

Abstract
Alzheimer's disease (AD) is the most common neurodegenerative disease and the leading cause of dementia in the elderly. Accumulating evidence supports soluble amyloid-β (Aβ) oligomers as the leading candidate for the causative agent in AD and synapses as the primary site of Aβ oligomer action. However, the molecular and cellular mechanisms by which Aβ oligomers cause synaptic dysfunction and cognitive impairments remain poorly understood. Using primary cultures of rat hippocampal neurons as a model system, we show that the partitioning defective-1 (PAR-1)/microtubule affinity-regulating kinase (MARK) family kinases act as critical mediators of Aβ toxicity on synapses and dendritic spines. Overexpression of MARK4 led to tau hyperphosphorylation, reduced expression of synaptic markers, and loss of dendritic spines and synapses, phenotypes also observed after Aβ treatment. Importantly, expression of a non-phosphorylatable form of tau with the PAR-1/MARK site mutated blocked the synaptic toxicity induced by MARK4 overexpression or Aβ treatment. To probe the involvement of endogenous MARK kinases in mediating the synaptic toxicity of Aβ, we employed a peptide inhibitor capable of effectively and specifically inhibiting the activities of all PAR-1/MARK family members. This inhibitor abrogated the toxic effects of Aβ oligomers on dendritic spines and synapses as assayed at the morphological and electrophysiological levels. Our results reveal a critical role for PAR-1/MARK kinases in AD pathogenesis and suggest PAR-1/MARK inhibitors as potential therapeutics for AD and possibly other tauopathies where aberrant tau hyperphosphorylation is involved.
INTRODUCTION
Despite the identification of amyloid plaques (AP) and neurofibrillary tangles (NFT) as pathological hallmarks of Alzheimer's disease (AD), the memory deficits in AD patients do not correlate well with AP or NFT burden; instead, loss of synaptic markers is a better predictor of clinical symptoms and disease progression, lending support to the notion that AD is a disease of synaptic failure (1). Synapses and dendritic spines are dynamic structures whose plasticity is thought to underlie learning and memory and contribute to AD pathogenesis (2). Accumulating evidence supports that amyloid-β (Aβ) is the primary agent causative of synaptic and spine pathology in AD (3–5). For example, rare genetic mutations cause familial AD by altering the production or metabolism of Aβ (6,7), the soluble pool of which correlates with disease progression and severity (5), and depletion of soluble Aβ in mouse AD models rescues the cognitive deficits (8). Moreover, synthetic Aβ oligomers or those purified from cultured cells or brain samples can induce neuritic degeneration, neurotransmission defects and spine loss (9–12). The detailed cellular and biochemical mechanisms involved, however, remain poorly defined.
Recent studies have supported tau as a major mediator of Aβ toxicity. In mouse models, intracranial injection of Aβ or crossing an APP transgene into tau transgenic animals promoted NFT pathology (13–15), and antibody-based removal of Aβ reduced hyperphosphorylated tau and rescued behavioral and pathological defects in an APP/Psn/tau 3xTg AD mouse model (16). Moreover, removal of tau relieved Aβ-induced neurotoxicity in culture (17) and prevented Aβ-induced behavioral deficits in an h-APP Tg mouse model (18). Together, these studies support the critical involvement of tau in mediating the toxicity of Aβ in AD pathogenesis.
It is not clear how tau abnormality arises in AD. Current efforts have focused on the role of tau hyperphosphorylation (19,20). A number of kinases and phosphatases are shown to regulate tau phosphorylation (21), but few of them have been examined for roles in linking amyloid with tau pathology. A strong case has been made for partitioning defective-1 (PAR-1)/microtubule affinity-regulating kinase (MARK). PAR-1 was originally identified for its role in regulating cell polarity (22). PAR-1 and its mammalian homolog MARK phosphorylate tau associate with NFT (23,24). In Drosophila models, PAR-1-mediated phosphorylation is required for conferring tau toxicity (25). Elevation of PAR-1/MARK-mediated tau phosphorylation (at the 12E8 sites) was observed in AD patients and mouse AD models (26,27). Activated Drosophila PAR-1 also directly phosphorylates the postsynaptic density protein 95 (PSD-95) homolog discs large (Dlg), impairing its postsynaptic localization (28), which might be mechanistically related to the synaptic loss of PSD-95 seen in early stages of AD in patients and mouse models (29,30).
The studies described above strongly implicate PAR-1/MARK kinases as critical players in AD. Consistent with this hypothesis, a recent genome-wide association study suggested a potential link of MARK4 to late onset AD (31,32), an observation requiring further validation, and PAR-1/MARK kinases are activated by APP or Aβ oligomers in Drosophila or mammalian neurons, respectively (33,34). To more rigorously test the pathogenic role of PAR-1/MARK kinases and to validate them as potential drug target, loss-of-function analysis in mammalian systems is critically needed. However, functional redundancy among at least four members of the mammalian PAR-1/MARK kinase family presents a considerable challenge for a genetic loss-of-function approach. In this study, we describe a pharmacological approach using a specific peptide inhibitor to probe the importance of PAR-1/MARK in mediating the synaptic and dendritic toxicity of Aβ in primary cultures of rat hippocampal neurons. Our results provide compelling evidence that PAR-1/MARK kinases are critically involved in mediating the synaptic toxicity of Aβ by promoting the aberrant phosphorylation of tau and PSD-95, and that inhibition of PAR-1/MARK kinases represents a viable therapeutic approach.
