomal pathway. Interestingly, Rab7 has well-characterized roles in vesicle trafficking in neurons and the regulation of neuronal process morphology. Knockdown of rab7 in mouse cortical neurons impairs neuronal migration and neurite morphology (71), and rab7-dependent vesicle trafficking has been shown to be required for the intracellular transport of neuritogenic growth factors and their receptors (72,73). Interestingly, dominant mutations in rab7 cause an inherited form of neuropathy (74), and expression of these dominant mutant forms of rab7 in cultured mammalian neurons impair neurite outgrowth (75). Thus, we hypothesize that LRRK2 and rab7 may cooperate in the maintenance of neuritic processes by linking effects on microtubule dynamics to the trafficking of endolysosomal structures.
A key goal in the search for novel therapies for PD is the development of compounds that target mutant LRRK2 alleles. Such an approach requires an understanding of the mechanisms by which the mutant protein exerts toxicity. A point mutation can reduce or abolish the endogenous functions of the protein (hypomorph/amporh), inhibit the endogenous functions of the remaining wild-type copy (dominant-negative) or exert gain-of-function effects that reflect either an increase in the normal functions of the protein (hypermorph) or novel functions not shared with the wild-type protein (neomorph). lrrkGS retains at least some endogenous functions of wild-type lrrk, as evidenced by the ability of lrrkGS to restore female fertility in lrrk null mutants. However, the GS mutation appears to cause at least partial loss of lrrk functions since lrrkGS, unlike lrrkWT, is unable to antagonize the activity of rab7 in regulating lysosome positioning. In fact, lrrkGS causes a phenotype consistent with rab7 activation, thus behaving precisely opposite to lrrkWT with respect to the genetic relationship with rab7 (Fig. 9A versus B). However, this is not a dominant-negative effect of lrrkGS, as lrrk loss-of-function on its own has no effect on lysosome distribution. Thus, we conclude that the GS mutation causes both partial loss-of-function and neomorphic effects with respect to rab7. Interestingly, this altered functional relationship is accompanied by altered binding to Rab7, in which LrrkWT, but not LrrkGS, binds preferentially to the inactive form of Rab7. These data suggest the possibility that this differential binding may at least partially explain the different genetic relationships that LrrkWT and LrrkGS have with Rab7 (i.e. LrrkWT negatively regulates Rab7, while LrrkGS promotes Rab7-dependent functions). One intriguing hypothesis that this result suggests is that LrrkWT may preferentially stabilize the inactive form of Rab7 by direct binding, but that LrrkGS may lack this ability. These results have important implications for the development of PD therapies targeting this allele.
MATERIALS AND METHODS
Drosophila genetics and stocks
The follicle cell-specific driver CY2-GAL4 was a gift from Celeste Berg, and UAS-Lamp1:GFP from Helmut Kramer. The lrrke03680 allele was obtained from the Harvard Drosophila stock center. UAS-Rab transgenic flies have been previously published (76), and were obtained from the Bloomington Drosophila Stock Center. Rab7CA is Rab7Q67L, and Rab7DN is Rab7T22N. All other lines, including the lrrk deficiency, Df(3R)BSC141, are from the Bloomington Drosophila Stock Center. For experiments involving transgenic flies, multiple lines were generated (Rainbow Transgenic Flies) and tested for each transgene. Drosophila strains were maintained in a 25°C humidified incubator. Two copies of the CY2-GAL4 driver were used for all follicle cell experiments aside from fertility rescue experiments.
Molecular biology
For UAS-lrrk, an NheI–KpnI fragment of the lrrk genomic DNA (BACR26M03) was cloned into pUASt. For the CaSpeR-lrrk genomic rescue, EcoRI–SalI fragments of the lrrk genomic DNA were cloned into the CaSpeR4 vector. For CaSpeR-lrrk-9myc, nine copies of myc were fused in frame just upstream of the lrrk stop codon by polymerase chain reaction (PCR). All cloned PCR products were confirmed by sequencing. pMT-lrrk-9myc was generated by subcloning lrrk-9myc into the pMT vector, and pMT-lrrkGS-9myc by subcloning the Sac1 fragment from UAS-lrrkGS into pMT-lrrk-9myc. UAS-lrrkGS was generated by mutating G1914 to S via site-specific mutagenesis.
