

Short abstract
EASE is a customizable software application for rapid biological interpretation of gene lists that result from the analysis of microarray, proteomics, SAGE and other high-throughput genomic data.
Abstract
EASE is a customizable software application for rapid biological interpretation of gene lists that result from the analysis of microarray, proteomics, SAGE and other high-throughput genomic data. The biological themes returned by EASE recapitulate manually determined themes in previously published gene lists and are robust to varying methods of normalization, intensity calculation and statistical selection of genes. EASE is a powerful tool for rapidly converting the results of functional genomics studies from 'genes' to 'themes'.
Rationale
High-density microarray and proteomics technologies have enabled the discovery of global patterns of biological responses to experimental or natural perturbations [1]. Much work has addressed the issues of data normalization and statistical selection of the genes that are significantly modulated or clustered on the basis of expression profiles [2]. The net result of these efforts is one or more lists of genes. Unfortunately, little work has addressed the issue of rapidly identifying biological themes in such lists [3]. Most investigators currently annotate genes one-at-a-time using internet-based databases or manual literature searches. After this tedious process, it can still be a struggle to identify the most salient biological themes in order to make sense of the results and researchers have no systematic way to prioritize these themes for further analysis. A parallel issue in interpreting such data is how to exploit the ever-expanding flood of functional genomic data and tools. We developed the Expression Analysis Systematic Explorer (EASE) to automate the process of biological theme determination for lists of genes and to serve as a customizable gateway to online analysis tools. This is the first report to show that the highest-ranking themes derived by a computational method can recapitulate manually derived themes in previously published microarray, proteomics and SAGE results, and to provide evidence that these themes are stable to varying methods of gene selection.
EASE performs three basic functions with any list of genes. The first is theme discovery, defined as the identification of terms or phrases that describe a statistically significant number of genes in the list with respect to the number of genes described by the term or phrase in the population of genes from which the list derived. The second is customizable linking to online tools, and the third is creation of descriptive annotation tables. Each of these functions uses a system of tab-delimited text files that are simple to customize and update. EASE is an easy-to-use, customizable tool that allows investigators to systematically mine the mass of functional information associated with data generated by microarray, proteomics or SAGE studies.
EASE uses customizable text files for theme discovery, annotation and linking to online tools
To analyze a gene list, EASE first maps the gene identifiers to a standardized gene accession (SGA) system via a simple text file in the \Data\Convert\ directory. The default SGA system used by EASE is LocusLink numbers [4]. Upon conversion to the SGA system, EASE maps the genes to biological categories within various classification systems. Each system is specified in a text file in the \Data\Class\ directory that maps many-to-many relationships between genes and gene categories within the classification system. Similarly, EASE maps genes to annotation fields specified in files of the \Data\ directory. Users can therefore utilize any system of identifying genes with any custom annotation fields or categorical systems by creating the associated text files in the appropriate directory, as outlined in the help files of EASE. EASE comes equipped with an automated update routine that downloads and parses public annotation data sources and installs a LocusLink-based system of files, thereby allowing researchers to use EASE with the most up-to-date annotation information.
EASE constructs hyperlinks to definitions for various categorical systems and the gene categories therein with configuration files in the \Data\Class\URL data\ directory. EASE can also load the genes in the current gene list into various online tools by using simple URL configuration text files in the \Links\ directory. Both types of configuration files are text files that are simple to create or modify to facilitate the addition of new links to online tools and definitions for new categorical systems added by the user.
For theme discovery via category over-representation analysis, EASE uses the three systems of the Gene Ontology [5] as default categorization systems. However, any set of custom or public systems can be simultaneously analyzed, including SWISS-PROT [6] and PIR keywords [7], transcription factor regulation, protein domains, pathway membership, chromosomal location, membership in previously published gene lists, and MeSH headings or keywords extracted from gene-associated literature. EASE calculates over-representation with respect to the total number of genes assayed and annotated within each system to allow for side-by-side comparisons of categories from categorization systems with varying levels of annotation. The conversion of gene identifiers to an SGA system such as LocusLink numbers is essential to the over-representation analysis to ensure that a single gene represented by more than one identifier (typical of GenBank) receives only one 'vote' for each of its categories.