RESULTS
Overexpression of MARK4 induces defects in synapses and dendritic spines
We used cultured rat primary hippocampal neurons to test whether the PAR-1/MARK family kinases mediate AD-related neurotoxicity in mammals. In addition to being the relevant cell type for studying AD pathogenesis, these neurons offer excellent cellular resolution for studying synaptic and dendritic spine morphologies and for electrophysiological characterization. As expected, we found that overexpression of Drosophila PAR-1 or mammalian MARK4 (31) in hippocampal neurons resulted in hyperphosphorylation of tau at the phosphorylation sites recognized by the 12E8 antibody (Supplementary Material, Fig. S1), sites known to be phosphorylated by PAR-1/MARK (23,25). This was accompanied by a number of phenotypes including loss of dendritic spines (Fig. 1A–C and F), the delocalization of PSD-95 from synapses (Fig. 1A and F) and decreased expression of other synaptic markers such as GluR1, a subunit of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors (Fig. 1B and F), and the presynaptic protein Synapsin I (Fig. 1C and F). In addition, we also observed defects in mitochondria localization in PAR-1 or MARK4 overexpressing neurons (data not shown). The kinase-dead form of MARK4 (MARK4-KD) had no such effect, indicating that kinase activity is required for inducing the observed toxic effects. Both MARK4-WT and MARK4-KD exhibited relatively uniform distribution in the dendrites (Fig. 1A–C, green signals).
Figure 1.
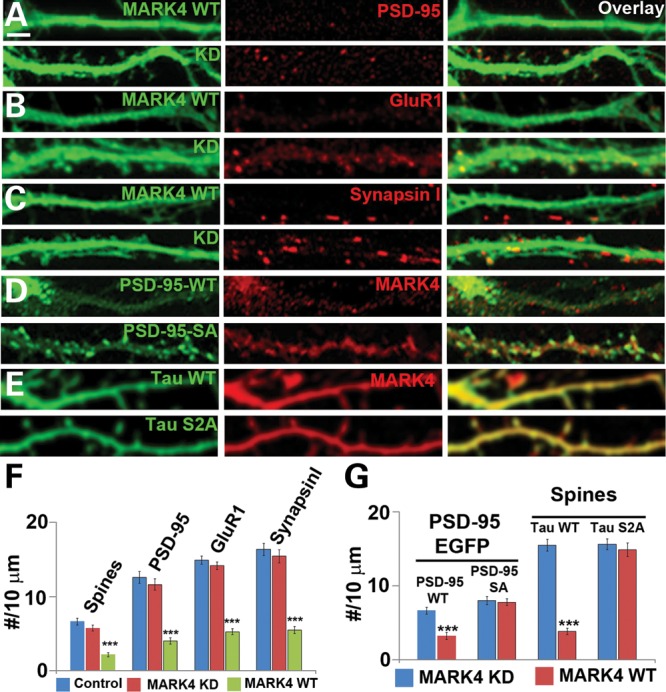
Overexpression of MARK4 causes synaptic and spine abnormality in rat hippocampal neurons. (A–C) Effects of MARK4-WT or MARK4-KD overexpression on postsynaptic PSD-95 clusters (A), AMPA receptor GluR1 clusters (B), presynaptic Synapsin I clusters (C) and spine numbers (A–C). Data quantification is shown in (F). (D) Differential response of PSD-95-S561A and PSD-95-WT synaptic localization to MARK4-WT overexpression. (E) Effect of h-tau-WT or h-tau-S2A co-expression on MARK4-WT overexpression-induced spines loss. (F) Quantification of spine number and PSD-95 (A), GluR1 (B) and Synapsin I (C) clusters in neurons transfected with MARK4-WT or MARK4-KD (***P< 0.001 in Student's t-test). (G) (Left) Quantification of PSD-95 clusters in neurons co-transfected with MARK4-WT or MARK4-KD and EGFP-tagged PSD-95-WT or PSD-95-S561A (***P< 0.001 in Student's t-test). (Right) Quantification of spine number in neurons co-transfected with MARK4-WT or MARK4-KD and h-tau-WT or h-tau-S2A (***P< 0.001 in Student's t-test). Scale bar, 5 μm.
Previous studies in Drosophila showed that PAR-1 could regulate the postsynaptic localization of Dlg through direct phosphorylation (28). The phosphorylation site is conserved in PSD-95, the mammalian homolog of Dlg. To test whether MARK4 may directly act on PSD-95 to regulate its synaptic localization, we generated a mutant form of PSD-95 in which the conserved PAR-1/MARK target site is mutated to Ala (PSD-95-SA). Both the WT and SA mutant forms of PSD-95 were fused with GFP to facilitate visualization. In the same expression vector, shRNAs targeting endogenous PSD-95 but not the exogenous PSD-95-GFP were also expressed (35). Thus, the observed effects were largely free of potential interference from endogenous PSD-95. Although PSD-95-WT-EGFP was delocalized from the dendritic spines by MARK4 and was present uniformly in the dendritic shaft, PSD-95-SA-EGFP effectively resisted the effect of MARK4 and maintained its punctate synaptic localization (Fig. 1D and G), consistent with MARK4 directly regulating PSD-95 localization in mammalian hippocampal neurons through phosphorylation.