Lysotracker staining and tracer uptake assay
For Lysotracker staining, ovaries were dissected and incubated for 15 min in 100 nm Lysotracker red DND-99 (Molecular Probes), washed briefly, then mounted and imaged immediately, all in Schneider's Drosophila Medium (Gibco). For nocodazole experiments, freshly dissected egg chambers were incubated for 1 h in 50 mm nocodazole (Sigma) plus 0.2% dimethyl sulfamethoxazole (DMSO), or DMSO alone as a vehicle, in Schneider's Drosophila Medium prior to Lysotracker staining. For dextran uptake assays, freshly dissected egg chambers were incubated in 0.5 mm Texas Red-dextran (3000 MW, Invitrogen) in Schneider's Drosophila Medium (Gibco) for 15 min, washed and then incubated for 30 min at 25°C prior to fixation in 4% paraformaldehyde. All follicle cell images shown are from stage 12 egg chambers unless otherwise stated.
Quantification of lysosome clustering
Follicle cell lysosome clusters were defined from confocal images of Lysotracker-stained stage 12 egg chambers as any particle larger than 1.0 μm2 in cross-sectional area using the Analyze Particles function in ImageJ software (NIH), while individual lysosomes were defined as particles 0.2–0.9 μm2 in area. These parameters correlated with categorization based on qualitative visual analysis with greater than 99% concordance. The total cross-sectional area of Lysotracker-positive signal residing in a cluster was then divided by the total area residing in clusters plus individual lysosomes for all main body follicle cells visualized in a 63× field from a single egg chamber, typically encompassing 30–35 cells. This figure was then averaged over a total of at least six individual stage 12 egg chambers per genotype. For determination of average cluster size, the same procedure was followed, and the average cluster size as measured by the Analyze Particles function in ImageJ was determined for each egg chamber and then averaged over multiple egg chambers per genotype as above. The two-tailed Student's t-test was used to determine statistical significance. Error bars represent standard error of the mean.
Immunofluorescence and confocal microscopy
Freshly eclosed females were maintained on wet yeast paste for 24 h prior to ovary dissection, and individual stage 11–13 egg chambers were hand dissected following fixation. All tissues were fixed in either 3.7% formaldehyde or 4% paraformaldehyde in phosphate buffered saline (PBS). PBS + 0.1% Triton X-100 + 2% bovine serum albumin was used for blocking and antibody incubations. The following primary antibodies were used for immunocytochemistry: mouse anti-Myc (DSHB, 9E10, 1:10), rabbit anti-Rab7 (a generous gift from Yashodhan Chinchore and Patrick Dolph, 1:100) and guinea pig anti-Hrs (a gift from Hugo Bellen, 1:100). Images were obtained with a Zeiss LSM5 confocal microscope.
Female fertility tests
Single 0–3-day-old females were placed in a vial supplemented with dry yeast along with three sibling males and maintained at 25°C. After 4 days, the flies were removed and progeny were allowed to develop at 25°C for an additional 13 days, at which time the number of adult progeny per vial was counted. The two-tailed Student's t-test was used to determine statistical significance. Error bars represent standard error of the mean.
S2 cell culture and transfection
S2 cells were cultured in Schneider's Drosophila Medium (Gibco) + 10% fetal bovine serum (Invitrogen) + 1% penicillin/streptomycin (Invitrogen). For immunoprecipitations, cells were plated on 10 cm dishes at a density of 9 × 106. Transfections were performed 24 h later using the Qiagen Effectene kit according to the manufacturer's recommendations. pMT-lrrk-9myc was transfected along with pAC-Gal4 and pUASp-Rab:YFP constructs (obtained from Matthew Scott) as indicated. pMT-lrrk-9myc expression was induced by adding 0.5 mm copper sulfate 24 h after transfection, and cells were harvested 24 h later.
Lysate preparation, immunoprecipitation and western blotting
Cells were lysed in 800 ml of radioimmunoprecipitation assay buffer (Upstate) containing protease inhibitor cocktail (Roche), and immunoprecipitations performed using Invitrogen Dynabeads according to the manufacturer's instructions. A rabbit anti-GFP antibody (Invitrogen) was used for immunoprecipitation. Bound proteins were eluted at 72°C for 10 min in 1× sodium dodecyl sulfate sample buffer containing 5% 2-mercaptoethanol, and were resolved on a 6 or 10% polyacrylamide gel. Proteins were transferred to an Immobilon membrane (Millipore), and labeled with mouse anti-Myc (Millipore, 1:1000), mouse anti-GFP (Roche, 1:5000) or rabbit anti-actin (Sigma, 1:2000) and goat anti-mouse or anti-rabbit horseradish peroxidase. Blots were developed using SuperSignal West Pico Chemiluminescent Substrate (Pierce).