The user has a choice of two statistical measures of over-representation - the one-tailed Fisher exact probability or a variant thereof - which is referred to as the 'EASE score'. The Fisher exact probability for over-representation is calculated using the Gaussian hypergeometric probability distribution that describes sampling without replacement from a finite population consisting of two types of elements [8]. In the case of microarray data, EASE defines this population of elements as the set of genes on the microarray annotated within a given gene-classification system. For each possible classification within the system, the two types of elements are: genes that belong to that classification; and genes that do not. Given the number of genes of each type within the finite population, it is possible to calculate the exact probability of randomly sampling a given number of genes and observing a specific number that belong to the classification. The one-tailed Fisher exact probability of over-representation is calculated by summing this probability with all probabilities for situations in which there is a greater number of genes within the classification. For example, assume a microarray contains 1,000 genes annotated within the Biological Process branch of the Gene Ontology, and five of these genes fall within the classification Apoptosis. The likelihood of observing four Apoptosis genes by random chance in a gene list containing 50 genes annotated within Biological Process is calculated by summing the hypergeometric probabilities of observing 4 out of 50 genes and 5 out of 50 genes. Note that 6 (or greater) out of 50 genes is not possible, since there are only five such genes on the whole microarray. This example highlights why the Fisher exact probability is more appropriate than methods describing sampling with replacement such as the chi-square and Z-score statistics.
It is easier to see why the Fisher exact probability ascribes a higher significance to the observation in the example after considering the following: as each Apoptosis gene is added to the gene list, Apoptosis genes become increasingly rare in the remaining population of genes not on the list. The Fisher exact probability takes this effect of finite populations into account whereas the chi-square and Z-score statistics do not. The Fisher exact probability is also more appropriate than a ratio-of-ratios type metric, wherein the ratio of genes in a category on the gene list is compared to the ratio of genes in that category within the population. This is because ratio-of-ratios tend to underestimate the significance of high-frequency categories. This problem is exemplified by observing 75 genes within some category out of 100 genes on a list when the background is 6,000 genes out of 10,000. The probability of observing such a situation by random chance using the Fisher exact probability is almost 1 in 1,000 (p = 0.0012), yet the ratio-of-ratios method only detects a modest 1.25-fold increase in proportion. Ratio-of-ratios are also prone to 'granularity effects' with low-frequency categories, in which the observation of a single gene in some rare category can have a large ratio enrichment compared to the population, yet be of little significance. This situation is exemplified by observing a single gene within some category on a list of 50 genes when there are 60 such genes on a 10,000-gene microarray. The Fisher exact probability tells us that this seemingly interesting 3.3-fold enrichment based on the ratio of ratios actually had a greater than one in four (p = 0.26) chance of occurring simply due to random chance.
The EASE score is offered as a conservative adjustment to the Fisher exact probability that weights significance in favor of themes supported by more genes. The theoretical basis of the EASE score lies in the concept of jackknifing a probability. The stability of any given statistic can be ascertained by a procedure called jackknifing, in which a single data point is removed and the statistic is recalculated many times to give a distribution of probabilities that is broad if the result is highly variable and tight if the result is robust [9]. The EASE score is calculated by penalizing (removing) one gene within the given category from the list and calculating the resulting Fisher exact probability for that category. It therefore represents the upper bound of the distribution of jackknife Fisher exact probabilities and has advantages in terms of penalizing the significance of categories supported by few genes. For example, assume a list of 206 genes is selected from a population of 13,679 genes. If there is only one gene in the population in some rare category, X, and that gene happens to appear on the list of 206 genes, the Fisher exact would consider category X significant (p = 0.0152). At the same time, the Fisher exact probability would deem a more common category, Y, with 787 members in the population and 20 members on the list, as slightly less significant (p = 0.0154). From the perspective of global biological themes, however, a theme based on the presence of a single gene is neither global nor stable and is rarely interesting. If the single gene happens to be a false positive, then the significance of the dependent theme is entirely false. However, the EASE score for these two situations is p = 1 for category X and p < 0.0274 for category Y, and thus the EASE score eliminates the significance of the 'unstable' category X while only slightly penalizing the significance of the more global theme Y. By extrapolating between these two extremes, the EASE score penalizes the significance of categories supported by fewer genes and thus favors more robust categories than the Fisher exact probability.
EASE comes equipped with: files for specifying genes as LocusLink numbers, gene symbols, GenBank, SWISS-PROT, Flybase [10], MGI [11], or RGD [12] accessions, UniGene clusters [13] or Affymetrix probe set identifiers [14]; annotation fields from LocusLink; population files consisting of all LocusLink numbers for selected species and for various Affymetrix GeneChips and other selected microarrays; and classification systems derived from the Gene Ontology, KEGG [15], BBID [16] and SWISS-PROT as well as classification systems parsed from LocusLink (including the Proteome HumanPSD database's 'At-a-Glance' [17]), chromosome location, PFAM [18] and SMART [19] protein domains. Furthermore, most of these data files can be updated from their original internet sources at any time by clicking the 'Update with the Most Recent Online Data' button (Figure 1). Storage of these data to local tab-delimited text files allows for quick access and removes any concern regarding the transmission of confidential research results over the internet.