Expression of a non-phosphorylatable form of tau blocks PAR-1/MARK toxicity
We were interested in identifying the key substrates through which PAR-1/MARK overexpression leads to synaptic and dendritic spine toxicity. Previous studies showed that PAR-1 overexpression in Drosophila photoreceptors caused neurotoxicity through phosphorylation of tau in the microtubule-binding domain, and that a mutant form of tau with the PAR-1 site mutated (tauS2A) not only was itself non-toxic but also could block PAR-1 overexpression-induced neurotoxicity (25). Co-transfection of a similar phosphorylation-mutant form of tau (h-tau-SA) blocked the toxic effects of MARK4 in rat hippocampal neurons, including the spine loss, whereas wild-type human tau (h-tau-WT) failed to do so (Fig. 1E and G). This is consistent with tau being a major mediator of MARK4 toxicity on synapses and dendritic spines.
Expression of non-phosphorylatable form of tau blocks Aβ toxicity
We further tested whether phosphorylation of tau by MARK might mediate the toxicity of Aβ on neuronal synapses and dendritic spines. Although MARK activation and elevation of tau phosphorylation at PAR-1/MARK target sites occurred in hippocampal neurons after Aβ oligomer treatment (11,34), it is not clear whether these events were disease-causing or simply a compensatory, protective response. Indeed, overexpression of MARK2 was reported to be able to rescue tau-induced synapse and spine loss (36). Consistent with previous reports (11,34), treatment of rat hippocampal neurons with synthetic Aβ, prepared using a well-characterized procedure that enriches for Aβ oligomers (37), resulted in increased tau phosphorylation at the 12E8 sites (Fig. 2A), suggesting that Aβ treatment had activated MARK kinases. Increased phosphorylation of tau at a site recognized by the PHF-1 phospho-tau antibody was also observed (data not shown). Aβ treatment also caused losses of synaptic marker expression and dendritic spines as seen in the PAR-1/MARK overexpression condition (Fig. 2B–E and G). Strikingly, in neurons transfected with h-tau-S2A but not h-tau-WT, the toxic effect of Aβ in causing spine loss was ameliorated (Fig. 2F and G). The toxic effects of Aβ on the density of synaptic marker PSD-95 and GluR1 clusters were also rescued by h-tau-S2A (Fig. 2H). These results support the notion that phosphorylation of tau by PAR-1/MARK family kinases is a primary downstream event by which Aβ exerts its toxicity on hippocampal synapses and dendritic spines.
Figure 2.
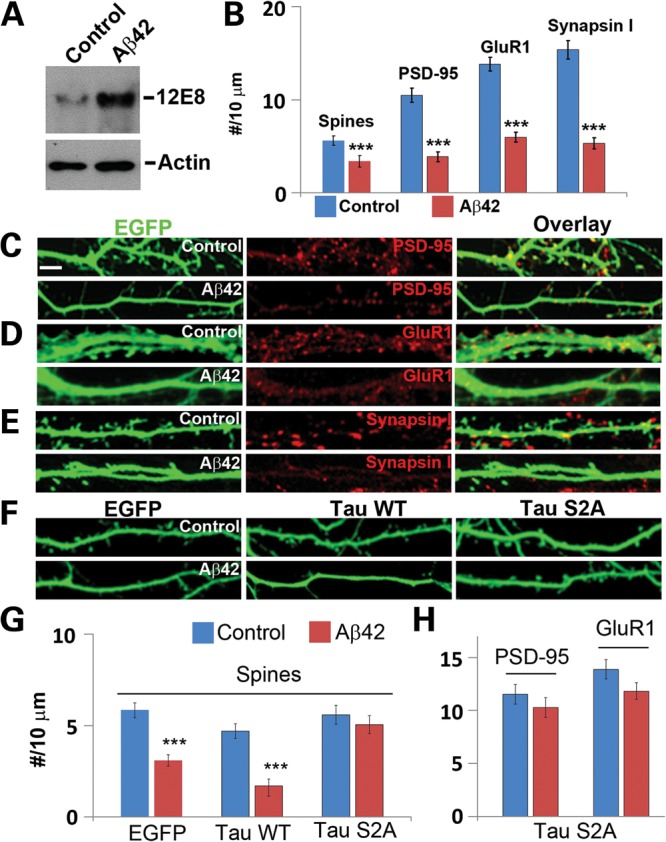
Synaptic and spine toxicity induced by Aβ oligomers and rescue of Aβ-induced spine loss by h-tau-S2A. (A) Aβ treatment increased endogenous tau phosphorylation at the 12E8 sites. Hippocampal neurons at 13 days in vitro were treated with Aβ oligomers for 12h and used for western blot analysis. Actin serves as loading control. (B–E) Effects of Aβ treatment on PSD-95 clusters (C), GluR1 clusters (D), Synapsin I clusters (E) and spine number (B) in EGFP-transfected neurons. Data quantification is shown in (B) (***P< 0.001 in Student's t-test). (F–H) Resistance of h-tau-S2A-transfected neurons to Aβ-induced loss of spines (F and G) and PSD-95 and GluR1 clusters (H). Scale bar, 10 μm.