FUNDING
This work was supported by the National Institute of Health (NIH) NRSA predoctoral fellowship and the Medical Scientist Training Program (MSTP) to M.W.D., a scholarship from the China Scholarship Council to T.Z., as well as grants and funds from NIH (R01, K02), the Glenn Family Foundation, the Alfred P. Sloan Foundation, the Klingenstein Fund (Robert H. Ebert Clinical Scholar) and the McKnight Foundation of Neuroscience to M.G.
ACKNOWLEDGEMENTS
We are very grateful to H. Kramer, P. Dolph, C. Berg, H. Bellen and M. Scott for fly strains, antibodies and DNA constructs, M. Lone for experimental assistance and to E. Dell'Angelica and B.A. Hay for discussions.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Paisan-Ruiz C., Jain S., Evans E.W., Gilks W.P., Simon J., van der Brug M., Lopz de Munain A., Aparicio S., Gil A.M., Khan N., et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. doi:10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 2.Zimprich A., Biskup S., Leitner P., Lichtner P., Farrer M., Lincoln S., Kachergus J., Hulihan M., Uitti R.J., Calne D.B., et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. doi:10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 3.Satake W., Nakabayashi Y., Mizuta I., Hirota Y., Ito C., Kubo M., Kawaguchi T., Tsunoda T., Watanabe M., Takeda A., et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nat. Genet. 2009;41:1303–1307. doi: 10.1038/ng.485. doi:10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- 4.Simon-Sanchez J., Schulte C., Bras J.M., Sharma M., Gibbs J.R., Berg D., Paisan-Ruiz C., Lichtner P., Scholz S.W., Hernandez D.G., et al. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat. Genet. 2009;41:1308–1312. doi: 10.1038/ng.487. doi:10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lesage S., Ibanez P., Lohmann E., Pollak P., Tison F., Tazir M., Leutenegger A.L., Guimaraes J., Bonnet A.M., Agid Y., et al. G2019S LRRK2 mutation in French and North African families with Parkinson's disease. Ann. Neurol. 2005;58:784–787. doi: 10.1002/ana.20636. doi:10.1002/ana.20636. [DOI] [PubMed] [Google Scholar]
- 6.Hardy J. Genetic analysis of pathways to Parkinson disease. Neuron. 2010;68:201–206. doi: 10.1016/j.neuron.2010.10.014. doi:10.1016/j.neuron.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dawson T.M., Ko H.S., Dawson V.L. Genetic animal models of Parkinson's disease. Neuron. 2010;66:646–661. doi: 10.1016/j.neuron.2010.04.034. doi:10.1016/j.neuron.2010.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bosgraaf L., Van Haastert P.J. Roc, a Ras/GTPase domain in complex proteins. Biochim. Biophys. Acta. 2003;1643:5–10. doi: 10.1016/j.bbamcr.2003.08.008. doi:10.1016/j.bbamcr.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 9.Cookson M.R. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson's disease. Nat. Rev. Neurosci. 2010;11:791–797. doi: 10.1038/nrn2935. doi:10.1038/nrn2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith W.W., Pei Z., Jiang H., Moore D.J., Liang Y., West A.B., Dawson V.L., Dawson T.M., Ross C.A. Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc. Natl Acad. Sci. USA. 2005;102:18676–18681. doi: 10.1073/pnas.0508052102. doi:10.1073/pnas.0508052102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kanao T., Venderova K., Park D.S., Unterman T., Lu B., Imai Y. Activation of FoxO by LRRK2 induces expression of proapoptotic proteins and alters survival of postmitotic dopaminergic neuron in Drosophila. Hum. Mol. Genet. 2010;19:3747–3758. doi: 10.1093/hmg/ddq289. doi:10.1093/hmg/ddq289. [DOI] [PubMed] [Google Scholar]
