

Abstract
Maternal immune activation (MIA) is a risk factor for the development of schizophrenia and autism. Infections during pregnancy activate the mother’s immune system and alter the fetal environment, with consequential effects on CNS function and behavior in the offspring, but the cellular and molecular links between infection-induced altered fetal development and risk for neuropsychiatric disorders are unknown. We investigated the immunological, molecular, and behavioral effects of MIA in the offspring of pregnant Sprague-Dawley rats given an intraperitoneal (0.25 mg/kg) injection of lipopolysaccharide (LPS) on gestational day 15. LPS significantly elevated pro-inflammatory cytokine levels in maternal serum, amniotic fluid, and fetal brain at 4 h, and levels decreased but remained elevated at 24 h. Offspring born to LPS-treated dams exhibited reduced social preference and exploration behaviors as juveniles and young adults. Whole genome microarray analysis of the fetal brain at 4 h post maternal LPS was performed to elucidate the possible molecular mechanisms by which MIA affects the fetal brain. We observed dysregulation of 3,285 genes in restricted functional categories, with increased mRNA expression of cellular stress and cell death genes and reduced expression of developmentally-regulated and brain-specific genes, specifically those that regulate neuronal migration of GABAergic interneurons, including the Distal-less (Dlx) family of transcription factors required for tangential migration from progenitor pools within the ganglionic eminences into the cerebral cortex. Our results provide a novel mechanism by which MIA induces the widespread down-regulation of critical neurodevelopmental genes, including those previously associated with autism.
Keywords: Maternal immune activation, Inflammation, Cytokine, Lipopolysaccharide, Schizophrenia, Autism, Microarray, Bioinformatics, Cortical interneurons, GABA
1. Introduction
There is growing evidence that the etiology of a number of neuropsychiatric disorders has its roots in the prenatal environment such that insults to the fetus during critical times of development may alter fetal brain structure and function thereby acting as risk factors for disorders later in life (Bilbo and Schwarz, 2009; Patterson, 2002; Schlotz and Phillips, 2009). Maternal infections of both viral and bacterial origin have been linked with later development in the offspring of psychiatric disorders including autism (Atladottir et al., 2010; Ciaranello and Ciaranello, 1995) and schizophrenia (Brown et al., 2004). The maternal immune activation (MIA) animal model (Meyer et al., 2009b) is used to study how an insult directed at the maternal host can have adverse effects on the fetus leading to behavioral changes later in life, specifically within the domains of sensorimotor function, exploration, and social behaviors (Meyer et al., 2009a; Nouel et al., 2011).
Whereas the exact mechanisms by which MIA disrupts fetal brain development resulting in characteristic behavioral changes remain unclear, it is believed that the induction of the maternal cytokine response and the cascade of downstream cellular and molecular events are responsible for the altered brain development and increased risk for atypical developmental outcomes (Jonakait, 2007). Cytokines are a class of secreted polypeptides that have diverse functions (Benveniste, 1998; Patterson and Nawa, 1993; Rothwell and Hopkins, 1995). Although cytokines were first described in terms of their role in the regulation of the inflammatory immune response, there is evidence, primarily from in vitro studies, that they modulate normal developmental processes such as glial and neuronal commitment, proliferation, differentiation and survival, neurite outgrowth, and synaptogenesis (Deverman and Patterson, 2009). In the MIA model, maternally generated cytokines are thought to penetrate the placental barrier to gain access to the fetus (Dahlgren et al., 2006; Nawa and Takei, 2006). In addition, the placenta itself reacts strongly to MIA both morphologically and immunologically and likely contributes to fetal distress (Abrahams and Geschwind, 2008; Carpentier et al., 2011; Fatemi et al., 2011; Girard et al., 2010; Hauguel-de Mouzon and Guerre-Millo, 2006; Hsiao and Patterson, 2011). Given the ability for cytokines to affect CNS developmental processes, the presence of inflammatory molecules in the fetal brain may adversely affect the survival of neuronal subpopulations (Araujo and Cotman, 1995; Jarskog et al., 1997; Keohane et al.; Marx et al., 2001; Neumann et al., 2002; Qiu et al., 1998; Zhao et al., 2001).
Rodent models have been developed to determine the behavioral and biological effects of MIA on offspring (for review (Boksa, 2010; Meyer et al., 2009a)). In these studies, lipopolysaccharide (LPS) or the synthetic, double-stranded RNA polyriboinosinic–polyribocytidilic acid (polyI:C) are administered to pregnant dams to mimic the immunestimulating actions of live bacterial or viral infections, respectively. Both agents trigger similar immune responses in the dam. We used LPS, a large lipid-polysaccharide complex released from the outer cell wall of gram-negative bacteria that binds to the toll-like receptor 4 (TLR4) expressed on the surface of certain cell types. The resulting activation of the transcription factor NF-κB induces pro-inflammatory cytokine expression (Palsson-McDermott and O'Neill, 2004). In the MIA model, the maternal LPS itself is not detected in the fetus (Ashdown et al., 2006). Rather, the maternal LPS increases pro-inflammatory cytokine mRNA expression and/or proteins in the maternal serum, amniotic fluid, and placenta (Ashdown et al., 2006; Cai et al., 2000; Gayle et al., 2004; Girard et al., 2010; Golan et al., 2005; Liverman et al., 2006; Ning et al., 2008; Urakubo et al., 2001). Whether or not these cytokines are produced locally within the fetal brain is less clear. Increased cytokine gene expression by fetal brain has not been shown in the maternal LPS model except in some studies that used a much higher dose of LPS than what was used in our study (Cai et al., 2000; Lasala and Zhou, 2007). Little is known about the acute biological effects of maternal LPS on the fetal brain aside from one report of maternal LPS-induced gene changes in the fetal brain (Liverman et al., 2006) and a recent report showing some structural changes and perturbed neural development in rat cerebral cortex at gestational day 18 following LPS administered maternally at gestational days 15 and 16 (Ghiani et al., 2011). Thus, although it has been demonstrated that MIA alters behavioral and biological outcomes in juvenile and adult offspring, the mechanisms by which this immune reaction ultimately disrupt brain and behavioral development remain unclear (Boksa, 2010). The purpose of the following study was to determine the effects of maternal LPS exposure on the profiles of MIA-induced cytokine levels, fetal gene expression, and social and investigative behavior of offspring in order to better understand the mechanisms by which MIA is a risk factor for atypical offspring development.
2. Materials and methods
2.1. Animals
Male and female Sprague-Dawley rats (Harlan, Indianapolis, IN) were housed separately until mating. Daily vaginal smears were collected to monitor the estrous cycle of the females, which underwent a single night of in-house mating. The presence of a sperm plug the following morning represented gestational day 0. Weights were obtained to confirm pregnancy. Offspring (control n = 17; LPS n = 17) used for behavioral testing were weaned on postnatal day 21 and separated into same-sex housing. Testing of male (control n = 6; LPS n = 9) and female (control n = 11; LPS n = 8) rats from multiple independent litters began on postnatal day 24 and concluded on postnatal 55. All animals were housed under standard light conditions (12 h light/12 h dark with lights on between 6 a.m. and 6 p.m.). All behavioral tests were done during lights-on periods. Food and water were available ad lib. All experimental protocols were approved by the NIMH Intramural Research Program Animal Care and Use Committee.
2.2. Maternal Immune Activation
Pregnant rats received an intraperitoneal injection of either Escherichia coli 05:B55 lipopolysaccharide (LPS; 0.25 mg/kg; Sigma, St. Louis, MO), diluted (1 mg/ml) in phosphate-buffered saline (PBS), or equivalent volume of PBS vehicle on gestational day 15. This dose was chosen based on our initial pilot work to determine a dose that still leads to postnatal behavioral changes in the offspring while minimizing spontaneous abortions or stillbirths commonly observed following maternal LPS treatment. Dams were monitored daily following the injection for signs of vaginal bleeding, weight loss, or sickness behavior, and following birth, proper pup retrieval and feeding behavior were assessed. Pups were assessed for general health by non-invasive visual inspection.
2.3. Maternal serum, amniotic fluid, and fetal brain collection
Four or 24 hours following maternal LPS or saline injections, pregnant dams were anesthetized using isoflurane and decapitated. Whole trunk blood was collected from decapitated dams in a sterile 1.5 ml polypropylene Eppendorf tubes and allowed to clot at room temperature for 30 min. Samples were then centrifuged in a refrigerated centrifuge at 4°C for 15 min at 2,000 × g. The resulting supernatant (serum) was collected via Pasteur pipette and transferred into a new Eppendorf tube and stored at −80°C until analysis. A midline incision was made into the peritoneal cavity to expose the embryos. Embryos were removed and placed in cold PBS. Amniotic fluid was removed from each amniotic sac using a 1 ml syringe with a 23g needle and collected in 0.2 ml certified PCR tubes. Fetal brains were removed and placed either in empty PCR-certified, DNase- and RNase-free 1.5 ml Eppendorf tubes for immunoaffinity capillary electrophoresis cytokine analysis, or 1.5 ml Eppendorf tubes containing 1 ml TRIzol (Invitrogen, USA) for gene expression studies. Maternal serum, amniotic fluid, and fetal brains were collected from multiple dams at the two post-injection time points for protein and gene expression studies. Samples were collected such that approximately half of each litter was used for cytokine protein work and the remaining half was used for microarray and qualitative real-time polymerase chain reaction (qRT-PCR) work, which allowed for the use of matched fetal littermates across the studies. This was possible given the large litter size of the Sprague-Dawley rat. For the 4 h analysis, serum was collected from 7 control dams and 7 LPS dams. Amniotic fluid (control n = 28; LPS n = 26) and fetal brains (control n = 19 for protein and n = 10 for gene expression; LPS n = 25 for protein and n = 9 for gene expression) were collected across multiple dams. For the 24 h analysis, serum was collected from 6 control dams and 6 LPS dams and amniotic fluid (control n = 24; LPS n = 20) and fetal brain (control n = 11 for protein and n = 10 for gene expression; LPS n = 10 for protein and n = 10 for gene expression) were collected across multiple dams.