A PAR-1/MARK inhibitor effectively blocked Aβ toxicity
Next we wished to assess the involvement of endogenous MARK kinases in mediating Aβ toxicity. To circumvent potential functional redundancy among at least four mammalian MARK family members, we used a peptide inhibitor (MKI) derived from the CagA protein of Helicobacter pylori, which can specifically bind with high affinity to the substrate-binding sites of all MARKs by mimicking the natural substrates of these kinases (38). We first confirmed that expression of an MKI-GFP fusion protein in rat hippocampal neurons effectively attenuated MARK4-mediated phosphorylation of both endogenous tau (Fig. 3A) and transfected human tau at the 12E8 sites (Fig. 3B). Phosphorylation of tau at the PHF-1 site was also reduced by MKI (Fig. 3A). It is possible that the PHF-1 site is also targeted by PAR-1/MARKs in vivo, or that the phosphorylation of tau at the 12E8 sites is a prerequisite for PHF-1 site phosphorylation, as the 12E8 sites were previously shown to be required for tau phosphorylation at other sites (25). Supporting the specificity of the MKI effect on PAR-1/MARK, under similar experimental conditions MKI-EGFP did not affect the phosphorylation of acetyl-CoA carboxylase (ACC) by AMP-activated kinase (AMPK) (Fig. 3C), which is closely related to PAR-1/MARK on the protein kinase phylogenetic tree (39). MARK4 overexpression-induced synaptic defects including spine loss (Fig. 3D and E) and reduction of PSD-95 and GluR1 puncta density were all effectively blocked by MKI-EGFP (Fig. 3E).
Figure 3.
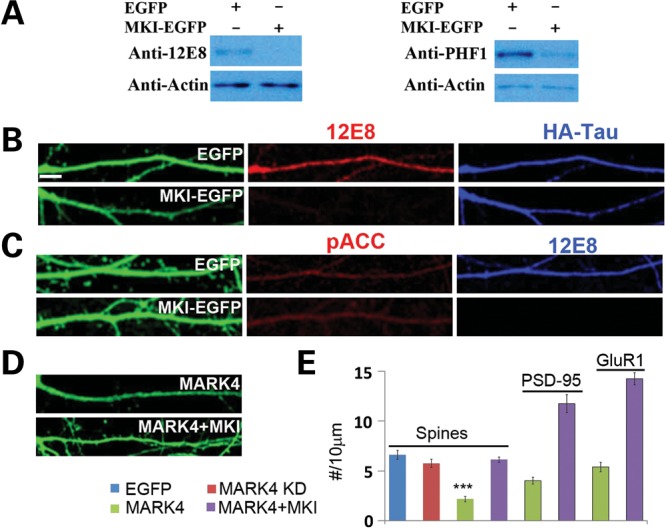
Inhibition of PAR-1/MARK-mediated tau phosphorylation and MARK4 overexpression-induced synaptic and spine toxicity by MKI. (A) MKI-EGFPinhibited phosphorylation of endogenous tau as assessed by western blot analysis using phosphor-specific antibodies 12E8 and PHF-1 (A). (B) MKI-EGFP inhibited phosphorylation of HA-tagged exogenous h-tau-WT at the 12E8 sites. Robust 12E8 staining (red) is seen in EGFP-transfected control neurons. Total tau is stained with anti-HA (blue). (C) No effect of MKI-EGFP on AMPK kinase activity as detected with the phospho-acetyl-CoA carboxylase (Ser79) (pACC) antibody. EGFP-transfected neurons serve as control. (D and E) MKI-EGFP blocked MARK4-overexpression-induced loss of spines (D and E) and PSD-95 and GluR1 clusters (E). Data quantification is shown in (E) (***P< 0.001 in Student's t-test). Scale bar, 10 μm.
To test the function of MARK kinases in mediating the toxicity of Aβ on synapses and dendritic spines, we next examine the effect of MKI in modulating the toxicity of Aβ. Remarkably, MKI-GFP expression effectively blocked the morphological defects caused by Aβ, such as the loss of dendritic spines (Fig. 4A–C), and the reduction in the density of PSD-95 (Fig. 4A and C) and GluR1 puncta (Fig. 4B and C).
Figure 4.

Rescue of Aβ-induced synaptic and dendritic spine defects by MKI at the morphological and electrophysiological levels. (A–C) Effects of MKI-EGFP on Aβ-induced loss of PSD-95 (A) and GluR1 (B) clusters and spine numbers. Neurons transfected with MKI-EGFP or EGFP were treated with Aβ oligomer or mock-treated with solvent as control. The cytoplasmic GFP signals allowed detection of dendritic spines, and PSD-95 or GluR1 clusters were detected by immunostaining. Quantification of data is shown in (C) (***P< 0.001 in Student's t-test). (D) Sample traces of synaptic activity (mEPSCs) recorded in neurons from four different conditions (control-EGFP, Aβ, MKI-EGFP + Aβ and MKI-EGFP alone) held at −70 mV. Summary graphs from four to six culture sets plotting normalized mEPSC frequency and mEPSC amplitude are shown beneath the traces. Scale bar, 5 μm.