Figure 1.
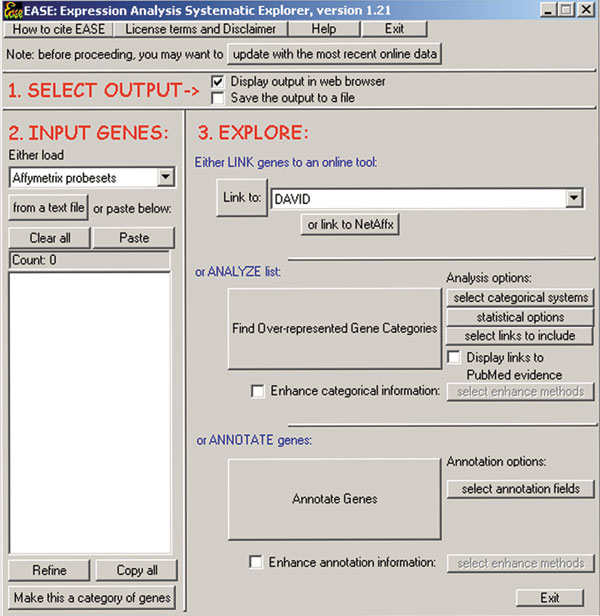
The EASE user interface is designed for quick annotation and analysis of gene lists. Gene identifiers are pasted into the 'INPUT GENES' section, and the processes of linking to online tools, over-representation analysis or annotation are launched with buttons in the 'EXPLORE' section. Annotation data can be automatically retrieved from the internet and stored into local data files by clicking the 'update with the most recent online data' button.
Exploring a gene list with EASE
The core function of EASE is to annotate or analyze a list of genes input as gene identifiers, and display the result in the system web-browser or save the result in a tab-delimited text or Microsoft Excel [20] format. The identifiers can be loaded from a text file or pasted into EASE from another application. Upon input of identifiers, the user can generate an annotation table by clicking the 'Annotate Genes' button (Figure 1). The user can also link to any number of online tools such as DAVID [21] via the 'Link to:' list box; this function automatically loads the information specific to the current gene list into the online tool, thereby allowing EASE to serve as a convenient interface to these resources.
The identification of biological themes in the gene list is initiated by clicking the 'Find over-represented gene categories' button. This function returns an output of all gene categories ranked by over-representation, with associated probabilities, counts used in the probability calculation, associated genes from the original list and links to various online tools for these genes. The most significantly over-represented categories that result from this analysis are deemed 'biological themes' of the gene list. The user can optionally limit these analyses to any particular set of gene categories to answer questions such as 'what is special about the mitochondrial genes on my list compared to all mitochondrial genes on the microarray?' The user can further use the 'Refine' functionality of EASE to remove specific genes from the original list and enable an over-representation analysis of the remaining genes exclusively. These two functions can be applied repeatedly until the gene list is thoroughly characterized. EASE also allows for comparisons of gene lists at a thematic level, whereby the results are expressed in terms of gene categories over-represented in one list compared to all lists combined.
Calculating statistics on thousands of gene categories can lead to a few seemingly significant probabilities due simply to chance [22]. To address this multiple comparison issue, EASE can calculate a wide variety of probability corrections including Bonferroni-type methods, false-discovery rate (FDR) and bootstrap methods [23]. The FDR and bootstrap methods are performed by iteratively running over-representation analyses on gene lists randomly picked from the population to estimate the true probability of observing a given categorical enrichment in a given list by chance given the multiple comparison issue. Nevertheless, the power of EASE is most appropriately viewed as an exploratory tool to direct the attention of the researcher to enriched biological themes by prioritizing functional categories based on the significance of over-representation.
EASE themes are robust and recapitulate manually determined themes
The published gene lists of Kayo et al. [24], Wu et al. [25] and Gnatenko et al. [26] were analyzed with EASE to test its ability to generate themes in comparison to manually determined themes from microarray, proteomic and SAGE data, respectively. In the Kayo study, the authors generated four gene lists corresponding to genes up- and downregulated in primate muscle in response to aging or caloric restriction. These gene lists were analyzed with the categorical over-representation function of EASE, using EASE scores that were corrected for multiplicity using 10,000 bootstrap iterations. All significant (p < 0.05) categories resulting from each list were compared to the themes manually determined and published by Kayo et al. [24] (Table 1).
Table 1.