- 12.Imai Y., Gehrke S., Wang H.Q., Takahashi R., Hasegawa K., Oota E., Lu B. Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J. 2008;27:2432–2443. doi: 10.1038/emboj.2008.163. doi:10.1038/emboj.2008.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gehrke S., Imai Y., Sokol N., Lu B. Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature. 2010;466:637–641. doi: 10.1038/nature09191. doi:10.1038/nature09191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sakaguchi-Nakashima A., Meir J.Y., Jin Y., Matsumoto K., Hisamoto N. LRK-1, a C. elegans PARK8-related kinase, regulates axonal-dendritic polarity of SV proteins. Curr. Biol. 2007;17:592–598. doi: 10.1016/j.cub.2007.01.074. [DOI] [PubMed] [Google Scholar]
- 15.Ho C.C., Rideout H.J., Ribe E., Troy C.M., Dauer W.T. The Parkinson disease protein leucine-rich repeat kinase 2 transduces death signals via Fas-associated protein with death domain and caspase-8 in a cellular model of neurodegeneration. J. Neurosci. 2009;29:1011–1016. doi: 10.1523/JNEUROSCI.5175-08.2009. doi:10.1523/JNEUROSCI.5175-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaleel M., Nichols R.J., Deak M., Campbell D.G., Gillardon F., Knebel A., Alessi D.R. LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson's disease mutants affect kinase activity. Biochem. J. 2007;405:307–317. doi: 10.1042/BJ20070209. doi:10.1042/BJ20070209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parisiadou L., Xie C., Cho H.J., Lin X., Gu X.L., Long C.X., Lobbestael E., Baekelandt V., Taymans J.M., Sun L., et al. Phosphorylation of ezrin/radixin/moesin proteins by LRRK2 promotes the rearrangement of actin cytoskeleton in neuronal morphogenesis. J. Neurosci. 2009;29:13971–13980. doi: 10.1523/JNEUROSCI.3799-09.2009. doi:10.1523/JNEUROSCI.3799-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gandhi P.N., Wang X., Zhu X., Chen S.G., Wilson-Delfosse A.L. The Roc domain of leucine-rich repeat kinase 2 is sufficient for interaction with microtubules. J. Neurosci. Res. 2008;86:1711–1720. doi: 10.1002/jnr.21622. doi:10.1002/jnr.21622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gillardon F. Leucine-rich repeat kinase 2 phosphorylates brain tubulin-beta isoforms and modulates microtubule stability—a point of convergence in parkinsonian neurodegeneration? J. Neurochem. 2009;110:1514–1522. doi: 10.1111/j.1471-4159.2009.06235.x. doi:10.1111/j.1471-4159.2009.06235.x. [DOI] [PubMed] [Google Scholar]
- 20.Lin X., Parisiadou L., Gu X.L., Wang L., Shim H., Sun L., Xie C., Long C.X., Yang W.J., Ding J., et al. Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson's-disease-related mutant alpha-synuclein. Neuron. 2009;64:807–827. doi: 10.1016/j.neuron.2009.11.006. doi:10.1016/j.neuron.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.West A.B., Moore D.J., Biskup S., Bugayenko A., Smith W.W., Ross C.A., Dawson V.L., Dawson T.M. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl Acad. Sci. USA. 2005;102:16842–16847. doi: 10.1073/pnas.0507360102. doi:10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Biskup S., Moore D.J., Celsi F., Higashi S., West A.B., Andrabi S.A., Kurkinen K., Yu S.W., Savitt J.M., Waldvogel H.J., et al. Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann. Neurol. 2006;60:557–569. doi: 10.1002/ana.21019. doi:10.1002/ana.21019. [DOI] [PubMed] [Google Scholar]
- 23.Gloeckner C.J., Kinkl N., Schumacher A., Braun R.J., O'Neill E., Meitinger T., Kolch W., Prokisch H., Ueffing M. The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum. Mol. Genet. 2006;15:223–232. doi: 10.1093/hmg/ddi439. doi:10.1093/hmg/ddi439. [DOI] [PubMed] [Google Scholar]
- 24.Hatano T., Kubo S., Imai S., Maeda M., Ishikawa K., Mizuno Y., Hattori N. Leucine-rich repeat kinase 2 associates with lipid rafts. Hum. Mol. Genet. 2007;16:678–690. doi: 10.1093/hmg/ddm013. doi:10.1093/hmg/ddm013. [DOI] [PubMed] [Google Scholar]