We further examined the effect of MKI-EGFP at the functional level by performing electrophysiological analysis. Aβ treatment caused a reduction in the frequency but not in the amplitude of AMPAR-mediated miniature excitatory postsynaptic current (mEPSC) (Fig. 4D). The reduction in the mEPSC frequency suggested that Aβ is causing a reduction in synapse number, whereas the unchanged mEPSC amplitude indicated that the postsynaptic AMPA receptor composition and/or activity of the remaining synapses were probably not affected. The reduction of mEPSC frequency caused by Aβ was effectively rescued by MKI-EGFP (Fig. 4D). We note that neurons expressing MKI-EGFP alone also showed reduced mEPSC frequency but not amplitude (Fig. 4D), suggesting that the PAR-1/MARK family kinases have a normal physiological function in hippocampal neurons and that its appropriate level is important for synaptic function.
DISCUSSION
The amyloid and tau lesions are pathological hallmarks of AD that are present in virtually all AD cases. The relationship between the two, however, remains unresolved. Accumulating evidence supports the toxic oligomers formed by the Aβ peptide as the disease-causing agents, and synapses and dendritic spines as their major sites of action. Here we show that Aβ acts through the PAR-1/MARK family kinases to impinge on tau to affect synaptic function and dendritic spine morphogenesis. This conclusion is supported by the suppression of Aβ toxicity by either the co-expression of a phospho-mutant form of tau that can no longer be phosphorylated by PAR-1/MARK, or the specific inhibition of PAR-1/MARK with a peptide inhibitor. Our findings are consistent with the hypothesis that tau abnormality is a major downstream pathogenic event in the signaling cascade initiated by Aβ (6), and the observation that removal of endogenous tau attenuates learning and memory deficits in AD models (18).
One major question concerns how tau abnormality, specifically its hyperphosphorylation, leads to synaptic and dendritic spine pathology. Despite tau being best known as an axonal protein, phospho-tau was shown to accumulate in dendritic spines, where it may affect the synaptic trafficking and/or anchoring of glutamate receptors, thereby influencing postsynaptic function (40). It is not clear whether phospho-tau is being actively transported to the dendritic compartment, or that due to its compromised ability to bind microtubules, especially after being phosphorylated by PAR-1/MARK, phospho-tau simply diffuses from axon to other cellular compartments. Dendritic tau was recently found to recruit fyn kinase, facilitating fyn-mediated phosphorylation of GluR2 and stabilizing the interaction between GluR2 and PSD-95 (41). This potentially promotes excitotoxicity and represents a possible mechanism by which dendritic tau mediates Aβ toxicity. It remains to be determined whether dendritic tau has a normal physiological function. Since Aβ-induced spine loss was preventable by taxol (34), tau-related microtubule destabilization and trafficking defects may also mediate Aβ toxicity.
Much of the molecular events linking Aβ to tau pathologies remain to be elucidated. The implication of PAR-1/MARK in this process offers a new entry point to further dissect the signaling process. Several kinases have been shown to act upstream of PAR-1/MARK (42), including LKB1 (33). It would be interesting to test the involvement of these kinases in mediating Aβ toxicity through the regulation of PAR-1/MARK. Previous studies have indicated that tau phosphorylation at the PAR-1/MARK target sites also responds to other stress stimuli including osmotic stress, oxidative stress, serum deprivation and glutamate-induced excitotoxicity (33,34), suggesting that other triggers of the disease may also impinge on the PAR-1/MARK-tau axis. Aβ could also induce reversible synapse loss by modulating an NMDA-type GluR-dependent signaling pathway (43), or use long-term depression-related signaling mechanisms to affect synaptic function and dendritic spine morphology (44). Whether these signaling pathways are connected to the PAR-1/MARK-tau axis will be important future research directions. It also remains to be determined whether Aβ oligomers act by directly activating transmembrane receptors, changing neuronal membrane properties or, simply, causing cellular stress (4,34).
Our results suggest that targeting PAR-1/MARK kinases represents a potential strategy to prevent the synaptic toxicity of Aβ, thereby preventing or treating the synaptic defects of AD. In vivo studies applying MKI to various AD mouse models with clear learning and memory deficits are needed to validate this therapeutic approach. One major concern is the potential side effects, given the myriad cellular functions of PAR-1/MARK kinases (42). This concern is warranted by our observation that although MKI restored normal neurotransmission to Aβ oligomer-treated neurons, it led to reduced mEPSC frequency in mock-treated neurons, suggesting that appropriate levels of PAR-1/MARK activity is important for neurotransmission. Thus, in Aβ oligomer-treated neurons, which possess elevated PAR-1/MARK activity (34), MKI tuned down kinase activity to a level compatible with normal neurotransmission. However, in normal neurons, reduction of PAR-1/MARK activity by MKI to below-threshold levels could compromise neurotransmission. It will therefore be necessary to adjust MKI dosages to achieve therapeutic effect without causing serious side effects. The same could be said for therapeutic approaches targeting Aβ or tau. For example, the production of Aβ is positively regulated by neuronal activity (45), and Aβ can in turn depress excitatory synaptic transmission (46). This negative feedback regulatory loop may normally serve to restrain neuronal hyperactivity, such that therapeutic approaches leading to too much removal of Aβ might have unwanted side effects as well.