2.4. Immunoaffinity Capillary Electrophoresis
Maternal serum, amniotic fluid, and fetal brain samples were analyzed using chip-based immunoaffinity capillary electrophoresis with laser-induced fluorescence detection, as previously described (Phillips and Wellner, 2006, 2007), to measure TNF-α, IL-6, and IL-1β. Briefly, primary antibodies (R&D Systems, Minneapolis, MN) were reconstituted to stock solutions of 1 µg/ml in 0.1M phosphate buffer, pH 7.4 and antibodies were reduced to F(Ab)’2 fragments using a Pierce Biotechnology F(Ab)’2 digestion kit according to the manufacturer’s instructions and further reduced to FAb fragments by incubation with 200 mM DTT solution. Equal parts of each digested FAb fragments were mixed and immobilized onto a 2-mm thiolderivatized glass disk. The disk was placed in the immunoaffinity port of a lab-on-a-chip and 200 nl of sample added to the port. The sample was allowed to reside in the immunoaffinity port for 3 min, during which time the immobilized FAb fragments captured their respective antigens. The sample was removed and the disk washed with 0.5 µl of 0.1 M phosphate buffer. AlexaFluor 633 laser dye (Molecular Probes, Eugene, OR, USA) was introduced into the immunoaffinity port, labeling the bound antigens. This reaction was allowed to take place for 2 min before again washing the port with 0.5 µl of phosphate buffer. Finally, the bound antigens were eluted from the FAbs by introducing 0.5 µl of acid buffer (0.1 M phosphate buffer titrated with 1N HCl to pH 1.0) and electrokinetically moving the released antigens into the separation channel where they were separated by applying a potential of 6 Kv, and the individual peaks measured in-channel by the fluorescence detector. The concentration of each separated peak was calculated from calibration curves constructed from known standards of each antigen run under identical conditions. Protein levels for serum and amniotic fluid were expressed as pg/ml fluid and as pg/µg extracted protein for fetal brain. All samples were run blind to the condition.
2.5. Microarray
For each time point, 4 h and 24 h post maternal injection, samples (4 h: n = 10 control, n = 9 LPS; 24 h: n = 10 control, n = 10 LPS) were prepared according to Affymetrix protocols (Affymetrix, Inc.). RNA quality and quantity was ensured using the Bioanalyzer (Agilent, Inc.) and NanoDrop respectively. Per RNA labeling, 200 ng of total RNA was used in conjunction with the Affymetrix recommended protocol for the GeneChip 1.0 ST chips. The hybridization cocktail containing the fragmented and labeled cDNAs was hybridized to the Affymetrix Rat GeneChip® 1.0 ST chips. The chips were washed and stained by the Affymetrix Fluidics Station using the standard format and protocols as described by Affymetrix. The probe arrays were stained with streptavidin phycoerythrin solution (Molecular Probes, Carlsbad, CA) and enhanced by using an antibody solution containing 0.5 mg/ml of biotinylated anti-streptavidin (Vector Laboratories, Burlingame, CA). An Affymetrix Gene Chip Scanner 3000 was used in conjunction with Affymetrix GeneChip Operation Software to generate probe-level data for 29,215 rat gene fragments per hybridized cRNA. Cel files generated by the Affymetrix AGCC program were imported into the Affymetrix Expression Consol program and RMA (Robust Multichips Analysis) normalization was performed to generate the .Chp files. The statistical programming language R was used (http://cran.r-project.org/). RMA (http://www.bioconductor.org/) was employed for probe-level data summarization and normalization. Data quality was assessed by visual inspection by Tukey box plot, covariance-based Principal Component Analysis (PCA), scatter plot, and correlation-based Heat Map (Supplementary Fig. 1). System noise was defined as the lowest observed mean data value at which the LOWESS fit of the normalized data (CV~mean) changes from non-linear to linear. Gene fragments not having at least one data value greater than system noise were discarded. Remaining data were floored to system noise and subjected to the Welch-modified t-test on a gene fragment by gene fragment basis with a Benjamin-Hochberg correction and false discovery rate of p < 0.05 and fold change threshold ≥ 1.3. Enriched biological functions for the selected gene fragments were determined using Ingenuity Pathways Analysis (IPA; Ingenuity, Inc.). Supervised analyses were conducted using separate gene fragment lists based on regulation direction (up versus down-regulation).
IPA is an accepted bioinformatics tool used to analyze microarray data. Of several tools available for mining array data, IPA uses a high percentage of curation by expert manual review (approximately 90% versus 10% natural language mining) of published literature and additional sources of information (see http://www.ingenuity.com/science/knowledge_base.html).
2.6. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Quantitative RT-PCR was performed on a subset of the samples (n = 7 control, n = 7 LPS) from the 4 h time point to validate selected candidate genes from microarray data. Total RNA was isolated using the RNeasy Mini Kit that includes reagents and columns (Qiagen, Valencia, CA) according to the manufacturer’s protocol. Briefly, 200 µl chloroform was added to homogenized tissue and centrifuged for 10 min at 10,000 rpm. The aqueous solution was transferred to a fresh Eppendorf tube and gently vortexed while 200 µl of ethanol was added. Solution was transferred to a Qiagen column and centrifuged for 5 min at 3,500 rpm. Liquid was transferred back to column and spin was repeated. Liquid was discarded and 750 µl RW1 buffer was added to the column and centrifuged for 5 min at 3,500 rpm. Liquid was discarded and 500 µl RPE buffer was added to the column and centrifuged for 10 min at 3,500 rpm. The liquid was discarded and this step was repeated. The column was placed in a new Eppendorf tube and centrifuged for 1 min at max speed to remove excess liquid. The column was placed in a collection tube and 30 µl DEPC-treated water was added and centrifuged for 5 min at 5,000 rpm. Total RNA was quantified on a spectrophotometer (NanoDrop, Thermo Scientific, USA) and reverse transcribed using a Superscript III First Strand cDNA Synthesis Kit (Invitrogen, USA). Two-step real-time qRT-PCR with 2× SYBR Green Master Mix (Bio-Rad, Hercules, CA) was performed using the Bio-Rad iQ5 iCycler. The endogenous -tubulin gene was used to normalize quantification of the mRNA target. All primers were validated and sequenced prior to this experiment and are shown in Table 1. Fold differences in gene expression were calculated using the ΔΔct method.
Table 1.
Primer Sequences used for qRT-PCR validation of selected genes from 4 h microarray
| Gene | Forward (5’-3’) | Reverse (5'-3') |
|---|---|---|
| APOLD1 | CATCGTCTTCTTCGGCTCGC | GGAGTTCCTGAGGTTGTGACCAC |
| AUT2 | ACGAGAAGCCCTTACCAATG | GCAGAGATTGCGGTCATTTT |
| BNIP3 | AAAGGGGGTATTTTCTCAGCA | ACGCCTTCCAATGTAGATCC |
| β-TUB | GTCGGACACTGTGGTGGAGC | TGGTCAGAGGTGCGAAGCC |
| CASP8 | CAAAGCACGGGAGGACATAC | GCTGTGCAATCACTGAAGGA |
| CAV1 | TGAAGATGTGATTGCGGAAC | CAGCAAGCGGTAAAACCAAT |
| CELSR3 | ACAGCCTCTGCACTTGGAC | AGGAGTGACCCTCGCTTCTG |
| DLX1 | TACCTAGCTCTGCCTGAGAGG | TCTTGAACTTGGAGCGTTTGT |
| DLX2 | AGTATCTGGCCCTGCCTGAG | ATCTCGCCACTCTTCCACATC |
| DLX5 | ACTGACGCAAACACAGGTGA | GCGAGTTACACGCCATAGG |
| ERAF | TCTTTGATGACCCTCTCATGC | CTCTTTCAGAGCTCCGTCTTG |
| GABBR1 | CAAGAGCGTGTCCACTGAAA | TGGAAAGGATCATGGTCACA |
| GPC2 | TACTTCGAGTCCCGATGTCC | ATCATGCATGTCAAGGTCCTG |
| IL-1β | CTGTGACTCGTGGGATGATG | AGACAGCACGAGGCATTTTT |
| IL-6 | TACCCCAACTTCCAATGCTC | GGTTTGCCGAGTAGACCTCA |
| MECP2 | GGACGCGAAAGCTTAAACAG | AGGAGGTGTCTCCCACCTTT |
| NAV1 | CCGGAGGATAGGACAGTCAA | GTCTCTCGATCTGGCGACTC |
| NEFM | GAGATCGCCGCATATAGGAA | AGGGCTGTCGGTGTGTGTA |
| PCADGB6 | TGAAGTGTAGCGCACCTTTG | CGGTGTTAGCGTCTCAGGAT |
| PLXNA2 | GCCACGGAGTTCAATATGCT | ACCTTGTAGGCCAGTCGTTG |
| PLXNA3 | CTCCATGCCAATGACTTCAA | AAGAGGCATCTCGGTCCAG |
| PLXN4A | CGAACAAGCTGCTCTACGC | TACGCGTTCATGTCCTGGT |
| POU3F4 | TTTCCTCAAGTGTCCCAAGC | CCTGGCGGAGTCATTCTTT |
| PDCD10 | AATGCAGAGGAAGCGTCATT | CTCCAAAGGTGGGAAGTGAA |
| SEMA4C | GCTGCAGGTGTTTGACCAG | CAGCGACAGCTGAACCAGT |
| TNFα | TCTCAAAACTCGAGTGACAAGC | GGTTGTCTTTGAGATCCATGC |
2.7. Behavioral Testing
Social Preference
Following the protocol established by (Walker et al., 2007) one day prior to testing, rats were brought into the testing room and placed in the testing apparatus for a 15-min habituation period. Social interaction was measured in each animal on postnatal 25, 30, 35, 45, and 55 using a three-chambered apparatus (Habitest, Coulbourn Instruments, Whitehall, PA) in which preference for home cage territory or an unknown stimulus rat of the same sex was examined over the course of a 15-min testing session. The three chambers included a central start box, a side home box, and side social box, each measuring 20.5 × 10.5 × 12.2 cm. The home box contained a tray with bedding from the home cage of the test subject, and the social box contained a same-sexed, same-aged, novel stimulus rat. Once the test subject was placed in the start box, the guillotine doors, which separated the start box from the side boxes, were removed allowing for investigation of the side boxes. Entry from the start box into the social and home boxes was measured by the breakage of photobeams that surround the chambers. The number of entries into the two chambers and the total time spent in each of the three chambers were recorded and analyzed by the GraphicState, Coulbourn Instruments computer program. Each chamber was cleaned with 70% ethanol between each run. Novel stimulus rats were used on the five testing dates. So that effective comparisons could be made between groups, a measure of social preference was established, hereafter called the “social index” (S.I.). The S.I. was derived according to the following formula: (S-H)/900, where S represents the time spent in the social chamber, H represents time spent in the home chamber, and 900 represents the total test time (sec).
Exploration
One day prior to testing, rats were brought into the testing room and placed in the testing apparatus for a 15-min habituation period. Exploratory behaviors were measured in each animal on postnatal day 25, 30, 35, 45, and 55. Subjects were placed in a box (40 × 40 × 40 cm) with 16 holes, 1 cm in diameter that are cupped at the bottom and filled with fresh bedding. The apparatus contained two rows of photocells arranged around the perimeter of the chamber to record locomotion along the plane of the chamber floor and nose-hole entries below the floor. The total numbers of movements and nose-hole pokes and the total times of movement, non-locomotor behavior, and ambulation (total movement time - non-locomotor time/60) were measured by the TruScan software. Exploration was operationally defined as the number of nose-hole pokes per minute of ambulation during a 10-min observation period (Walker et al., 2007). This allows for the separation of spontaneous movements from motivated exploration. The experimental box was thoroughly cleaned with 70% ethanol between testing sessions.