- 25.Vitte J., Traver S., Maues De Paula A., Lesage S., Rovelli G., Corti O., Duyckaerts C., Brice A. Leucine-rich repeat kinase 2 is associated with the endoplasmic reticulum in dopaminergic neurons and accumulates in the core of Lewy bodies in Parkinson disease. J. Neuropathol. Exp. Neurol. 2010;69:959–972. doi: 10.1097/NEN.0b013e3181efc01c. doi:10.1097/NEN.0b013e3181efc01c. [DOI] [PubMed] [Google Scholar]
- 26.Alegre-Abarrategui J., Christian H., Lufino M.M., Mutihac R., Venda L.L., Ansorge O., Wade-Martins R. LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum. Mol. Genet. 2009;18:4022–4034. doi: 10.1093/hmg/ddp346. doi:10.1093/hmg/ddp346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berger Z., Smith K.A., Lavoie M.J. Membrane localization of LRRK2 is associated with increased formation of the highly active LRRK2 dimer and changes in its phosphorylation. Biochemistry. 2010;49:5511–5523. doi: 10.1021/bi100157u. doi:10.1021/bi100157u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shin N., Jeong H., Kwon J., Heo H.Y., Kwon J.J., Yun H.J., Kim C.H., Han B.S., Tong Y., Shen J., et al. LRRK2 regulates synaptic vesicle endocytosis. Exp. Cell Res. 2008;314:2055–2065. doi: 10.1016/j.yexcr.2008.02.015. doi:10.1016/j.yexcr.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 29.Higashi S., Moore D.J., Yamamoto R., Minegishi M., Sato K., Togo T., Katsuse O., Uchikado H., Furukawa Y., Hino H., et al. Abnormal localization of leucine-rich repeat kinase 2 to the endosomal-lysosomal compartment in lewy body disease. J. Neuropathol. Exp. Neurol. 2009;68:994–1005. doi: 10.1097/NEN.0b013e3181b44ed8. doi:10.1097/NEN.0b013e3181b44ed8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Piccoli G., Condliffe S.B., Bauer M., Giesert F., Boldt K., De Astis S., Meixner A., Sarioglu H., Vogt-Weisenhorn D.M., Wurst W., et al. LRRK2 controls synaptic vesicle storage and mobilization within the recycling pool. J. Neurosci. 2011;31:2225–2237. doi: 10.1523/JNEUROSCI.3730-10.2011. doi:10.1523/JNEUROSCI.3730-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xiong Y., Coombes C.E., Kilaru A., Li X., Gitler A.D., Bowers W.J., Dawson V.L., Dawson T.M., Moore D.J. GTPase activity plays a key role in the pathobiology of LRRK2. PLoS Genet. 2010;6:e1000902. doi: 10.1371/journal.pgen.1000902. doi:10.1371/journal.pgen.1000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.MacLeod D., Dowman J., Hammond R., Leete T., Inoue K., Abeliovich A. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron. 2006;52:587–593. doi: 10.1016/j.neuron.2006.10.008. doi:10.1016/j.neuron.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 33.Tong Y., Yamaguchi H., Giaime E., Boyle S., Kopan R., Kelleher R.J., 3rd, Shen J. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proc. Natl Acad. Sci. USA. 2010;107:9879–9884. doi: 10.1073/pnas.1004676107. doi:10.1073/pnas.1004676107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lessing D., Bonini N.M. Maintaining the brain: insight into human neurodegeneration from Drosophila melanogaster mutants. Nat. Rev. Genet. 2009;10:359–370. doi: 10.1038/nrg2563. doi:10.1038/nrg2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dodson M.W., Guo M. Pink1, Parkin, DJ-1 and mitochondrial dysfunction in Parkinson's disease. Curr. Opin. Neurobiol. 2007;17:331–337. doi: 10.1016/j.conb.2007.04.010. doi:10.1016/j.conb.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 36.Guo M. What have we learned from Drosophila models of Parkinson's disease. Prog. Brain Res. 2010;184:3–16. doi: 10.1016/S0079-6123(10)84001-4. [DOI] [PubMed] [Google Scholar]
- 37.Lee S.B., Kim W., Lee S., Chung J. Loss of LRRK2/PARK8 induces degeneration of dopaminergic neurons in Drosophila. Biochem. Biophys. Res. Commun. 2007;358:534–539. doi: 10.1016/j.bbrc.2007.04.156. doi:10.1016/j.bbrc.2007.04.156. [DOI] [PubMed] [Google Scholar]