The four gene lists analyzed by Kayo et al. [24] along with the total time needed for initial analysis are shown for the manual and EASE analyses
| Total time for analysis | Themes of genes downregulated with caloric restriction | Themes of genes upregulated with caloric restriction | Themes of genes downregulated with aging | Themes of genes upregulated with aging | |
| Manual analysis by Kayo et al. [24] | Approximately 200 hours | Energy metabolism, mitochondrial bioenergetics | Structural proteins, cytoskeletal proteins | Energy metabolism, mitochondrial electron transport, oxidative phosphorylation | [Inflamation/immune function], [oxidative stress] |
| EASE | Approximately 15 minutes | Mitochondrion, electron transport, mitochondrial membrane, inner membrane, primary active transporter, mitochondrial electron transport chain, oxidoreductase, hydrogen ion transporter, mitochondrial inner membrane, monovalent inorganic cation transporter, energy pathways, [carrier], ion transporter, [cytoplasm] | Extracellular matrix, [calmodulin binding], [morphogenesis], structural molecule, [development], microfibril, cytoskeleton | Inner membrane, hydrogen ion transporter, [intracellular], monovalent inorganic cation transporter, metabolism, mitochondrial membrane, mitochondrial inner membrane, primary active transporter, energy pathways, mitochondrion, ion transporter, [carrier], cation transporter | [Gas transport], [oxygen transport] |
For each list, the major biological themes as determined by Kayo et al. [24] are shown, as well as all significant (p < 0.05) gene categories as determined by EASE score corrected for multiplicity with the bootstrap function using 10,000 random trials. The time shown for EASE represents initial analysis before running the bootstrap analysis. Bootstrap analysis took an additional 2 hours. Themes that differ between Kayo et al.'s themes and the EASE results are in square brackets.
The initial EASE analysis successfully discovered the same themes as Kayo et al. in three of four gene lists in less than 15 minutes, with the 10,000-iteration bootstrap corrections requiring an additional 2 hours per list. In contrast, the manual analysis by Kayo et al. required approximately 200 hours of gene annotation and literature reading, (R. Weindruch, personal communication.) EASE also uncovered new and potentially interesting themes including the upregulation of calmodulin-binding and morphogenesis genes with caloric restriction and the upregulation of hemoglobin components within aging muscle. The disparate results for the list of genes upregulated with aging is due to the lack of relative enrichment for 'inflammation/immunity' genes in the list of genes upregulated with aging (7.5%) relative to all 'inflammation/immunity' genes on the HuGeneFL microarray (8.9%). Therefore, any random list of the same size would be expected to result in about the same number of 'immunity/inflammatory response' genes as the Kayo list. Similarly, no significant enrichment was detected for the 'stress response/oxidative stress' theme (8.3% vs 7.8% on list and microarray, respectively). Nevertheless, the discovery of all manual themes by EASE for the majority of gene lists demonstrates the power of EASE to dramatically reduce the time required to interpret microarray results while adding a statistical measure of confidence to the interpretation.
To test the ability of EASE to find themes similar to manually determined themes from proteomics research, a previously published list of proteins was analyzed with EASE. Wu et al. developed a method for the simultaneous analysis of membrane and soluble proteins from a crude brain homogenate using tandem mass spectrometry proteomics. The authors identified a total of 1,610 proteins in the crude lysate and postulated that 454 of the proteins were transmembrane proteins on the basis of the use of a sequence-analysis algorithm. These 454 proteins were analyzed with respect to the entire set of 1,610 identified proteins using the categorical over-representation function of EASE. As shown in Table 2, the top-scoring theme from the EASE analysis, SWISS-PROT keyword 'transmembrane', matches well with the predicted 'transmembrane' theme of these 454 proteins.
Table 2.
EASE analysis of proteomics and SAGE data
| Proteomics* | SAGE† | |
| Manually determined theme | Transmembrane | Mitochondrial genes |
| Top EASE theme | Transmembrane (SWISS-PROT keyword) | Mitochondrion (chromosome) |
*Themes of proteins predicted to have transmembrane domains versus all proteins in a brain homogenate. †Themes of SAGE tags identified in human platelets versus all human genes in LocusLink. The predicted 454 transmembrane proteins in a proteomics study by Wu et al. [25] were analyzed with the categorical over-representation function of EASE for enriched themes with respect to the total of 1,610 proteins identified in a crude brain homogenate. Similarly, the SAGE tags detected by Gnatenko et al. [26] in a SAGE-based study of human platelets were analyzed for enriched themes against a background of all human genes in LocusLink. In both cases the manually determined theme is compared to the most significant EASE category as determined by EASE score. The categorical system from which the category derives is shown in parentheses.