Olfaction
Basic olfactory skills (the ability to recover a hidden piece of food) were assessed on postnatal day 40 following the Hidden Cookie Task established by (Yang and Crawley, 2009). To familiarize the rats with the olfactory stimulus, a small cookie (honey-flavored Teddy Graham, Nabisco) was placed in the home cages overnight for three consecutive days (postnatal day 36–38). A fresh cookie was placed each night. On the fourth night (postnatal day 39), food pellets were removed from the chow hopper overnight for a single night of food deprivation. The next morning, testing began by placing the subject in a clean rat cage with 3 cm-deep fresh bedding for a 5-min acclimation period. The subject was then transferred to an empty clean cage while the experimenter buried a cookie 1 cm beneath the surface in a random corner of the cage. The subject was immediately returned to the testing cage, and latency to find the cookie was recorded. A cut-off latency time of 600 sec was used. A new, clean testing cage with fresh bedding and a new cookie were used for each subject, and the chow hoppers were filled with food pellets immediately following task completion.
2.10. Statistical Analyses
All data other than the social preference data were normally distributed. Therefore, for the social data, log10 transformed S.I. scores were analyzed using the proper statistical approach. Social and exploration behaviors were analyzed using a 2-way mixed ANOVA design with Student Newman-Keuls post hoc test. Olfaction data were analyzed using pair-wise Student’s t tests. Cytokine data were analyzed using a 2-way between subjects MANOVA design with treatment and time as independent variables and the proteins as multiple dependent variables. The three compartments (serum, amniotic fluid, and brain) were analyzed separately. Effects were considered statistically significant when p < 0.05. Statistical significance for changes in relative gene expression (qRT-PCR) was determined by using an independent samples Student’s t-tests comparing the mean Δct values for control and LPS groups for each target gene. Statistical significance was defined as p < 0.05. Gene expression data are presented as fold changes. Statistical analyses were performed using SPSS 17.0 software.
3. Results
3.1. Maternal LPS treatment increases cytokines in maternal serum
A 2(treatment × time) between subjects MANOVA revealed a significant main effect of treatment (F(3,19) = 429.76, p < 0.001) and time (F(3,19) = 137.19, p < 0.001), which was qualified by a treatment × time interaction (F(3,19) = 137.13, p < 0.001) for the pro-inflammatory cytokines. Dams exposed to LPS had significantly greater levels of TNFα, IL-1β, and IL-6 than control dams at both 4 h and 24 h post injection; however, the expression at 24 h was significantly less than expression at 4 h (Fig. 1A, D, G).
Figure 1.

Maternal LPS elevates cytokine levels in maternal fluids and fetal brain. Pro-inflammatory cytokines in maternal serum (4 h: control n = 7; LPS n = 7 and 24 h: control n = 6; LPS n = 6), amniotic fluid (4 h: control n = 28; LPS n = 26 and 24 h: control n = 24; LPS n = 20), and fetal brain (4 h control n = 19; LPS n = 25 and 24 h: control n = 11; LPS n = 10) at 4 and 24 h post maternal LPS (0.25 mg/kg) or saline administration on gestational day 15. Immunoaffinity electrophoresis analysis of fluid and tissue samples shows that cytokine protein levels are significantly elevated across the three maternal-fetal compartments at both time-points in offspring born to LPS-treated dams. Results are expressed as means ± SEM whereby * represents comparisons between LPS and control, p < 0.05 and # represents comparisons between LPS across time, p < 0.05 (2-way between subjects MANOVA).
3.2. Maternal LPS treatment increases cytokines in amniotic fluid
Cytokine protein levels in amniotic fluid paralleled the levels measured in serum but with a reduced magnitude. Levels in amniotic fluid were approximately half those in serum. For the pro-inflammatory cytokines, a 2(treatment × time) between subjects MANOVA revealed a significant main effect of treatment (F(3,92) = 443.48, p < 0.001) and time (F(3,92) = 194.51, p < 0.001), which was qualified by a treatment × time interaction (F(3,92) = 194.15, p < 0.001). Fluid harvested from embryos belonging to dams exposed to LPS had significantly greater levels of TNFα, IL-1β, and IL-6 than control amniotic fluid at both 4 h and 24 h post injection. However, the expression at 24 h was significantly less than expression at 4 h (Fig. 1B, E, H).
3.3. Maternal LPS treatment increases cytokines in fetal brain
The pattern of pro-inflammatory cytokine protein levels in fetal brain matched the respective patterns of these proteins in serum and amniotic fluid, albeit with a greatly reduced relative magnitude. Levels in brain were approximately one-tenth those in amniotic fluid. A 2(treatment × time) between subjects MANOVA revealed a significant main effect of treatment (F(3,59) = 305.53, p < 0.001) and time (F(3,59) = 68.78, p < 0.001), which was qualified by a treatment × time interaction (F(3,59) = 68.47, p < 0.001). Fetal brains from LPS-exposed dams had significantly greater levels of TNFα, IL-1β, and IL-6 than control fetal brains at both 4 h and 24 h post injection. However, the levels at 24 h were significantly lower than at 4 h (Fig. 1C, F, I).
3.4. Maternal immune activation dysregulates gene expression in fetal brain at 4 h post maternal LPS treatment
To investigate molecular changes in fetal brain following maternal LPS treatment, we conducted a microarray experiment with follow-up qRT-PCR validation at 4 h post LPS and in a separate experiment (data not shown) with different samples again at 24 h post LPS to examine gene expression changes in fetal brain in response to MIA. The raw microarray data files were deposited in the NCBI Gene Expression Omnibus at http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=djqzbyaakgsgoxw&acc=GSE34058.
At the 24 h time point, no sample outliers were present, and no gene fragments were found to have a FDR multiple comparison-corrected p < 0.05. Inspection of effect sizes between control and LPS fragments by rotated volcano plot revealed low to no differences between them. To validate the 24 h microarray data, qRT-PCR was performed on a subset of the most down-regulated genes from the 4 h time point to ensure that they were not missed on the 24 h microarray. There were no changes in gene expression between groups, further supporting the transient nature of the gene changes reported at 4 h (data not shown).
At the 4 h time point, 29,215 gene fragment expression measurements were generated per sample; no sample outliers were observed (Supplementary Fig. 1). Measurement data for 8,161 gene fragments were discarded as noise, leaving 21,054 gene fragments for significance testing. Using a fold change threshold of ≥ 1.3, a total of 3,285 genes differed significantly in their expression levels between LPS (n = 9) and control (n = 10) groups at 4 h post treatment (Supplementary Table 1). Of the 3,285 gene fragments, 2,192 were up-regulated and 1,093 were down-regulated. The top 5 most up-regulated genes in terms of fold change were: apolipoprotein L domain containing 1 (2.92), alpha-hemoglobin stabilizing protein (2.77), arrestin domain containing 3 (2.47), hemoglobin gamma A (2.31), and BCL2/adenovirus E1B 19kDa interacting protein (2.28). The top 5 most down-regulated genes were: ischemia related factor vof 16 (−3.56), distal-less homeobox 5 (−2.02), ribosomal protein L7a (−1.89), distal-less homeobox 1 (− 1.82), and autism susceptibility candidate 2 (−1.82). Using Ingenuity Pathways Analysis software, the 3,285 genes were segregated by regulation direction and underwent a supervised analysis to determine significantly enriched biological functions for the up-regulated (2,192) versus down-regulated (1,093) gene fragments. There were 73 and 71 significantly enriched functions for the up-regulated and down-regulated genes, respectively. “Cell cycle” represented the most enriched function for the up-regulated gene set (Table 2). “Nervous system development and function” represented the most enriched function for the down-regulated gene set, and the specific categories and numbers of genes per category within that particular function are shown in Table 3.
Table 2.
Top 10 enriched biological functions determined by Ingenuity Pathways Analysis (IPA) for all up-regulated genes at 4 h post maternal LPS (0.25 mg/kg) administration.
| Significance −log(p-value) |
# Genes |
IPA Enriched Function |
|---|---|---|
| 11.38 | 190 | Cell cycle |
| 11.38 | 167 | Cellular assembly and organization |
| 11.38 | 163 | DNA replication, recombination, repair |
| 11.02 | 156 | Protein synthesis |
| 6.63 | 36 | Gene expression |
| 6.37 | 433 | Cancer |
| 6.22 | 176 | Infectious disease |
| 5.64 | 54 | RNA post-transcriptional modification |
| 5.29 | 389 | Neurological disease |
| 5.22 | 165 | Gastrointestinal disease |
Table 3.
Top 10 enriched biological functions and corresponding “nervous system development and function” subcategories determined by Ingenuity Pathways Analysis (IPA) for all down-regulated genes at 4 h post maternal LPS (0.25 mg/kg) administration.
| Signifiance −log(p- value) |
# Genes |
IPA Enriched Function | Category | # Genes |
|
|---|---|---|---|---|---|
| 13.31 | 161 | Nervous system development/function |  |
Neurogenesis | 78 |
| 10.59 | 79 | Cell-cell signaling | Guidance of neurites | 47 | |
| 9.30 | 134 | Tissue development | Neurological process of cells | 34 | |
| 8.80 | 124 | Cellular development | Differentiation of neurons | 32 | |
| 8.71 | 140 | Gene expression | Growth of neurites | 31 | |
| 8.46 | 222 | Neurological disease | Development of brain | 29 | |
| 7.27 | 105 | Psychological disorders | Neurotransmission | 27 | |
| 6.67 | 126 | Cellular assembly/organization | Neurite outgrowth | 23 | |
| 5.77 | 329 | Genetic disorder | Migration of neurons | 20 | |
| 5.73 | 55 | Cellular movement | Quantity of neurons | 18 |
The function categories generated by bioinformatics analysis tools like IPA generate lists of genes that appear redundantly in multiple categories. Most genes lack dedicated and exclusive functions, and thus fall into more than one function or functional category. To better estimate the contributions of redundant versus exclusive genes, we further analyzed the data to determine the numbers of uniquely mapped genes per function. Within each enriched function and subcategory between 0–34% of the total number of mapped genes were unique to that specific function, and on average 11% of the total number of mapped genes were unique (Supplementary Table 1).
Examples of significantly up-regulated genes are shown in Table 4. Included in this domain are genes associated with cellular stress and cell death pathways. Examples of significantly down-regulated genes are shown in Table 5. These genes reside in categories such as neurogenesis, neurotransmission, migration of neurons, and neurite outgrowth. Remarkably, down-regulated genes that showed some of the largest fold changes in the microarray included three Distal-less (Dlx) genes and several others related to migration of cortical interneurons. The direction and magnitude of all changes were confirmed by qRT-PCR analysis for a selected subset of up and down-regulated genes.