- 38.Wang D., Tang B., Zhao G., Pan Q., Xia K., Bodmer R., Zhang Z. Dispensable role of Drosophila ortholog of LRRK2 kinase activity in survival of dopaminergic neurons. Mol. Neurodegener. 2008;3:3. doi: 10.1186/1750-1326-3-3. doi:10.1186/1750-1326-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee S., Liu H.P., Lin W.Y., Guo H., Lu B. LRRK2 kinase regulates synaptic morphology through distinct substrates at the presynaptic and postsynaptic compartments of the Drosophila neuromuscular junction. J. Neurosci. 2010;30:16959–16969. doi: 10.1523/JNEUROSCI.1807-10.2010. doi:10.1523/JNEUROSCI.1807-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu Z., Wang X., Yu Y., Li X., Wang T., Jiang H., Ren Q., Jiao Y., Sawa A., Moran T., et al. A Drosophila model for LRRK2-linked parkinsonism. Proc. Natl Acad. Sci. USA. 2008;105:2693–2698. doi: 10.1073/pnas.0708452105. doi:10.1073/pnas.0708452105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin C.H., Tsai P.I., Wu R.M., Chien C.T. LRRK2 G2019S mutation induces dendrite degeneration through mislocalization and phosphorylation of tau by recruiting autoactivated GSK3ss. J. Neurosci. 2010;30:13138–13149. doi: 10.1523/JNEUROSCI.1737-10.2010. doi:10.1523/JNEUROSCI.1737-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spradling A.C. In: The Development of Drosophila Melanogaster. Arias M.B.a.A.M., editor. I. Plainview, NY: Cold Spring Harbor Laboratory Press; 1993. pp. 1–70. [Google Scholar]
- 43.Chavrier P., Parton R.G., Hauri H.P., Simons K., Zerial M. Localization of low molecular weight GTP binding proteins to exocytic and endocytic compartments. Cell. 1990;62:317–329. doi: 10.1016/0092-8674(90)90369-p. doi:10.1016/0092-8674(90)90369-P. [DOI] [PubMed] [Google Scholar]
- 44.Lombardi D., Soldati T., Riederer M.A., Goda Y., Zerial M., Pfeffer S.R. Rab9 functions in transport between late endosomes and the trans Golgi network. EMBO J. 1993;12:677–682. doi: 10.1002/j.1460-2075.1993.tb05701.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rohrer J., Schweizer A., Russell D., Kornfeld S. The targeting of Lamp1 to lysosomes is dependent on the spacing of its cytoplasmic tail tyrosine sorting motif relative to the membrane. J. Cell Biol. 1996;132:565–576. doi: 10.1083/jcb.132.4.565. doi:10.1083/jcb.132.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pulipparacharuvil S., Akbar M.A., Ray S., Sevrioukov E.A., Haberman A.S., Rohrer J., Kramer H. Drosophila Vps16A is required for trafficking to lysosomes and biogenesis of pigment granules. J. Cell Sci. 2005;118:3663–3673. doi: 10.1242/jcs.02502. doi:10.1242/jcs.02502. [DOI] [PubMed] [Google Scholar]
- 47.Wucherpfennig T., Wilsch-Brauninger M., Gonzalez-Gaitan M. Role of Drosophila Rab5 during endosomal trafficking at the synapse and evoked neurotransmitter release. J. Cell Biol. 2003;161:609–624. doi: 10.1083/jcb.200211087. doi:10.1083/jcb.200211087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Komada M., Masaki R., Yamamoto A., Kitamura N. Hrs, a tyrosine kinase substrate with a conserved double zinc finger domain, is localized to the cytoplasmic surface of early endosomes. J. Biol. Chem. 1997;272:20538–20544. doi: 10.1074/jbc.272.33.20538. doi:10.1074/jbc.272.33.20538. [DOI] [PubMed] [Google Scholar]
- 49.Urbe S., Mills I.G., Stenmark H., Kitamura N., Clague M.J. Endosomal localization and receptor dynamics determine tyrosine phosphorylation of hepatocyte growth factor-regulated tyrosine kinase substrate. Mol. Cell Biol. 2000;20:7685–7692. doi: 10.1128/mcb.20.20.7685-7692.2000. doi:10.1128/MCB.20.20.7685-7692.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bache K.G., Brech A., Mehlum A., Stenmark H. Hrs regulates multivesicular body formation via ESCRT recruitment to endosomes. J. Cell Biol. 2003;162:435–442. doi: 10.1083/jcb.200302131. doi:10.1083/jcb.200302131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Urbe S., Huber L.A., Zerial M., Tooze S.A., Parton R.G. Rab11, a small GTPase associated with both constitutive and regulated secretory pathways in PC12 cells. FEBS Lett. 1993;334:175–182. doi: 10.1016/0014-5793(93)81707-7. doi:10.1016/0014-5793(93)81707-7. [DOI] [PubMed] [Google Scholar]