To determine whether EASE can detect themes similar to manually determined themes from SAGE research, a previously published list of SAGE tags was analyzed with EASE. Gnatenko et al. [26] identified transcripts expressed in human platelets via SAGE and determined that transcripts originating from the mitochondrial chromosome predominated. As shown in Table 2, the top-scoring theme from the EASE analysis is 'Mitochondrion' from the system of gene classification by chromosome.
Having shown that EASE can rapidly identify themes similar to those in published articles, we investigated the robustness of themes within a given study following the analysis of gene lists generated using various gene-selection methods. A typical problem in the analysis of microarray data is that different methods of array normalization and statistical selection of genes can lead to strikingly different lists of genes from the same experiment. As there is currently little consensus on the ideal method to select differentially expressed genes, this issue tends to lead the microarray researcher to ask the difficult question, 'which of these gene lists is correct?' We reasoned that the underlying biological phenomenon under study is most likely being captured within the functional context of the genes within each of the lists, despite the lists having different genes. We tested this hypothesis by looking at biological themes identified by EASE.
Eight methods using different combinations of chip-to-chip normalization protocols, gene-intensity calculations and statistical significance tests were used to select genes upregulated in peripheral blood mononuclear cells following HIV-1 viral rebound in the plasma of six HIV-infected patients discontinuing antiviral drug therapy for one month. The raw data for this analysis comes from a microarray-based study of the in vivo effects of HIV infection and HAART (G.D., Jun Yang, B.T.S., D.A.H., Randy Stevens, Joseph Adelsberger, Julie Metcalf, Robin Dewar, Igor Sidorov, Dimiter Dimitrov, et al., unpublished work). Gene expression was assayed with the Affymetrix HuGeneFL microarray. One of four different normalization protocols was applied: MAS 4 (Microarray Suite 4, Affymetrix Inc.); dChip [27]; rank-remapping (D.H., unpublished work); and non-parametric local fitting [28]. Gene-expression intensity was determined using either MAS 4 average difference method or dChip MBEI. Significantly upregulated genes were identified using either a paired student T statistic (t > 2.2) or significance analysis of microarrays (SAM; d > 2.2) [29]. Significantly over-represented functional categories (p < 0.05) for each of the eight lists were identified using EASE scores corrected for multiplicity using 10,000 bootstrap iterations (Tables 3, 4).
Table 3.
EASE themes are consistent despite the poor overlap of gene lists derived from the same experiment by various analytical methods
| Method | A | B | C | D | E | F | G | H | |||
| Normalization | MAS 4 | MAS 4 | dChip | dChip | Rank remap | Rank remap | NP | NP | |||
| Intensity calculation | MAS 4 | MAS 4 | dChip | dChip | dChip | dChip | dChip | dChip | |||
| Method | Normalization | Intensity calculation | Gene selection | t-test | SAM | t-test | SAM | t-test | SAM | t-test | SAM |
| A | MAS 4 | MAS 4 | t-test | [72] | 72 (60%) | 20 (7%) | 17 (12%) | 18 (8%) | 19 (12%) | 24 (8%) | 22 (10%) |
| B | MAS 4 | MAS 4 | SAM | [120] | 27 (9%) | 22 (12%) | 25 (9%) | 25 (13%) | 34 (10%) | 31 (12%) | |
| C | dChip | dChip | t-test | [220] | 81 (36%) | 130 (48%) | 70 (27%) | 105 (29%) | 71 (22%) | ||
| D | dChip | dChip | SAM | [86] | 56 (27%) | 55 (40%) | 49 (18%) | 47 (27%) | |||
| E | Rank remap | dChip | t-test | [180] | 95 (50 %) | 109 (35%) | 76 (32%) | ||||
| F | Rank remap | dChip | SAM | [105] | 68 (24%) | 67 (59%) | |||||
| G | NP | dChip | t-test | [242] | 154 (59%) | ||||||
| H | NP | dChip | SAM | [173] |
Gene lists resulting from the same experiment can differ greatly as a result of selection criteria. Eight different methods were used to select genes upregulated in peripheral blood mononuclear cells (PBMCs) of HIV patients after discontinuation of antiretroviral drug therapy. The various lists resulted from four different array-to-array normalizations, two different methods of intensity calculation and two methods of statistical selection of genes (see text). The values in square brackets on the diagonal show the total number of genes yielded by each method. The unbracketed numbers give the absolute number of genes shared by any two methods, with the percentage of genes in both lists from the combined gene lists in parentheses. MAS 4, Affymetrix analysis software version 4.0; dChip, dChip software [8]; Rank remap, unpublished method of D.A.H; NP, nonparametric method of Sidirov et al. [9]; t-test, Student T statistic; SAM, statistical analysis of microarrays software [10].
Table 4.