Table 4.
Selected up-regulated genes from 4 h microarray ranked by fold change (FC)
| Gene Name | Gene Symbol |
Array FC |
Array p-value |
qRT- PCR FC |
qRT-PCR p-value |
|---|---|---|---|---|---|
| Top 5 up-regulated genes | |||||
| Apolipoprotein L domain containing 1 | Apold1 | 2.92 | < 0.001 | 6.51 | 0.001 |
| Alpha-hemoglobin stabilizing protein | Ahsp | 2.77 | < 0.001 | 4.62 | 0.002 |
| Arrestin domain containing 3 | Arrdc3 | 2.47 | < 0.0003 | ||
| Hemoglobin, gamma A | Hbg1 | 2.31 | < 0.0003 | ||
| BCL2/adenovirus E1B 19KDa interacting protein 3 | Bnip3 | 2.28 | < 0.001 | 5.16 | 0.001 |
| Additional up-regulated genes | |||||
| Caveolin 1 | Cac1 | 2.11 | < 0.001 | 4.83 | 0.005 |
| Programmed cell death 10 | Pdcd10 | 1.81 | < 0.001 | 3.13 | 0.003 |
| Caspase 8 | Casp8 | 1.53 | < 0.001 | 2.57 | 0.001 |
| Apoptosis-inducing factor 1 | Siva1 | 1.47 | < 0.001 | ||
| Programmed cell death 6 | Pdcd6 | 1.45 | < 0.001 | ||
| Defender against cell death | Dad1 | 1.44 | < 0.002 | ||
| Programmed cell death 5 | Pdcd5 | 1.39 | < 0.001 | ||
| Heat Shock 70kDa Protein 13 | Hspa13 | 1.37 | < 0.001 | ||
| Heat Shock 10kDa protein 1 | Hspe1 | 1.33 | < 0.001 | ||
| Caspase 3 | Casp3 | 1.32 | < 0.001 | ||
| Heat Shock Transcription Factor 2 | Hsf2 | 1.32 | < 0.001 | ||
| Heat Shock 70kDa Protein 14 | Hspa14 | 1.31 | |||
Table 5.
Selected down-regulated genes from 4 h microarray ranked by fold change (FC)
| Gene Name | Gene Symbol |
Array FC |
Array p-value |
qRT- PCR FC |
qRT- PCR p-value |
|---|---|---|---|---|---|
| Top 5 down-regulated genes | |||||
| Ischemia related factor vof-16 | Vof16 | −3.56 | < 0.006 | ||
| Distal-less 5 | Dlx5 | −2.02 | < 0.0001 | −2.95 | 0.0378 |
| Ribosomal protein L7a | Rpl7a | −1.89 | < 0.0006 | ||
| Distal-less 1 | Dlx1 | −1.82 | < 0.0001 | −4.27 | 0.0001 |
| Autism susceptibility candidate 2 | Aut2 | −1.82 | < 0.0001 | −4.52 | 0.0035 |
| GABA-specific genes | |||||
| Distal-less 2 | Dlx2 | −1.74 | < 0.0001 | −4.67 | 0.0043 |
| Cadherin, EGF LAG seven-pass cadherin G-type receptor 3 | Celsr3 | −1.67 | < 0.0001 | −2.87 | 0.0024 |
| γ-aminobutyric acid (GABA) B receptor, 1 | Gabbr1 | −1.51 | < 0.0001 | −3.15 | 0.0064 |
| Distal-less 6 | Dlx6 | −1.43 | < 0.0001 | ||
| Mannosyl-glycoproteinb-1,6-N-acetyl-glucosaminyl Transferaseisozyme b | Mgat55B | −1.48 | < 0.0001 | ||
| LIM homeobox 6 | Lhx6 | −1.47 | < 0.0001 | ||
| Glutamate decarboxylse 2 (65 kDa) | Gad2 | −1.45 | < 0.0002 | ||
| Glutamate decarboxylse 1 (67 kDa) | Gad1 | −1.44 | < 0.0002 | ||
| Distal-less 6 | Dlx6 | −1.43 | < 0.0001 | ||
| Aristaless-related homeobox | Arx | −1.37 | < 0.0001 | ||
| Additional down-regulated genes | |||||
| Glypican 2 | Gpc2 | −1.76 | < 0.0001 | −2.00 | 0.0050 |
| Semaphorin 4C | Sema4C | −1.52 | < 0.0001 | −2.83 | 0.0027 |
| Semaphorin 6C | Sema6C | −1.46 | < 0.0001 | ||
| Semaphorin 6B | Sema6B | −1.35 | < 0.0001 | ||
| Plexin 4A | Plxn4A | −1.64 | < 0.0001 | −2.38 | 0.0056 |
| Plexin A3 | PlxnA2 | −1.56 | < 0.0001 | −5.07 | 0.0021 |
| Plexin A2 | PlxnA3 | −1.56 | < 0.0001 | −5.07 | 0.002 |
| Plexin A1 | PlxnA1 | −1.45 | < 0.0001 | ||
| Protocadherin GB6 | PcdhgB6 | −1.60 | < 0.0001 | −2.74 | 0.0024 |
| Protocadherin 8 | Pcadh8 | −1.40 | < 0.0001 | ||
| Protocadherin 10 | Pcadh10 | −1.32 | < 0.0004 | ||
| POU class 3 homeobox 4 | Pou3f4 | −1.51 | < 0.0001 | −1.87 | 0.0014 |
| POU class 3 homeobox 2 | Pou3f2 | −1.34 | < 0.0001 | ||
| POU class 3 homeobox 3 | Pou3f3 | −1.33 | < 0.0001 | ||
| Neurofilament medium polypeptide | Nefm | −1.64 | < 0.0001 | −3.31 | 0.0001 |
| Neuron navigator 1 | Nav1 | −1.55 | < 0.0001 | −2.14 | 0.0055 |
3.5. Maternal LPS treatment does not impact maternal care behaviors or result in sex differences in postnatal behavioral performance of the offspring
No differences in maternal care were observed between saline-treated and LPS-treated dams. Normal feeding was observed as evidenced by the presence of equal amounts of milk in the stomachs of each pup. The lack of maternal neglect supported the rationale to refrain from cross-fostering pups.
Statistical analyses revealed no significant main effects of sex or a sex × treatment interaction on any of the behavioral parameters of interest: social index main effect (F(1,30) = 1.60, p = N.S.; interaction F(1,30) = 0.63, p = N.S., total # chamber entries main effect (F(1,64) = 0.16, p = N.S.; interaction F(1,64) = 0.001, p = N.S.; exploration main effect (F(1,30) = 0.33 p = N.S.; interaction F(1,30) = 0.31, p = N.S., ambulation main effect (F(1,30) = 1.45, p = N.S., interaction F(1,30) = 0.89, p = N.S. Therefore, males and females were collapsed into a single group for behavioral analyses.
3.6. Postnatal social and exploration behaviors are altered in offspring born to LPS-treated dams
To test the effects of LPS-induced maternal immune activation on postnatal behavioral development, offspring underwent behavioral testing across multiple times beginning post-weaning at postnatal day 25 and concluding in young adulthood at postnatal day 55.
Social Preference
A 2(trial × treatment) mixed ANOVA design revealed a significant main effect of treatment (F(1, 32) = 36.52, p < 0.001), although the main effect of trial (F(4,128) = 1.75, p = N.S.) and the interaction between trial and treatment (F(4,128) = 0.96, p = N.S.) failed to reach significance (Fig. 2A). Student Newman-Keul’s post-hoc analyses showed that social preference as measured by the S.I. was significantly reduced on all trials (postnatal day 25, 30, 35, 45, and 55) in offspring born to LPS-exposed dams. Breaking down the S.I. into its individual components (time investigating the social and home chambers) allowed for a deeper understanding of the social behaviors in the two groups. Examination of time spent investigating the social chamber revealed a significant main effect of treatment (F(1,32) = 33.1, p < 0.001) and a trial × treatment interaction (F(4,178) = 3.17, p < 0.001). The main effect of trial failed to reach significance (F(4,128) = 1.4, p = N.S.). Post-hoc analyses showed that LPS offspring spent significantly less time relative to control offspring investigating the social chamber on all trials (postnatal day 25, 30, 35, 45, and 55). Examination of time spent investigating the home chamber revealed significant main effects for treatment (F(1,32) = 17.03, p < 0.001) and trial (F(4,128) = 4.50, p < 0.01). The trial × treatment interaction failed to reach significance (F(4,128) = 0.67, p = N.S.). Post-hoc analyses showed that the offspring born to LPS-exposed dams investigated the home chamber significantly more than the control offspring on the first four trials (postnatal day 25, 30, 35, and 45) with no difference in home chamber investigation time on the last trial (postnatal day 55).
Figure 2.

Postnatal social and exploration behaviors are altered in offspring born to dams maternally injected with LPS compared to saline. Dams were injected with 0.25 mg/kg LPS on gestational day 15. Offspring underwent behavioral testing on four separate testing dates beginning on postnatal day 25 and concluding on postnatal day 55. On the automated 3-chambered social preference task in which rats are given the choice to investigate a novel stimulus rat (social chamber), or a non-social, empty side (home chamber), offspring born to LPS-treated dams have reduced levels of social investigation across each testing date, reflected by their lower social index score (A), which measures the preference for investigation of the social vs. home chamber. LPS offspring also have reduced exploration as defined by number of nose-hole pokes/minute ambulation within an open field (C). These deficits are not due to overall locomotor defects or periods of immobility, which is reflected by the lack of differences in number of overall chamber (both social and home) entries (B) or ambulation in the open field (D). Results (n = 17 per group) are expressed as means ± SEM, and all comparisons are made between LPS treatment group and matched controls whereby *p < 0.05. (2-way mixed ANOVA followed by Student Newman-Keul’s post hoc test).
Total numbers of entries into the social and home chambers were also analyzed as a control measure to ensure that the differences in social preference were not driven by locomotor defects or freezing behaviors, which would be manifested as an overall reduction in the number of entries into both chambers. Collapsing the number of entries into the social and home chambers into a single measure of chamber entries revealed no main effect of trial (F(4,264) = 1.67 p = N.S.), treatment (F(1,66) = 0.45 p = N.S.) or trial × treatment interaction (F(4, 264) = 0.11 p = N.S.) (Fig. 2B) suggesting that reduced sociability it not a consequence of overall reduced motor exploration of the apparatus.