- 52.Ullrich O., Reinsch S., Urbe S., Zerial M., Parton R.G. Rab11 regulates recycling through the pericentriolar recycling endosome. J. Cell Biol. 1996;135:913–924. doi: 10.1083/jcb.135.4.913. doi:10.1083/jcb.135.4.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pilling A.D., Horiuchi D., Lively C.M., Saxton W.M. Kinesin-1 and Dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol. Biol. Cell. 2006;17:2057–2068. doi: 10.1091/mbc.E05-06-0526. doi:10.1091/mbc.E05-06-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kondylis V., Spoorendonk K.M., Rabouille C. dGRASP localization and function in the early exocytic pathway in Drosophila S2 cells. Mol. Biol. Cell. 2005;16:4061–4072. doi: 10.1091/mbc.E04-10-0938. doi:10.1091/mbc.E04-10-0938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bucci C., Thomsen P., Nicoziani P., McCarthy J., van Deurs B. Rab7: a key to lysosome biogenesis. Mol. Biol. Cell. 2000;11:467–480. doi: 10.1091/mbc.11.2.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jordens I., Fernandez-Borja M., Marsman M., Dusseljee S., Janssen L., Calafat J., Janssen H., Wubbolts R., Neefjes J. The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors. Curr. Biol. 2001;11:1680–1685. doi: 10.1016/s0960-9822(01)00531-0. doi:10.1016/S0960-9822(01)00531-0. [DOI] [PubMed] [Google Scholar]
- 57.Cantalupo G., Alifano P., Roberti V., Bruni C.B., Bucci C. Rab-interacting lysosomal protein (RILP): the Rab7 effector required for transport to lysosomes. EMBO J. 2001;20:683–693. doi: 10.1093/emboj/20.4.683. doi:10.1093/emboj/20.4.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Johansson M., Rocha N., Zwart W., Jordens I., Janssen L., Kuijl C., Olkkonen V.M., Neefjes J. Activation of endosomal dynein motors by stepwise assembly of Rab7-RILP-p150Glued, ORP1L, and the receptor betalll spectrin. J. Cell Biol. 2007;176:459–471. doi: 10.1083/jcb.200606077. doi:10.1083/jcb.200606077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Korolchuk V.I., Saiki S., Lichtenberg M., Siddiqi F.H., Roberts E.A., Imarisio S., Jahreiss L., Sarkar S., Futter M., Menzies F.M., et al. Lysosomal positioning coordinates cellular nutrient responses. Nat. Cell Biol. 2011;13:453–460. doi: 10.1038/ncb2204. doi:10.1038/ncb2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Taylor J.P., Mata I.F., Farrer M.J. LRRK2: a common pathway for parkinsonism, pathogenesis and prevention? Trends Mol. Med. 2006;12:76–82. doi: 10.1016/j.molmed.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 61.Bonifati V. LRRK2 low-penetrance mutations (Gly2019Ser) and risk alleles (Gly2385Arg)-linking familial and sporadic Parkinson's disease. Neurochem. Res. 2007;32:1700–1708. doi: 10.1007/s11064-007-9324-y. doi:10.1007/s11064-007-9324-y. [DOI] [PubMed] [Google Scholar]
- 62.Greggio E., Jain S., Kingsbury A., Bandopadhyay R., Lewis P., Kaganovich A., van der Brug M.P., Beilina A., Blackinton J., Thomas K.J., et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol. Dis. 2006;23:329–341. doi: 10.1016/j.nbd.2006.04.001. doi:10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 63.West A.B., Moore D.J., Choi C., Andrabi S.A., Li X., Dikeman D., Biskup S., Zhang Z., Lim K.L., Dawson V.L., et al. Parkinson's disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum. Mol. Genet. 2007;16:223–232. doi: 10.1093/hmg/ddl471. doi:10.1093/hmg/ddl471. [DOI] [PubMed] [Google Scholar]
- 64.Chu Y., Dodiya H., Aebischer P., Olanow C.W., Kordower J.H. Alterations in lysosomal and proteasomal markers in Parkinson's disease: relationship to alpha-synuclein inclusions. Neurobiol. Dis. 2009;35:385–398. doi: 10.1016/j.nbd.2009.05.023. doi:10.1016/j.nbd.2009.05.023. [DOI] [PubMed] [Google Scholar]