EASE identifies the same themes in spite of the notable variability in gene lists resulting from different methods of data normalization and gene selection criteria
| Method | A | B | C | D | E | F | G | H | ||
| Normalization | MAS 4 | MAS 4 | dChip | dChip | Rank remap | Rank remap | NP | NP | ||
| Intensity calculation | MAS 4 | MAS 4 | dChip | dChip | dChip | dChip | dChip | dChip | ||
| Gene selection | t-test | SAM | t-test | SAM | t-test | SAM | t-test | SAM | System | Category |
| <0.0001 (1) | 0.0002 (1) | 0.0004 (1) | <0.0001 (1) | 0.0005 (2) | <0.0001 (4) | <0.0001 (1) | <0.0001 (1) | SWISS-PROT keyword | Interferon induction | |
| 0.0006 (2) | 0.0027 (2) | 0.0383 (4) | < 0.0001 (2) | <0.0001 (1) | <0.0001 (1) | <0.0001 (2) | <0.0001 (3) | Biological process | Response to biotic stimulus | |
| 0.0036 (5) | 0.0061 (3) | NS (5) | < 0.0001 (4) | 0.0005 (3) | <0.0001 (2) | <0.0001 (4) | <0.0001 (4) | Biological process | Immune response | |
| 0.0031 (4) | 0.0081 (4) | NS (7) | < 0.0001 (5) | 0.0006 (4) | <0.0001 (3) | <0.0001 (3) | <0.0001 (2) | Biological process | Defense response | |
| 0.0333 (6) | NS (7) | 0.0329 (3) | < 0.0001 (3) | 0.0128 (6) | <0.0001 (5) | 0.0003 (5) | <0.0001 (5) | Biological process | Response to external stimulus | |
| 0.0008 (3) | 0.0101 (5) | 0.0308 (2) | < 0.0001 (6) | 0.0078 (5) | 0.0002 (6) | 0.0558 (8) | 0.0071 (8) | Molecular function | Antiviral response protein |
The six dominant categories over-represented in the genes upregulated in PBMCs of HIV patients one month after discontinuing therapy all implicate a vigorous immune response. EASE scores (corrected for multiplicity with the bootstrap function using 10,000 random trials) are shown along with the rank (in parentheses) of each category in each of the eight lists. The category outside of the top five for each list is indicated in italics. NS, p > 0.05.
Table 3 demonstrates the fact that different gene-selection methods can lead to strikingly different gene lists from the same experiment. The percentage of genes overlapping in any two lists was highly variable, and ranged from 7% to 60%. In spite of this striking variation, the top five biological themes returned by EASE for each of the eight gene lists were virtually the same; all derived from a group of six categories that implicate a vigorous interferon-induced immune response in patients with rebounding HIV viral loads (Table 4). The conversion of genes to themes with EASE allowed the underlying biological phenomenon of the experiment to be determined despite substantial differences in gene-list content resulting from the use of various normalization, gene intensity and statistical selection methods.
EASE has capabilities not found in similar software
Recent reports have introduced software packages designed to help biologists with the interpretation of genome-scale data, including MAPPfinder [30] and GoMiner [31]. MAPPfinder is an accessory program to GenMAPP [32], and is used to find the MAPP pathways most enriched for the genes in given gene list using a z-score metric. Since MAPPfinder reports a z-score with no correction for the multiple comparison problem, MAPPfinder does not give an accurate probability of over-representation, especially for rare categories. In the publication introducing MAPPfinder, no evidence is given for the ability of MAPPfinder to find biological themes similar to those of human annotators.
GoMiner is a program for visualizing the genes on a list within the context of the structure of the Gene Ontology. Such an analysis leaves finding the most significant categories to visual inspections; that is, the user must manually scan the entire tree/DAG visualization to find the most over-represented categories, and no correction is offered to address the multiple comparison problem. Hence it is impossible to be certain that there are any significant categories within a list using GoMiner. As evidence for the utility of GoMiner, the authors show the GoMiner results for a particular Gene Ontology term 'apoptosis regulator' within the data of their own microarray study of cancer cell lines. As the authors make no claims as to whether or not this was the highest-scoring or most significant term determined by GoMiner, the reader cannot ascertain how 'apoptosis regulator' ranks among an unknown number of categories with high enrichment scores, nor can the reader determine whether the highest-ranking term, as determined by GoMiner, is relevant to the biology of this experiment.