MATERIALS AND METHODS
Hippocampal neuronal culture and transfection
Rat E18 hippocampal neuron primary cultures were prepared as described (47). Cultured neurons (10-day in vitro) in 12-well plates were transfected with 2–3 μg of plasmid/well, using a CalPhos Mammalian Transfection Kit (Clontech, San Jose, CA, USA). Fluorescence immunocytochemistry was performed 3 days after transfection. Preparation and concentration of lentiviral particles were performed as described before (47). Viral transfection of hippocampal neurons was carried out at 10 days after in vitro culture. The next day, culture medium was replaced with 50% fresh medium plus 50% conditioned medium.
Aβ oligomer preparation
Commercial Aβ-42 peptide was purchased from rPeptide (Bogart, GA, USA). Aβ oligomer was prepared following a published protocol (37). Briefly, Aβ-42 was dissolved in hexafluoroisopropanol (HFIP) at 4 μg/μl and aliquoted into 0.6 ml tubes. Peptide films were obtained by evaporating HFIP with a Speed VAC and stored at −80°C. Prior to use, the peptide was dissolved in phosphate buffer (pH 7.4) to 80 μm and incubated at 4°C for 24 h. Culture medium containing 5 μm Aβ was used to treat neurons.
Recombinant DNA construction
EGFP-tagged PSD-95-WT in the FHUGW lentivirus vector was described before (35). The S561A mutation was introduced by site-directed mutagenesis. Tau S2A mutations were introduced as described before (25), and subcloned into FHUGW with a C-terminal HA tag. EGFP- and HA-tagged human MARK4 constructs were obtained from Dr Gerard Drewes. MKI cDNA was obtained from Dr C. Erec Stebbins, and subcloned into FHUGW vector with a C-terminal EGFP tag.
Immunofluorescence analysis
Chicken anti-EGFP (1:4000) and rabbit anti-HA (1:3000) antibodies were purchased from Abcam (Cambridge, MA, USA); mouse anti-Myc (1:1500, 4A6) and mouse anti-PSD-95 (1:300) from Millipore (Billerica, MA, USA); rabbit anti-GluR1 (1:200) from Calbiochem (San Diego, CA, USA) and rabbit anti-Synapsin I (1: 1000) from Sigma (St Louis, MO, USA). Immunofluorescence analysis was done essentially as described before (47). Confocal images were obtained using a Leica TCS SP5 confocal microscope or Zeiss inverted LSM510 confocal microscope (Carl Zeiss, Inc.) with 63× NA 1.4 objectives. Processed confocal images were used to make 2D projections. Dendritic spine number and clusters of PSD-95, GluR1 or Synapsin I were manually counted as the number along a 10 µm length of dendrite and presented as mean ± SEM. Each experiment was done at least three times and six to nine neurons were randomly selected for analysis each time. Between 15 and 30 dendritic processes were selected for quantification. Data were analyzed while blind to the conditions.
Electrophysiology in dissociated cultures
Whole-cell patch-clamp recordings were made from neurons at 13–14 DIV in voltage clamp mode using a Multiclamp 700B amplifier (Molecular Devices, Union City, CA, USA), digitized at 10 kHz and filtered at 4 kHz. Data were acquired and analyzed using AxographX (Axograph, Sydney). Whole-cell recording pipettes (3–5 MΩ) were filled with a solution containing (in mm) 135 CsMeSO4, 8 NaCl, 10 HEPES, 0.25 EGTA, 2 Mg2ATP, 0.3 Na3GTP, 0.1 spermine and 7 phosphocreatine. The bath solution contained (in mm): 140 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES-NaOH pH 7.4 and 10 glucose. Miniature AMPAR-mediated EPSCs were isolated by including D-APV (50 µm), picrotoxin (100 µm) and 1 μm tetrodotoxin in the bath solution. All recordings were performed at a holding potential of −70 mV at room temperature. mEPSCs were identified using a template with a threshold of −6 pA (2.5 × SD of the noise) and were individually proofread for accuracy. To plot summary graphs, the average frequency from the EGFP control cells from each culture preparation was normalized. Individual cells from all four conditions were then compared with this normalized average.
SUPPLEMENTARY MATERIAL
FUNDING
This project is supported by Dean's Postdoctoral Fellowship, Stanford University School of Medicine (W.Y.), Brain Disorders Award from the McKnight Endowment Fund for Neurosciences (B.L.) and National Institute of Health grants R01MH080378 (B.L.), R01AR054926 (B.L.) and R01MH063394 (R.M.).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Drs Gerard Drewes and C. Erec Stebbins for reagents, Dr Su Guo for reading the manuscript and members of the Lu and Malenka Laboratories for discussions.
Conflict of Interest statement. The authors declare that there is no conflict of interest.