Exploration
A 2(trial × treatment) mixed ANOVA design revealed significant main effects of treatment (F(1,32) = 12.9, p < 0.01) and trial (F(4,128) = 7.8, p < 0.01) for the level of exploration as defined by number of nose-hole pokes/min ambulation (Fig. 2C). The trial × treatment interaction failed to reach significance (F(4,128) = 1.75, p = N.S.). Post-hoc analyses showed that the LPS offspring displayed a significant reduction in their motivated exploration (number of nose-hole pokes/min. ambulation) across the first four trials (postnatal day 25, 30, 35, and 45). Breaking down level of exploration into its component parts (number of nose-hole pokes and ambulation time) and analyzing them separately revealed significant main effects of treatment (F(1,32) = 16.19, p < 0.001) and trial (F(4,128) = 4.98, p < 0.001) for number of nose-hole pokes. The trial × treatment interaction failed to reach significance (F(4,128) = 1.75, p = N.S.). Post-hoc analyses showed that offspring born to LPS-exposed dams had significantly fewer nose-hole pokes on the first four trials (postnatal day 25, 30, 35, and 45) compared to controls (Fig. 2C). By postnatal day 55 the LPS offspring had a similar level of nose-hole poking to controls, which resulted in similar overall levels of exploration for that particular trial. There was a main effect of trial on total horizontal ambulation time (F(4,128) = 11.79 p < 0.001) but there were no differences in ambulation time between groups (F(1,32) = 1.11 p = N.S.) or a trial × treatment interaction F(4, 128) = .053 p = N.S.) (Fig. 2D) suggesting that the reduced exploration by the LPS group is driven by their lack of nose-hole poking, rather that freezing or motor impairments that would reduce their total ambulation time. In the hidden cookie task, no significant differences in latency to retrieve the buried food existed between offspring born to saline or LPS-treated dams (t(17) = 0.03, p = N.S.) demonstrating that basic olfaction is intact in the LPS offspring.
4. Discussion
We used the maternal LPS model whereby the bacterial endotoxin was administered to pregnant dams on gestational day 15, which approximates the second trimester of human gestation (Meyer et al., 2009a), to examine the immunological, molecular, and behavioral consequences of MIA. The MIA model was established to test the hypothesis that activation of the maternal immune system, specifically the pro-inflammatory cytokines, permanently alters fetal brain development and increases risk for neurodevelopmental disorders (Jarskog et al., 1997; Meyer et al., 2005). The downstream mechanisms by which pro-inflammatory cytokines in the fetal environment perturb brain development remain unknown, but blocking the actions of the specific cytokines such as IL-6 or IL-1βprevents MIA-induced behavioral and physiological consequences in mouse offspring, offering further support for the cytokine hypothesis (Girard et al., 2010; Smith et al., 2007). We showed substantial increases in pro-inflammatory cytokine protein levels in maternal serum, amniotic fluid, and fetal brain following maternal LPS treatment. The profile of induction was identical across these three compartments with the only difference being the induction magnitude. The greatest induction was present in the maternal serum followed by progressive decreases in magnitude in the amniotic fluid and fetal brains. The successive magnitude reductions from serum to brain could be indicative of the maternal-fetal protective barriers in place that filter out complete passage of cytokines, such that a fraction of cytokines is passing from serum to amniotic fluid to brain. Our microarray and qRT-PCR analysis are consistent with other published data (Gayle et al., 2004; Ning et al., 2008; Urakubo et al., 2001) that fail to show changes in TNFα, IL-1βor IL-6 mRNA expression in fetal brain at any time post-LPS expression despite showing increases in cytokine protein levels, suggesting that the increased cytokines in fetal brain are not fetally derived but likely transferred into the fetal brain from maternal serum, amniotic fluid, and the placenta. The lack of a fetal immune response may be explained by the immaturity of the resident immune cell in the brain, the microglial cell, in early gestation. Microglia populate the embryonic brain during a surge of infiltration from external sources such as the yolk sac at about the time of the LPS injection in our study (Rigato et al., 2011). It is possible that they are not mature enough to respond to an immune challenge by making more cytokines of their own. Indeed, in preliminary studies, using CX3CR1-GFP heterozygous reporter mice, we find no evidence of microglial activation after maternal LPS injection given at the equivalent gestational age of day 13 (unpublished data).
A recent report by Ghiani et al. (2011) showed that following maternal LPS administered twice, on gestational days 15 and 16 in rats, there was evidence of activated microglia and astrocytes in fetal cortex two days later. In addition, both mRNA and protein levels of several inflammatory cytokines were elevated in the time window of 4–72 h following the LPS. A possible explanation for a fetal brain inflammatory response in that paradigm, not seen in our paradigm, may be attributable to the double injection procedure used in that study. Nevertheless, we emphasize that in our paradigm, despite the lack of evidence of gross changes in brain development and inflammation, the offspring displayed deficits in social and exploration behaviors as adults, indicating that the subtle changes occurring after a single maternal LPS injection are sufficient to cause altered behavior symptomatic of psychiatric disease.
The developing fetus relies on placental transfer of maternal nutrients including oxygen and glucose across the transporting epithelial membrane into the fetal capillary circulation (Desforges and Sibley, 2009). The nature of the insult imparted on the fetus by MIA can be surmised from our data showing induction of genes associated with hypoxia, oxidative, stress, and cell death (Table. 4). Histopathological (Carpentier et al., 2011; Fatemi et al., 2011; Girard et al., 2010) and oxidative stress activity (Paintlia et al., 2008) in the placenta following MIA can disrupt the maternal/placental transfer of nutrients to the fetus leading to transient fetal hypoxia. Carpentier et al. (2011) showed evidence consistent with reduced oxygen supply to the fetal brain following maternal LPS administration on gestational day 12.5 in the mouse. Our data support the likelihood that the fetal brain is undergoing cellular stress and imperiled cell viability mediated by MIA-induced hypoxia rather than by a cytokine-mediated inflammatory response. The up-regulation of genes associated with cellular stress, hypoxia, and pro-apoptosis genes suggests a plausible scenario whereby maternal LPS results in fetal oxygen deprivation leading to a transient threat to cell viability.
Many of the highly enriched canonical pathways for the up-regulated genes determined by Ingenuity Pathways Analysis fall under the pathway category “Cellular Stress and Injury,” and inspection of the affected molecules revealed a predominance of hypoxia-induced programmed cell death genes. Some of the most up-regulated genes belong to the BH3-only family of pro-cell death genes. The BH3 domain is present on upstream activators of cell death and facilitates death by binding pro-death effector molecules such as the Bcl-2 family members to initiate a pro-death signaling cascade (Ghiotto et al., 2009). Apolipoprotein L domain containing 1 (Apold1) and BCL2/adenovirus E1B 19KDa interacting protein 3 (Bnip3) code for BH3-only proteins that respond to hypoxic and inflammatory signals to induce programmed cell death via apoptotic and autophagic processes (Azad et al., 2008; Guo et al., 2001; Kothari et al., 2003; Wan et al., 2008; Zhaorigetu et al., 2008). Bnip3 is a prominent and early responding hypoxia-induced cell death gene (Chinnadurai et al., 2008), and silencing of the gene using an siRNA suppresses hypoxia-induced autophagy (Bellot et al., 2009). Members of the heat shock family of proteins, which respond to cellular stress signals including oxidative stress and hypoxia (Chi and Karliner, 2004; Tucker et al., 2011; Welch, 1992) were also significantly up-regulated in fetal brain further suggestive of intermittent fetal hypoxia. Caveolin1 (Cav1), while not a BH3-containing molecule, also regulates apoptosis (Gargalovic and Dory, 2003). Additional pro-apoptosis genes that were significantly up-regulated in the fetal brain following maternal LPS include a subset of the programmed cell death and caspase genes. Alpha-hemoglobin stabilizing protein, also called erythroid associated factor (Eraf) the second-most up-regulated gene, serves to protect against the production of reactive oxygen species by excessive levels of free α-hemoglobin (Feng et al., 2004; Weiss et al., 2005). In vivo and in vitro models show that cerebral ischemia induced either by artery occlusion or oxygen-glucose deprivation significantly up-regulates hemoglobin mRNA expression (He et al., 2009), and that excessive hemoglobin is toxic to cerebral cortical neurons (Wang et al., 2002).
The microarray results clearly indicated that maternal LPS can affect the fetal brain by down-regulating the expression of a number of genes that are critically important to CNS development. We showed that the most enriched biological function for down-regulated genes as determined by Ingenuity Pathways Analysis is “Nervous system development and function” with a number of down-regulated genes from the microarray data mapping to specific categories related to neuronal processes (Table 3). The genes with the greatest fold reduction were those that regulate neuronal migration. Specifically, these genes, mostly encoding transcription factors, guide migration and maturation of GABAergic interneurons. Cortical and hippocampal interneuron loss has been reported following maternal polyI:C (Meyer and Feldon, 2009), neonatal LPS (Feleder et al., 2009), and maternal LPS treatment (Nouel et al., 2011). In our study, expression levels of the GABA-synthesizing enzymes glutamic acid decarboxylase (Gad1 and Gad2) were significantly reduced in fetal brain, providing additional support for the involvement of this neurotransmitter system. Our results offer a molecular explanation by which MIA selectively affects the interneuron subpopulation. The targeting of such a large number of developmental genes with discrete roles in the GABA system is a novel finding and has direct implications for the basis for how maternal LPS can affect selective aspects of behavior in the adult offspring and has broader implications for the pathogenesis of schizophrenia and autism, both of which are characterized by GABA dysfunction (for review: (Rossignol, 2011; Rubenstein, 2010; Sgado et al., 2011).
The cerebral cortex is mainly populated by two main types of neurons-glutamatergic pyramidal projection neurons and GABAerigc interneurons. Interneurons serve a vital role in local circuit function. Their inhibitory outputs are responsible for maintaining a balance between excitation and inhibition, and a loss of GABAergic activity is responsible for unchecked excitatory neuronal firing that causes seizure activity (Cossart et al., 2005). The high co-morbidity between autism and epilepsy suggest a common underlying pathophysiological mechanism, such as disruptions in the functions and properties of GABAergic interneurons, as well as local cortical circuit formation that accounts for the co-occurrence (Di Cristo, 2007; Rubenstein, 2010; Sgado et al., 2011). The cognitive deficits that characterize both autism and schizophrenia are believed to result at least partially from an excitation/inhibition imbalance caused by a loss of GABAergic interneurons, supported by the clinical evidence for reduced protein and gene markers for GABAergic interneurons (Curley et al., 2011; Fatemi et al., 2002; Thompson Ray et al., 2011).