EASE contains several functions not found in MAPPfinder or GoMiner that are useful to a biologist exploring a list of genes. One is the ability to load a list of genes into new online tools when given simple configuration files for each tool. Another function is the ability to generate annotation tables with any number of descriptive fields, including fields listing all classifications of a particular gene within a selected classification system. Perhaps the most distinctive characteristic of EASE compared with these other tools is ease-of-use. EASE is available as a self-extracting distribution that is ready to be used as soon as it is unzipped. After unzipping, the user can simply paste a list of identifiers from their gene list into EASE and quickly find the most over-represented category of genes from many different classification systems. This paste-and-go functionality is not available within the other two programs, both of which require substantial time to create files of genes for gene lists and gene populations in an acceptable format. In the case of MAPPfinder, the GenMAPP program must create these files after loading a dataset, and in the case of GoMiner, the files must consist of HUGO symbols.
'Genes to Themes' with EASE: possible uses of the EASE method
EASE rapidly converts a list of genes into an ordered table of robust biological themes that summarize the biological result of the experiment. This method has immediate utility for finding themes that most differentiate lists of genes, for example upregulated versus downregulated in a single experiment, but could potentially be applied to compare the results of different experiments, even involving different species and/or technology platforms. The EASE method has proven useful for a SAGE analysis of cancer (W.D. Stein unpublished work), a proteomics analysis of samples enriched for integral membrane proteins (J. Blonder, unpublished work), and for microarray analyses of cancer (A. Domkowski, unpublished work; K. Akagi, personal communication), cataracts (M. Kantorow, unpublished work), type I diabetes (P. Jailwala, unpublished work) and immune function in HIV disease [33,34]. The EASE method also enables rapid assay for overlap between gene clusters identified in any number of experiments when the user creates gene-classification schema based on these clusters. EASE can potentially be used to facilitate the development of data-normalization and gene-selection criteria by observing the highest enrichment attained for EASE themes within a particular experiment in which the biological phenomenon is well characterized and confirmed. EASE allows investigators to fully exploit the potential of high-throughput functional genomics technologies to infer biological themes. A full-featured version of EASE is freely available to non-profit researchers for use on Windows operating systems [35] and an online version of the EASE over-representation function called EASEonline is available on the DAVID website [36].
A previous version of this manuscript was made available before peer review at http://genomebiology.com/2003/4/6/P4
References
- Heller MJ. DNA microarray technology: devices, systems, and applications. Annu Rev Biomed Eng. 2002;4:129–153. doi: 10.1146/annurev.bioeng.4.020702.153438. [DOI] [PubMed] [Google Scholar]
- Quackenbush J. Microarray data normalization and transformation. Nat Genet. 2002;32 Suppl:496–501. doi: 10.1038/ng1032. [DOI] [PubMed] [Google Scholar]
- Slonim DK. From patterns to pathways: gene expression data analysis comes of age. Nat Genet. 2002;32 Suppl:502–508. doi: 10.1038/ng1033. [DOI] [PubMed] [Google Scholar]
- Pruitt KD, Katz KS, Sicotte H, Maglott DR. Introducing RefSeq and LocusLink: curated human genome resources at the NCBI. Trends Genet. 2000;16:44–47. doi: 10.1016/s0168-9525(99)01882-x. [DOI] [PubMed] [Google Scholar]
- The Gene Ontology Consortium Gene Ontology: tool for the unification of biology. Nature Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch A, Boeckmann B. The SWISS-PROT protein sequence data bank. Nucleic Acids Res. 1991;19 Suppl:2247–2249. doi: 10.1093/nar/19.suppl.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CH, Huang H, Arminski L, Castro-Alvear J, Chen Y, Hu Z, Ledley RS, Lewis KC, Mewes H, Orcutt BC, et al. The Protein Information Resource: an integrated public resource of functional annotation of proteins. Nucleic Acids Res. 2002;30:35–37. doi: 10.1093/nar/30.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleiss JL. Statistical Methods for Rates and Proportions. New York: John Wiley; 1981. [Google Scholar]
- Tukey JW. Bias and confidence in not quite large samples. Ann Math Stat. 1958;29:614. [Google Scholar]