REFERENCES
- 1.Selkoe D.J. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 2.Penzes P., Cahill M.E., Jones K.A., VanLeeuwen J.E., Woolfrey K.M. Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 2011;14:285–293. doi: 10.1038/nn.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spires T.L., Meyer-Luehmann M., Stern E.A., McLean P.J., Skoch J., Nguyen P.T., Bacskai B.J., Hyman B.T. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J. Neurosci. 2005;25:7278–7287. doi: 10.1523/JNEUROSCI.1879-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reddy P.H., Beal M.F. Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer's disease. Trend. Mol. Med. 2008;14:45–53. doi: 10.1016/j.molmed.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Selkoe D.J. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav. Brain Res. 2008;192:106–113. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hardy J., Selkoe D.J. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 7.Holtzman D.M., Morris J.C., Goate A.M. Alzheimer's disease: the challenge of the second century. Sci. Transl. Med. 2011;3:77sr71. doi: 10.1126/scitranslmed.3002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Billings L.M., Oddo S., Green K.N., McGaugh J.L., LaFerla F.M. Intraneuronal Abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- 9.Yankner B.A., Dawes L.R., Fisher S., Villa-Komaroff L., Oster-Granite M.L., Neve R.L. Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer's disease. Science. 1989;245:417–420. doi: 10.1126/science.2474201. [DOI] [PubMed] [Google Scholar]
- 10.Walsh D.M., Klyubin I., Fadeeva J.V., Cullen W.K., Anwyl R., Wolfe M.S., Rowan M.J., Selkoe D.J. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 11.Jin M., Shepardson N., Yang T., Chen G., Walsh D., Selkoe D.J. Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl Acad. Sci. USA. 2011;108:5819–5824. doi: 10.1073/pnas.1017033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lambert M.P., Barlow A.K., Chromy B.A., Edwards C., Freed R., Liosatos M., Morgan T.E., Rozovsky I., Trommer B., Viola K.L., et al. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc. Natl Acad. Sci. USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lewis J., Dickson D.W., Lin W.L., Chisholm L., Corral A., Jones G., Yen S.H., Sahara N., Skipper L., Yager D., et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- 14.Gotz J., Chen F., van Dorpe J., Nitsch R.M. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293:1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- 15.Oddo S., Caccamo A., Shepherd J.D., Murphy M.P., Golde T.E., Kayed, R., Metherate R., Mattson M.P., Akbari Y., LaFerla F.M. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 16.Oddo S., Billings L., Kesslak J.P., Cribbs D.H., LaFerla F.M. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004;43:321–332. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 17.Rapoport M., Dawson H.N., Binder L.I., Vitek M.P., Ferreira A. Tau is essential to beta-amyloid-induced neurotoxicity. Proc. Natl Acad. Sci. USA. 2002;99:6364–6369. doi: 10.1073/pnas.092136199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roberson E.D., Scearce-Levie K., Palop J.J., Yan F., Cheng I.H., Wu T., Gerstein H., Yu G.Q., Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 19.Spires-Jones T.L., Stoothoff W.H., de Calignon A., Jones P.B., Hyman B.T. Tau pathophysiology in neurodegeneration: a tangled issue. Trend. Neurosci. 2009;32:150–159. doi: 10.1016/j.tins.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 20.Ittner L.M., Gotz J. Amyloid-beta and tau—a toxic pas de deux in Alzheimer's disease. Nat. Rev. Neurosci. 2011;12:65–72. doi: 10.1038/nrn2967. [DOI] [PubMed] [Google Scholar]
- 21.Lee V.M., Goedert M., Trojanowski J.Q. Neurodegenerative tauopathies. Annu. Rev. Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 22.Guo S., Kemphues K.J. par-1, a gene required for establishing polarity in C. elegans embryos, encodes a putative Ser/Thr kinase that is asymmetrically distributed. Cell. 1995;81:611–620. doi: 10.1016/0092-8674(95)90082-9. [DOI] [PubMed] [Google Scholar]
- 23.Drewes G., Ebneth A., Preuss U., Mandelkow E.M., Mandelkow E. MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell. 1997;89:297–308. doi: 10.1016/s0092-8674(00)80208-1. [DOI] [PubMed] [Google Scholar]
- 24.Chin J.Y., Knowles R.B., Schneider A., Drewes G., Mandelkow E.M., Hyman B.T. Microtubule-affinity regulating kinase (MARK) is tightly associated with neurofibrillary tangles in Alzheimer brain: a fluorescence resonance energy transfer study. J. Neuropathol. Exp. Neurol. 2000;59:966–971. doi: 10.1093/jnen/59.11.966. [DOI] [PubMed] [Google Scholar]
- 25.Nishimura I., Yang Y., Lu B. PAR-1 kinase plays an initiator role in a temporally ordered phosphorylation process that confers tau toxicity in Drosophila. Cell. 2004;116:671–682. doi: 10.1016/s0092-8674(04)00170-9. [DOI] [PubMed] [Google Scholar]
- 26.Augustinack J.C., Schneider A., Mandelkow E.M., Hyman B.T. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer's disease. Acta Neuropathol. 2002;103:26–35. doi: 10.1007/s004010100423. [DOI] [PubMed] [Google Scholar]