Immature interneurons arise from germinal zones within the ventral telencephalon, specifically the medial, lateral, and caudal ganglionic eminences, and they migrate in a tangential direction across the network of radial glial fibers to enter the cortex before differentiating into mature neurons (Cobos et al., 2007). Migrating interneurons express a number of specific genetic markers, namely transcription factors, which are essential for proper migration and differentiation. The distal-less (Dlx) homeobox genes are a class of six genes, four of which are brain-specific (Dlx1, 2, 5 and 6), whose expression is intimately linked with the expression of developing GABAergic interneurons (Anderson et al., 1997; Panganiban and Rubenstein, 2002). Point mutations of Dlx1, 2, and 6 have been detected in patients diagnosed with autism (Liu et al., 2009; Nakashima et al., 2010), which provides a genetic basis for GABAerigc dysfunction. All GABAergic interneurons require proper expression and function of Dlx1 and 2 for intact migration, and Dlx5 and 6 preferentially regulate the MGE-derived parvalbumin-expressing cortical interneurons (Wang et al., 2010b). Tangential migration from the ganglionic eminences to the cortex and hippocampus is virtually absent in double Dlx1/2 knock-out mice (Anderson et al., 1997; Cobos et al., 2005), whereas migration is fairly intact in single Dlx1 or Dlx2 knock-outs, but the mutants have fewer total numbers of cortical and hippocampal interneurons, abnormal neurite morphology, and increased seizure susceptibility (Cobos et al., 2007; Cobos et al., 2005; Mao et al., 2009). We are the first to report reduced Dlx gene expression in the MIA model. In our study, Dlx1, 2 and 5 were among the top 10 most significantly down-regulated genes in fetal brains at 4 h post maternal LPS. Additional transcription factors, Arx1, which is directly regulated by Dlx1/2, Lhx6, and Mgat5B, all of whose expression was reduced in our study, play specific roles in interneuron fate determination and migration (Alifragis et al., 2004; Butt et al., 2007; Cobos et al., 2006; Faux et al., 2010; Hernandez-Miranda et al., 2010; Huang, 2009; Metin et al., 2006).
There was a significant reduction in mRNA expression in the LPS fetal brains of a number of other developmental genes whose functions are not specifically associated with interneuron development but are involved in neuronal migration. The semaphorins are a large class of membrane-bound and secreted molecules, who along with their primary receptors the plexins and co-receptors the neuropilins, provide chemoattractant and chemorepulsive cues to migrating axons. Additional roles include the regulation of dendritic branching, synaptogenesis, and synaptic pruning (Mann et al., 2007; Tran et al., 2007). There has been one other report of gestational day 18 maternal LPS reducing semaphorin (Sema 5b) mRNA expression in the mouse fetal brain 6 h post treatment (Liverman et al., 2006). There have also been reports of semaphorin mutations in individuals with autism (Melin et al., 2006).
Whereas the semaphorins are the primary axon guidance molecules in the CNS, there are additional genes that influence migration, many of which were down-regulated by maternal LPS treatment. The class III POU domain transcription factors, all of whose members had reduced expression following maternal LPS, are important for radial migration of pyramidal neurons destined for the neocortex and hippocampus (Alvarez-Bolado et al., 1995; McEvilly et al., 2002; Sugitani et al., 2002). Neuron navigator 1 (Nav1) and glypican 2 (Gpc2) are developmentally-regulated genes, both of whose expression is restricted to the axonal growth cones of postmitotic, migrating neurons (Ivins et al., 1997; Martinez-Lopez et al., 2005). The protocadherin genes Celsr3 and Pcdhγb6 are enriched within growth cones and synapses, and they are known to regulate synapse formation and maintenance (Kallenbach et al., 2003; Morishita and Yagi, 2007; Zhou et al., 2008). Celsr3 mutant mice have a number of brain malformations such as a loss of several major axonal fiber tracts (Tissir et al., 2005; Zhou et al., 2008), stunted tangential migration evidenced by misguided cortical interneurons, and accumulation of interneurons at the corticostriatal boundary (Ying et al., 2009).
It is important to point out that the down-regulation of interneuron migration-related gene expression was transient; neither microarray nor qRT-PCR revealed expression differences at the 24 h time point (data not shown). We speculate that the transient hypoxic-like conditions produced a temporary disturbance in migration patterns of inhibitory neurons, leading to small but permanent changes in final position and function of the inhibitory system, which is consistent with current views of origins of several psychiatric diseases. Our behavioral results are similar to those of others who have found deficits in social interaction and exploration of an open field maze in offspring born to dams exposed to LPS (Hava et al., 2006; Kirsten et al., 2010a; Kirsten et al., 2010b; Wang et al.), polyI:C (Meyer et al., 2006; Meyer et al., 2008; Smith et al., 2007), influenza (Shi et al., 2003), or Borna disease virus (Pletnikov et al., 1999). Transient reductions in the gene expression of a number of key neuronal development genes in combination with disrupted signaling caused by circulating cytokines could lead to subtle defects in CNS development within certain brain regions responsible for the behavioral disturbances we see in the LPS offspring. At this point, the focus of investigation appears to be on the establishment of properly timed connectivity within the cerebral cortex. Further work is needed to delineate the mechanisms at an anatomical and physiological level. Already evidence is surfacing to show that improper cortical interneuron development and function may play a role in maladaptive social behaviors associated with autism and schizophrenia (Belforte et al., 2009; Geschwind and Levitt, 2007; Levitt, 2005). Our data provide a plausible mechanism by which infections during pregnancy can affect cortical interneuron development leading to risk for later psychiatric disorders. Some evidence exists supporting a selective vulnerability of cortical interneurons to perinatal hypoxia (Fagel et al., 2009). Thus, our data also provide potential therapeutic approach to at-risk conditions, namely introduction of procedures that can counter the hypoxic conditions generated in the fetus by maternal infections.
Research Highlight.
Maternal immune activation induces a maternal cytokine response and selectively targets the fetal expression of neuronal migration and hypoxia-induced genes
Supplementary Material
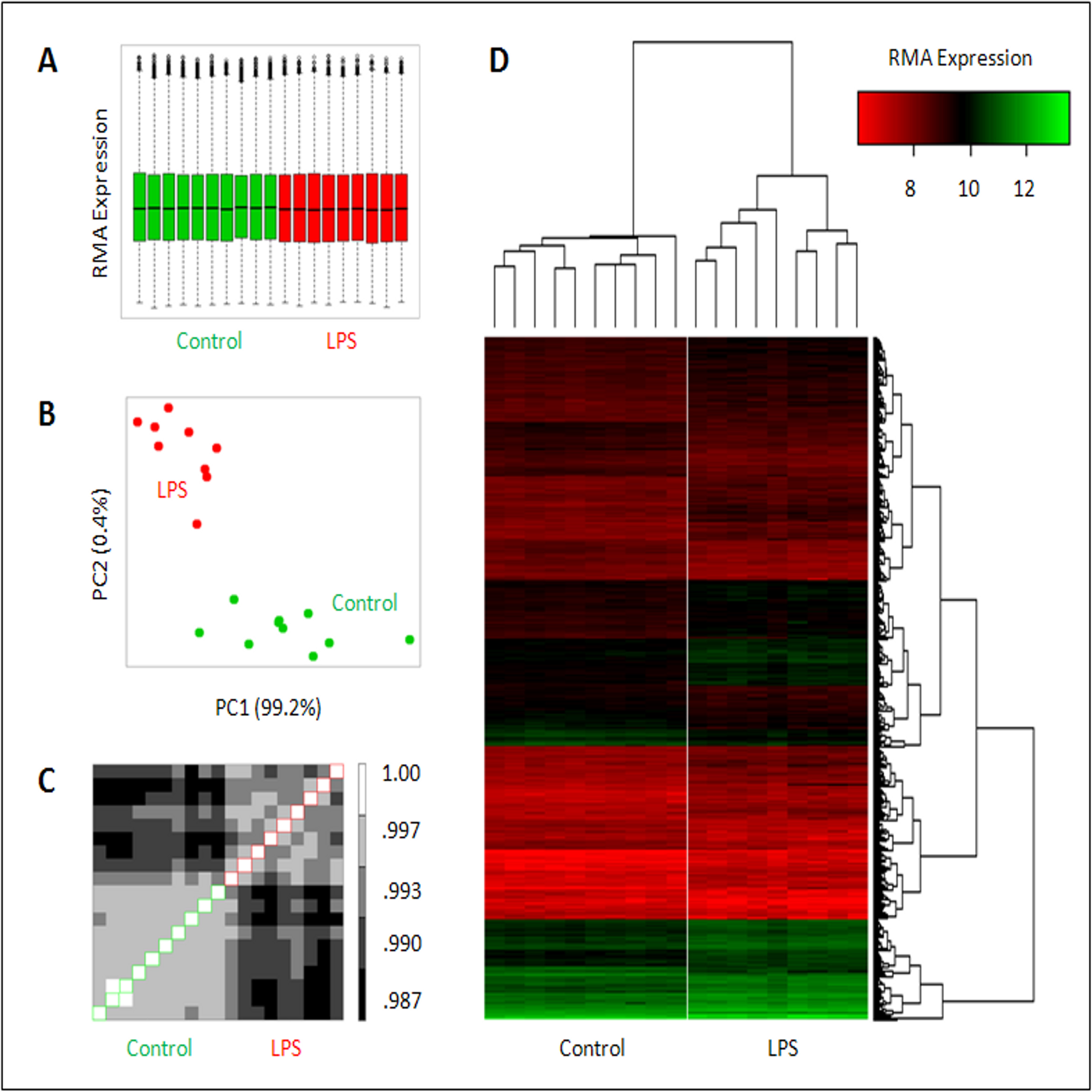
Analysis of microarray data shows goodness of fit within the two conditions and overall outcome for LPS versus saline. For the 4 h time-point samples, bioinformatic analysis shows A) Tukey box plot comparing the sample level distributions of Robust Multichip Average (RMA) normalized gene expression data, consisting of 29,215 gene probe measurements per distribution for 10 control samples (green) and 9 LPS samples (red) at 4 h post maternal LPS or saline injection (0.25 mg/kg); B) Covariance-based Principal Component Analysis (PCA) scatter-plot using RMA normalized gene expression data for 29,215 gene probes per sample; C) Pearson-based correlation Heat Map RMA using normalized gene expression data for 29,215 gene probes per sample; and D) RMA normalized gene expression based Heat Map depicting sample-to-sample relationships using gene probes (n = 3,285) selected as differentially changing between LPS and Control.
{kind=link}
First tab: Final gene list representing all significantly up- and down-regulated genes that met the significance testing and fold change set at ≥ 1.3. Second tab: List of the top 10 most enriched annotated functions as determined by Ingenuity Pathways Analysis (IPA) processing of the up-regulated genes at 4 h post maternal LPS. The list contains the function, numbers of mapped and uniquely mapped genes, and gene symbols. Third tab: List of the top 10 most enriched annotated functions as determined by IPA processing of the down-regulated genes at 4 h post maternal LPS. The list contains the function, numbers of mapped and uniquely mapped genes, and gene symbols. It additionally contains the top 10 subcategories belonging to the overall enriched function “Nervous System Development & Function.”