- The FlyBase Consortium The FlyBase database of the Drosophila genome projects and community literature. Nucleic Acids Res. 2003;31:172–175. doi: 10.1093/nar/gkg094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake JA, Richardson JE, Bult CJ, Kadin JA, Eppig JT, Mouse Genome Database Group MGD: The Mouse Genome Database. Nucleic Acids Res. 2003;31:193–195. doi: 10.1093/nar/gkg047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rat Genome Database http://rgd.mcw.edu/
- Wheeler DL, Church DM, Federhen S, Lash AE, Madden TL, Pontius JU, Schuler GD, Schriml LM, Sequeira E, et al. Database resources of the National Center for Biotechnology. Nucleic Acids Res. 2003;31:28–33. doi: 10.1093/nar/gkg033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Affymetrix, Inc. http://www.affymetrix.com
- Kanehisa M, Goto S, Kawashima S, Nakaya A. The KEGG databases at GenomeNet. Nucleic Acids Res. 2002;30:42–46. doi: 10.1093/nar/30.1.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker KG, White SL, Muller J, Engel J. BBID: the biological biochemical image database. Bioinformatics. 2000;16:745–746. doi: 10.1093/bioinformatics/16.8.745. [DOI] [PubMed] [Google Scholar]
- Proteome HumanPSD database http://www.incyte.com/documents/HumanPSD_Brochure.pdf
- Bateman A, Birney E, Cerruti L, Durbin R, Etwiller L, Eddy SR, Griffiths-Jones S, Howe KL, Marshall M, Sonnhammer EL. The Pfam protein families database. Nucleic Acids Res. 2002;30:276–280. doi: 10.1093/nar/30.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz J, Milpetz F, Bork P, Ponting CP. SMART, a simple modular architecture research tool: identification of signaling domains. Proc Natl Acad Sci USA. 1998;95:5857–5864. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Microsoft, Inc. http://www.microsoft.com/office/excel/default.asp
- Dennis G, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- Cui X, Churchill GA. Statistical tests for differential expression in cDNA microarray experiments. Genome Biol. 2003;4:210. doi: 10.1186/gb-2003-4-4-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efron B. Bootstrap methods: another look at the jackknife. Ann Statistics. 1979;7:1–26. [Google Scholar]
- Kayo T, Allison DB, Weindruch R, Prolla TA. Influences of aging and caloric restriction on the transcriptional profile of skeletal muscle from rhesus monkeys. Proc Natl Acad Sci USA. 2001;98:5093–5098. doi: 10.1073/pnas.081061898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CC, MacCoss MJ, Howell KE, Yates JR. A method for the comprehensive proteomic analysis of membrane proteins. Nat Biotechnol. 2003;21:532–538. doi: 10.1038/nbt819. [DOI] [PubMed] [Google Scholar]
- Gnatenko DV, Dunn JJ, McCorkle SR, Weissmann D, Perrotta PL, Bahou WF. Transcript profiling of human platelets using microarray and serial analysis of gene expression. Blood. 2003;101:2285–2293. doi: 10.1182/blood-2002-09-2797. [DOI] [PubMed] [Google Scholar]
- Li C, Wong WH. Model-based analysis of oligonucleotide arrays: Expression index computation and outlier detection. Proc Natl Acad Sci USA. 2001;98:31–36. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidorov IA, Hosack DA, Gee D, Yang J, Cam MC, Lempicki RA, Dimitrov DS. Oligonucleotide microarray data distribution and normalization. Inform Sci. 2002;146:65–71. [Google Scholar]
- Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doniger SW, Salomonis N, Dahlquist KD, Vranizan K, Lawlor SC, Conklin BR. MAPPFinder: using Gene Ontology and GenMAPP to create a global gene-expression profile from microarray data. Genome Biol. 2003;4:R7. doi: 10.1186/gb-2003-4-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeeberg BR, Feng W, Wang G, Wang MD, Fojo AT, Sunshine M, Narasimhan S, Kane DW, Reinhold WC, Lababidi S, et al. GoMiner: a resource for biological interpretation of genomic and proteomic data. Genome Biol. 2003;4:R28. doi: 10.1186/gb-2003-4-4-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlquist KD, Salomonis N, Vranizan K, Lawlor SC, Conklin BR. GenMAPP, a new tool for viewing and analyzing microarray data on biological pathways. Nat Genet. 2002;31:19–20. doi: 10.1038/ng0502-19. [DOI] [PubMed] [Google Scholar]
- Cicala C, Arthos J, Selig SM, Dennis G, Jr, Hosack DA, Van Ryk D, Spangler ML, Steenbeke TD, Khazanie P, Gupta N, et al. HIV envelope induces a cascade of cell signals in non-proliferating target cells that favor virus replication. Proc Natl Acad Sci USA. 2002;99:9380–9385. doi: 10.1073/pnas.142287999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun TW, Justement JS, Lempicki RA, Yang J, Dennis G, Jr, Hallahan CW, Sanford C, Pandya P, Liu S, McLaughlin M, et al. Gene expression and viral production in latently infected, resting CD4+ T cells in viremic versus aviremic HIV-infected individuals. Proc Natl Acad Sci USA. 2003;100:1908–1913. doi: 10.1073/pnas.0437640100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAVID: download EASE http://david.niaid.nih.gov/david/ease.htm
- DAVID: Database for Annotation, Visualization, and Integrated Discovery http://david.niaid.nih.gov [PubMed]