- 27.Perez M., Ribe E., Rubio A., Lim F., Moran M.A., Ramos P.G., Ferrer I., Isla M.T., Avila J. Characterization of a double (amyloid precursor protein-tau) transgenic: tau phosphorylation and aggregation. Neuroscience. 2005;130:339–347. doi: 10.1016/j.neuroscience.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Y., Guo H., Kwan H., Wang J.W., Kosek J., Lu B. PAR-1 kinase phosphorylates Dlg and regulates its postsynaptic targeting at the Drosophila neuromuscular junction. Neuron. 2007;53:201–215. doi: 10.1016/j.neuron.2006.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gylys K.H., Fein J.A., Yang F., Wiley D.J., Miller C.A., Cole G.M. Synaptic changes in Alzheimer's disease: increased amyloid-beta and gliosis in surviving terminals is accompanied by decreased PSD-95 fluorescence. Am. J. Pathol. 2004;165:1809–1817. doi: 10.1016/s0002-9440(10)63436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Almeida C.G., Tampellini D., Takahashi R.H., Greengard P., Lin M.T., Snyder E.M., Gouras G.K. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol. Dis. 2005;20:187–198. doi: 10.1016/j.nbd.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 31.Trinczek B., Brajenovic M., Ebneth A., Drewes G. MARK4 is a novel microtubule-associated proteins/microtubule affinity-regulating kinase that binds to the cellular microtubule network and to centrosomes. J. Biol. Chem. 2004;279:5915–5923. doi: 10.1074/jbc.M304528200. [DOI] [PubMed] [Google Scholar]
- 32.Seshadri S., Fitzpatrick A.L., Ikram M.A., DeStefano A.L., Gudnason V., Boada M., Bis J.C., Smith A.V., Carassquillo M.M., Lambert J.C., et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA. 2010;303:1832–1840. doi: 10.1001/jama.2010.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang J.W., Imai Y., Lu B. Activation of PAR-1 kinase and stimulation of tau phosphorylation by diverse signals require the tumor suppressor protein LKB1. J. Neurosci. 2007;27:574–581. doi: 10.1523/JNEUROSCI.5094-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zempel H., Thies E., Mandelkow E., Mandelkow E.M. Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J. Neurosci. 2010;30:11938–11950. doi: 10.1523/JNEUROSCI.2357-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu W., Schluter O.M., Steiner P., Czervionke B.L., Sabatini B., Malenka R.C. Molecular dissociation of the role of PSD-95 in regulating synaptic strength and LTD. Neuron. 2008;57:248–262. doi: 10.1016/j.neuron.2007.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thies E., Mandelkow E.M. Missorting of tau in neurons causes degeneration of synapses that can be rescued by the kinase MARK2/Par-1. J. Neurosci. 2007;27:2896–2907. doi: 10.1523/JNEUROSCI.4674-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Knowles J.K., Rajadas J., Nguyen T.V., Yang T., LeMieux M.C., Vander Griend L., Ishikawa C., Massa S.M., Wyss-Coray T., Longo F.M. The p75 neurotrophin receptor promotes amyloid-beta(1–42)-induced neuritic dystrophy in vitro and in vivo. J. Neurosci. 2009;29:10627–10637. doi: 10.1523/JNEUROSCI.0620-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nesic D., Miller M.C., Quinkert Z.T., Stein M., Chait B.T., Stebbins C.E. Helicobacter pylori CagA inhibits PAR1-MARK family kinases by mimicking host substrates. Nat. Struct. Mol. Biol. 2010;17:130–132. doi: 10.1038/nsmb.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lizcano J.M., Goransson O., Toth R., Deak M., Morrice N.A., Boudeau J., Hawley S.A., Udd L., Makela T.P., Hardie D.G., Alessi D.R. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004;23:833–843. doi: 10.1038/sj.emboj.7600110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoover B.R., Reed M.N., Su J., Penrod R.D., Kotilinek L.A., Grant M.K., Pitstick R., Carlson G.A., Lanier L.M., Yuan L.L., Ashe K.H., Liao D. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68:1067–1081. doi: 10.1016/j.neuron.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ittner L.M., Ke Y.D., Delerue F., Bi M., Gladbach A., van Eersel J., Wolfing H., Chieng B.C, Christie M.J., Napier I.A., et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- 42.Matenia D., Mandelkow E.M. The tau of MARK: a polarized view of the cytoskeleton. Trend. Biochem. Sci. 2009;34:332–342. doi: 10.1016/j.tibs.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 43.Shankar G.M., Bloodgood B.L., Townsend M., Walsh D.M., Selkoe D., Sabatini B.L. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hsieh H., Boehm J., Sato C., Iwatsubo T., Tomita T., Sisodia S., Malinow R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bero A.W., Yan P., Roh J.H., Cirrito J.R., Stewart F.R., Raichle M.E., Lee J.M., Holtzman D.M. Neuronal activity regulates the regional vulnerability to amyloid-beta deposition. Nat. Neurosci. 2011;14:750–756. doi: 10.1038/nn.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kamenetz F., Tomita T., Hsieh H., Seabrook G., Borchelt D., Iwatsubo T., Sisodia S., Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- 47.Yu W., Sun Y., Guo S., Lu B. The PINK1/Parkin pathway regulates mitochondrial dynamics and function in mammalian hippocampal and dopaminergic neurons. Hum. Mol. Genet. 2011;20:3227–3240. doi: 10.1093/hmg/ddr235. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.