Acknowledgement
The research was supported by the Intramural Research Program, National Institute of Mental Health, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet. 2008;9:341–355. doi: 10.1038/nrg2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alifragis P, Liapi A, Parnavelas JG. Lhx6 regulates the migration of cortical interneurons from the ventral telencephalon but does not specify their GABA phenotype. J Neurosci. 2004;24:5643–5648. doi: 10.1523/JNEUROSCI.1245-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Bolado G, Rosenfeld MG, Swanson LW. Model of forebrain regionalization based on spatiotemporal patterns of POU-III homeobox gene expression, birthdates, and morphological features. J Comp Neurol. 1995;355:237–295. doi: 10.1002/cne.903550207. [DOI] [PubMed] [Google Scholar]
- Anderson SA, Eisenstat DD, Shi L, Rubenstein JL. Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science. 1997;278:474–476. doi: 10.1126/science.278.5337.474. [DOI] [PubMed] [Google Scholar]
- Araujo DM, Cotman CW. Differential effects of interleukin-1 beta and interleukin-2 on glia and hippocampal neurons in culture. Int J Dev Neurosci. 1995;13:201–212. doi: 10.1016/0736-5748(94)00072-b. [DOI] [PubMed] [Google Scholar]
- Ashdown H, Dumont Y, Ng M, Poole S, Boksa P, Luheshi GN. The role of cytokines in mediating effects of prenatal infection on the fetus: implications for schizophrenia. Mol Psychiatry. 2006;11:47–55. doi: 10.1038/sj.mp.4001748. [DOI] [PubMed] [Google Scholar]
- Atladottir HO, Thorsen P, Ostergaard L, Schendel DE, Lemcke S, Abdallah M, Parner ET. Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. J Autism Dev Disord. 2010;40:1423–1430. doi: 10.1007/s10803-010-1006-y. [DOI] [PubMed] [Google Scholar]
- Azad MB, Chen Y, Henson ES, Cizeau J, McMillan-Ward E, Israels SJ, Gibson SB. Hypoxia induces autophagic cell death in apoptosis-competent cells through a mechanism involving BNIP3. Autophagy. 2008;4:195–204. doi: 10.4161/auto.5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belforte JE, Zsiros V, Sklar ER, Jiang Z, Yu G, Li Y, Quinlan EM, Nakazawa K. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat Neurosci. 2009;13:76–83. doi: 10.1038/nn.2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouyssegur J, Mazure NM. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29:2570–2581. doi: 10.1128/MCB.00166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benveniste EN. Cytokine actions in the central nervous system. Cytokine Growth Factor Rev. 1998;9:259–275. doi: 10.1016/s1359-6101(98)00015-x. [DOI] [PubMed] [Google Scholar]
- Bilbo SD, Schwarz JM. Early-life programming of later-life brain and behavior: a critical role for the immune system. Front Behav Neurosci. 2009;3:14. doi: 10.3389/neuro.08.014.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boksa P. Effects of prenatal infection on brain development and behavior: a review of findings from animal models. Brain Behav Immun. 2010;24:881–897. doi: 10.1016/j.bbi.2010.03.005. [DOI] [PubMed] [Google Scholar]
- Brown AS, Begg MD, Gravenstein S, Schaefer CA, Wyatt RJ, Bresnahan M, Babulas VP, Susser ES. Serologic evidence of prenatal influenza in the etiology of schizophrenia. Arch Gen Psychiatry. 2004;61:774–780. doi: 10.1001/archpsyc.61.8.774. [DOI] [PubMed] [Google Scholar]
- Butt SJ, Cobos I, Golden J, Kessaris N, Pachnis V, Anderson S. Transcriptional regulation of cortical interneuron development. J Neurosci. 2007;27:11847–11850. doi: 10.1523/JNEUROSCI.3525-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Pan ZL, Pang Y, Evans OB, Rhodes PG. Cytokine induction in fetal rat brains and brain injury in neonatal rats after maternal lipopolysaccharide administration. Pediatr Res. 2000;47:64–72. doi: 10.1203/00006450-200001000-00013. [DOI] [PubMed] [Google Scholar]
- Carpentier PA, Dingman AL, Palmer TD. Placental TNF-alpha signaling in illness-induced complications of pregnancy. Am J Pathol. 2011;178:2802–2810. doi: 10.1016/j.ajpath.2011.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi NC, Karliner JS. Molecular determinants of responses to myocardial ischemia/reperfusion injury: focus on hypoxia-inducible and heat shock factors. Cardiovasc Res. 2004;61:437–447. doi: 10.1016/j.cardiores.2003.11.033. [DOI] [PubMed] [Google Scholar]
- Chinnadurai G, Vijayalingam S, Gibson SB. BNIP3 subfamily BH3-only proteins: mitochondrial stress sensors in normal and pathological functions. Oncogene. 2008;27 Suppl 1:S114–S127. doi: 10.1038/onc.2009.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciaranello AL, Ciaranello RD. The neurobiology of infantile autism. Annu Rev Neurosci. 1995;18:101–128. doi: 10.1146/annurev.ne.18.030195.000533. [DOI] [PubMed] [Google Scholar]
- Cobos I, Borello U, Rubenstein JL. Dlx transcription factors promote migration through repression of axon and dendrite growth. Neuron. 2007;54:873–888. doi: 10.1016/j.neuron.2007.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobos I, Calcagnotto ME, Vilaythong AJ, Thwin MT, Noebels JL, Baraban SC, Rubenstein JL. Mice lacking Dlx1 show subtype-specific loss of interneurons, reduced inhibition and epilepsy. Nat Neurosci. 2005;8:1059–1068. doi: 10.1038/nn1499. [DOI] [PubMed] [Google Scholar]
- Cobos I, Long JE, Thwin MT, Rubenstein JL. Cellular patterns of transcription factor expression in developing cortical interneurons. Cereb Cortex. 2006;16 Suppl 1:i82–i88. doi: 10.1093/cercor/bhk003. [DOI] [PubMed] [Google Scholar]
- Cossart R, Bernard C, Ben-Ari Y. Multiple facets of GABAergic neurons and synapses: multiple fates of GABA signalling in epilepsies. Trends Neurosci. 2005;28:108–115. doi: 10.1016/j.tins.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Curley AA, Arion D, Volk DW, Asafu-Adjei JK, Sampson AR, Fish KN, Lewis DA. Cortical deficits of glutamic acid decarboxylase 67 expression in schizophrenia: clinical, protein, and cell type-specific features. Am J Psychiatry. 2011;168:921–929. doi: 10.1176/appi.ajp.2011.11010052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlgren J, Samuelsson AM, Jansson T, Holmang A. Interleukin-6 in the maternal circulation reaches the rat fetus in mid-gestation. Pediatr Res. 2006;60:147–151. doi: 10.1203/01.pdr.0000230026.74139.18. [DOI] [PubMed] [Google Scholar]
- Desforges M, Sibley CP. Placental nutrient supply and fetal growth. Int J Dev Biol. 2009;54:377–390. doi: 10.1387/ijdb.082765md. [DOI] [PubMed] [Google Scholar]
- Deverman BE, Patterson PH. Cytokines and CNS development. Neuron. 2009;64:61–78. doi: 10.1016/j.neuron.2009.09.002. [DOI] [PubMed] [Google Scholar]
- Di Cristo G. Development of cortical GABAergic circuits and its implications for neurodevelopmental disorders. Clin Genet. 2007;72:1–8. doi: 10.1111/j.1399-0004.2007.00822.x. [DOI] [PubMed] [Google Scholar]
- Fagel DM, Ganat Y, Cheng E, Silbereis J, Ohkubo Y, Ment LR, Vaccarino FM. Fgfr1 is required for cortical regeneration and repair after perinatal hypoxia. J Neurosci. 2009;29:1202–1211. doi: 10.1523/JNEUROSCI.4516-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, Emamian ES, Sidwell RW, Kist DA, Stary JM, Earle JA, Thuras P. Human influenza viral infection in utero alters glial fibrillary acidic protein immunoreactivity in the developing brains of neonatal mice. Mol Psychiatry. 2002;7:633–640. doi: 10.1038/sj.mp.4001046. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Folsom TD, Rooney RJ, Mori S, Kornfield TE, Reutiman TJ, Kneeland RE, Liesch SB, Hua K, Hsu J, Patel DH. The viral theory of schizophrenia revisited: Abnormal placental gene expression and structural changes with lack of evidence for H1N1 viral presence in placentae of infected mice or brains of exposed offspring. Neuropharmacology. 2011 doi: 10.1016/j.neuropharm.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faux C, Rakic S, Andrews W, Yanagawa Y, Obata K, Parnavelas JG. Differential gene expression in migrating cortical interneurons during mouse forebrain development. J Comp Neurol. 2010;518:1232–1248. doi: 10.1002/cne.22271. [DOI] [PubMed] [Google Scholar]
- Feleder C, Tseng KY, Calhoon GG, O'Donnell P. Neonatal intrahippocampal immune challenge alters dopamine modulation of prefrontal cortical interneurons in adult rats. Biol Psychiatry. 2009;67:386–392. doi: 10.1016/j.biopsych.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Gell DA, Zhou S, Gu L, Kong Y, Li J, Hu M, Yan N, Lee C, Rich AM, Armstrong RS, Lay PA, Gow AJ, Weiss MJ, Mackay JP, Shi Y. Molecular mechanism of AHSP-mediated stabilization of alpha-hemoglobin. Cell. 2004;119:629–640. doi: 10.1016/j.cell.2004.11.025. [DOI] [PubMed] [Google Scholar]
- Gargalovic P, Dory L. Cellular apoptosis is associated with increased caveolin-1 expression in macrophages. J Lipid Res. 2003;44:1622–1632. doi: 10.1194/jlr.M300140-JLR200. [DOI] [PubMed] [Google Scholar]
- Gayle DA, Beloosesky R, Desai M, Amidi F, Nunez SE, Ross MG. Maternal LPS induces cytokines in the amniotic fluid and corticotropin releasing hormone in the fetal rat brain. Am J Physiol Regul Integr Comp Physiol. 2004;286:R1024–R1029. doi: 10.1152/ajpregu.00664.2003. [DOI] [PubMed] [Google Scholar]
- Geschwind DH, Levitt P. Autism spectrum disorders: developmental disconnection syndromes. Curr Opin Neurobiol. 2007;17:103–111. doi: 10.1016/j.conb.2007.01.009. [DOI] [PubMed] [Google Scholar]