- 65.Dehay B., Bove J., Rodriguez-Muela N., Perier C., Recasens A., Boya P., Vila M. Pathogenic lysosomal depletion in Parkinson's disease. J. Neurosci. 2010;30:12535–12544. doi: 10.1523/JNEUROSCI.1920-10.2010. doi:10.1523/JNEUROSCI.1920-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Aharon-Peretz J., Rosenbaum H., Gershoni-Baruch R. Mutations in the glucocerebrosidase gene and Parkinson's disease in Ashkenazi Jews. N. Engl. J. Med. 2004;351:1972–1977. doi: 10.1056/NEJMoa033277. doi:10.1056/NEJMoa033277. [DOI] [PubMed] [Google Scholar]
- 67.Gan-Or Z., Giladi N., Rozovski U., Shifrin C., Rosner S., Gurevich T., Bar-Shira A., Orr-Urtreger A. Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology. 2008;70:2277–2283. doi: 10.1212/01.wnl.0000304039.11891.29. doi:10.1212/01.wnl.0000304039.11891.29. [DOI] [PubMed] [Google Scholar]
- 68.Ramirez A., Heimbach A., Grundemann J., Stiller B., Hampshire D., Cid L.P., Goebel I., Mubaidin A.F., Wriekat A.L., Roeper J., et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 2006;38:1184–1191. doi: 10.1038/ng1884. doi:10.1038/ng1884. [DOI] [PubMed] [Google Scholar]
- 69.Wurmser A.E., Sato T.K., Emr S.D. New component of the vacuolar class C-Vps complex couples nucleotide exchange on the Ypt7 GTPase to SNARE-dependent docking and fusion. J. Cell Biol. 2000;151:551–562. doi: 10.1083/jcb.151.3.551. doi:10.1083/jcb.151.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee B.D., Shin J.H., VanKampen J., Petrucelli L., West A.B., Ko H.S., Lee Y.I., Maguire-Zeiss K.A., Bowers W.J., Federoff H.J., et al. Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson's disease. Nat. Med. 2010;16:998–1000. doi: 10.1038/nm.2199. doi:10.1038/nm.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kawauchi T., Sekine K., Shikanai M., Chihama K., Tomita K., Kubo K., Nakajima K., Nabeshima Y., Hoshino M. Rab GTPases-dependent endocytic pathways regulate neuronal migration and maturation through N-cadherin trafficking. Neuron. 2010;67:588–602. doi: 10.1016/j.neuron.2010.07.007. doi:10.1016/j.neuron.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 72.Saxena S., Bucci C., Weis J., Kruttgen A. The small GTPase Rab7 controls the endosomal trafficking and neuritogenic signaling of the nerve growth factor receptor TrkA. J. Neurosci. 2005;25:10930–10940. doi: 10.1523/JNEUROSCI.2029-05.2005. doi:10.1523/JNEUROSCI.2029-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Deinhardt K., Salinas S., Verastegui C., Watson R., Worth D., Hanrahan S., Bucci C., Schiavo G. Rab5 and Rab7 control endocytic sorting along the axonal retrograde transport pathway. Neuron. 2006;52:293–305. doi: 10.1016/j.neuron.2006.08.018. doi:10.1016/j.neuron.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 74.Verhoeven K., De Jonghe P., Coen K., Verpoorten N., Auer-Grumbach M., Kwon J.M., FitzPatrick D., Schmedding E., De Vriendt E., Jacobs A., et al. Mutations in the small GTP-ase late endosomal protein RAB7 cause Charcot-Marie-Tooth type 2B neuropathy. Am. J. Hum. Genet. 2003;72:722–727. doi: 10.1086/367847. doi:10.1086/367847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cogli L., Progida C., Lecci R., Bramato R., Kruttgen A., Bucci C. CMT2B-associated Rab7 mutants inhibit neurite outgrowth. Acta Neuropathol. 2010;120:491–501. doi: 10.1007/s00401-010-0696-8. doi:10.1007/s00401-010-0696-8. [DOI] [PubMed] [Google Scholar]
- 76.Zhang J., Schulze K.L., Hiesinger P.R., Suyama K., Wang S., Fish M., Acar M., Hoskins R.A., Bellen H.J., Scott M.P. Thirty-one flavors of Drosophila rab proteins. Genetics. 2007;176:1307–1322. doi: 10.1534/genetics.106.066761. doi:10.1534/genetics.106.066761. [DOI] [PMC free article] [PubMed] [Google Scholar]