- Ghiani CA, Mattan NS, Nobuta H, Malvar JS, Boles J, Ross MG, Waschek JA, Carpenter EM, Fisher RS, de Vellis J. Early effects of lipopolysaccharide-induced inflammation on foetal brain development in rat. ASN Neuro. 2011;3 doi: 10.1042/AN20110027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiotto F, Fais F, Bruno S. BH3-only proteins: the death-puppeteer's wires. Cytometry A. 2009;77:11–21. doi: 10.1002/cyto.a.20819. [DOI] [PubMed] [Google Scholar]
- Girard S, Tremblay L, Lepage M, Sebire G. IL-1 receptor antagonist protects against placental and neurodevelopmental defects induced by maternal inflammation. J Immunol. 2010;184:3997–4005. doi: 10.4049/jimmunol.0903349. [DOI] [PubMed] [Google Scholar]
- Golan HM, Lev V, Hallak M, Sorokin Y, Huleihel M. Specific neurodevelopmental damage in mice offspring following maternal inflammation during pregnancy. Neuropharmacology. 2005;48:903–917. doi: 10.1016/j.neuropharm.2004.12.023. [DOI] [PubMed] [Google Scholar]
- Guo K, Searfoss G, Krolikowski D, Pagnoni M, Franks C, Clark K, Yu KT, Jaye M, Ivashchenko Y. Hypoxia induces the expression of the pro-apoptotic gene BNIP3. Cell Death Differ. 2001;8:367–376. doi: 10.1038/sj.cdd.4400810. [DOI] [PubMed] [Google Scholar]
- Hauguel-de Mouzon S, Guerre-Millo M. The placenta cytokine network and inflammatory signals. Placenta. 2006;27:794–798. doi: 10.1016/j.placenta.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Hava G, Vered L, Yael M, Mordechai H, Mahoud H. Alterations in behavior in adult offspring mice following maternal inflammation during pregnancy. Dev Psychobiol. 2006;48:162–168. doi: 10.1002/dev.20116. [DOI] [PubMed] [Google Scholar]
- He Y, Hua Y, Liu W, Hu H, Keep RF, Xi G. Effects of cerebral ischemia on neuronal hemoglobin. J Cereb Blood Flow Metab. 2009;29:596–605. doi: 10.1038/jcbfm.2008.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Miranda LR, Parnavelas JG, Chiara F. Molecules and mechanisms involved in the generation and migration of cortical interneurons. ASN Neuro. 2010;2:e00031. doi: 10.1042/AN20090053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao EY, Patterson PH. Activation of the maternal immune system induces endocrine changes in the placenta via IL-6. Brain Behav Immun. 2011;25:604–615. doi: 10.1016/j.bbi.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z. Molecular regulation of neuronal migration during neocortical development. Mol Cell Neurosci. 2009;42:11–22. doi: 10.1016/j.mcn.2009.06.003. [DOI] [PubMed] [Google Scholar]
- Ivins JK, Litwack ED, Kumbasar A, Stipp CS, Lander AD. Cerebroglycan, a developmentally regulated cell-surface heparan sulfate proteoglycan, is expressed on developing axons and growth cones. Dev Biol. 1997;184:320–332. doi: 10.1006/dbio.1997.8532. [DOI] [PubMed] [Google Scholar]
- Jarskog LF, Xiao H, Wilkie MB, Lauder JM, Gilmore JH. Cytokine regulation of embryonic rat dopamine and serotonin neuronal survival in vitro. Int J Dev Neurosci. 1997;15:711–716. doi: 10.1016/s0736-5748(97)00029-4. [DOI] [PubMed] [Google Scholar]
- Jonakait GM. The effects of maternal inflammation on neuronal development: possible mechanisms. Int J Dev Neurosci. 2007;25:415–425. doi: 10.1016/j.ijdevneu.2007.08.017. [DOI] [PubMed] [Google Scholar]
- Kallenbach S, Khantane S, Carroll P, Gayet O, Alonso S, Henderson CE, Dudley K. Changes in subcellular distribution of protocadherin gamma proteins accompany maturation of spinal neurons. J Neurosci Res. 2003;72:549–556. doi: 10.1002/jnr.10618. [DOI] [PubMed] [Google Scholar]
- Keohane A, Ryan S, Maloney E, Sullivan AM, Nolan YM. Tumour necrosis factor-alpha impairs neuronal differentiation but not proliferation of hippocampal neural precursor cells: Role of Hes1. Mol Cell Neurosci. 2010;43:127–135. doi: 10.1016/j.mcn.2009.10.003. [DOI] [PubMed] [Google Scholar]
- Kirsten TB, Taricano M, Florio JC, Palermo-Neto J, Bernardi MM. Prenatal lipopolysaccharide reduces motor activity after an immune challenge in adult male offspring. Behav Brain Res. 2010a;211:77–82. doi: 10.1016/j.bbr.2010.03.009. [DOI] [PubMed] [Google Scholar]
- Kirsten TB, Taricano M, Maiorka PC, Palermo-Neto J, Bernardi MM. Prenatal lipopolysaccharide reduces social behavior in male offspring. Neuroimmunomodulation. 2010b;17:240–251. doi: 10.1159/000290040. [DOI] [PubMed] [Google Scholar]
- Kothari S, Cizeau J, McMillan-Ward E, Israels SJ, Bailes M, Ens K, Kirshenbaum LA, Gibson SB. BNIP3 plays a role in hypoxic cell death in human epithelial cells that is inhibited by growth factors EGF and IGF. Oncogene. 2003;22:4734–4744. doi: 10.1038/sj.onc.1206666. [DOI] [PubMed] [Google Scholar]
- Lasala N, Zhou H. Effects of maternal exposure to LPS on the inflammatory response in the offspring. J Neuroimmunol. 2007;189:95–101. doi: 10.1016/j.jneuroim.2007.07.010. [DOI] [PubMed] [Google Scholar]
- Levitt P. Disruption of interneuron development. Epilepsia. 2005;46 Suppl 7:22–28. doi: 10.1111/j.1528-1167.2005.00305.x. [DOI] [PubMed] [Google Scholar]
- Liu X, Novosedlik N, Wang A, Hudson ML, Cohen IL, Chudley AE, Forster-Gibson CJ, Lewis SM, Holden JJ. The DLX1and DLX2 genes and susceptibility to autism spectrum disorders. Eur J Hum Genet. 2009;17:228–235. doi: 10.1038/ejhg.2008.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liverman CS, Kaftan HA, Cui L, Hersperger SG, Taboada E, Klein RM, Berman NE. Altered expression of pro-inflammatory and developmental genes in the fetal brain in a mouse model of maternal infection. Neurosci Lett. 2006;399:220–225. doi: 10.1016/j.neulet.2006.01.064. [DOI] [PubMed] [Google Scholar]
- Mann F, Chauvet S, Rougon G. Semaphorins in development and adult brain: Implication for neurological diseases. Prog Neurobiol. 2007;82:57–79. doi: 10.1016/j.pneurobio.2007.02.011. [DOI] [PubMed] [Google Scholar]
- Mao R, Page DT, Merzlyak I, Kim C, Tecott LH, Janak PH, Rubenstein JL, Sur M. Reduced conditioned fear response in mice that lack Dlx1 and show subtype-specific loss of interneurons. J Neurodev Disord. 2009;1:224–236. doi: 10.1007/s11689-009-9025-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Lopez MJ, Alcantara S, Mascaro C, Perez-Branguli F, Ruiz-Lozano P, Maes T, Soriano E, Buesa C. Mouse neuron navigator 1, a novel microtubule-associated protein involved in neuronal migration. Mol Cell Neurosci. 2005;28:599–612. doi: 10.1016/j.mcn.2004.09.016. [DOI] [PubMed] [Google Scholar]
- Marx CE, Jarskog LF, Lauder JM, Lieberman JA, Gilmore JH. Cytokine effects on cortical neuron MAP-2 immunoreactivity: implications for schizophrenia. Biol Psychiatry. 2001;50:743–749. doi: 10.1016/s0006-3223(01)01209-4. [DOI] [PubMed] [Google Scholar]
- McEvilly RJ, de Diaz MO, Schonemann MD, Hooshmand F, Rosenfeld MG. Transcriptional regulation of cortical neuron migration by POU domain factors. Science. 2002;295:1528–1532. doi: 10.1126/science.1067132. [DOI] [PubMed] [Google Scholar]
- Melin M, Carlsson B, Anckarsater H, Rastam M, Betancur C, Isaksson A, Gillberg C, Dahl N. Constitutional downregulation of SEMA5A expression in autism. Neuropsychobiology. 2006;54:64–69. doi: 10.1159/000096040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metin C, Baudoin JP, Rakic S, Parnavelas JG. Cell and molecular mechanisms involved in the migration of cortical interneurons. Eur J Neurosci. 2006;23:894–900. doi: 10.1111/j.1460-9568.2006.04630.x. [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J. Neural basis of psychosis-related behaviour in the infection model of schizophrenia. Behav Brain Res. 2009;204:322–334. doi: 10.1016/j.bbr.2008.12.022. [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J, Fatemi SH. In-vivo rodent models for the experimental investigation of prenatal immune activation effects in neurodevelopmental brain disorders. Neurosci Biobehav Rev. 2009a;33:1061–1079. doi: 10.1016/j.neubiorev.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J, Schedlowski M, Yee BK. Towards an immuno-precipitated neurodevelopmental animal model of schizophrenia. Neurosci Biobehav Rev. 2005;29:913–947. doi: 10.1016/j.neubiorev.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J, Yee BK. A review of the fetal brain cytokine imbalance hypothesis of schizophrenia. Schizophr Bull. 2009b;35:959–972. doi: 10.1093/schbul/sbn022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer U, Nyffeler M, Engler A, Urwyler A, Schedlowski M, Knuesel I, Yee BK, Feldon J. The time of prenatal immune challenge determines the specificity of inflammation-mediated brain and behavioral pathology. J Neurosci. 2006;26:4752–4762. doi: 10.1523/JNEUROSCI.0099-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer U, Nyffeler M, Yee BK, Knuesel I, Feldon J. Adult brain and behavioral pathological markers of prenatal immune challenge during early/middle and late fetal development in mice. Brain Behav Immun. 2008;22:469–486. doi: 10.1016/j.bbi.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Morishita H, Yagi T. Protocadherin family: diversity, structure, and function. Curr Opin Cell Biol. 2007;19:584–592. doi: 10.1016/j.ceb.2007.09.006. [DOI] [PubMed] [Google Scholar]
- Nakashima N, Yamagata T, Mori M, Kuwajima M, Suwa K, Momoi MY. Expression analysis and mutation detection of DLX5 and DLX6 in autism. Brain Dev. 2010;32:98–104. doi: 10.1016/j.braindev.2008.12.021. [DOI] [PubMed] [Google Scholar]
- Nawa H, Takei N. Recent progress in animal modeling of immune inflammatory processes in schizophrenia: implication of specific cytokines. Neurosci Res. 2006;56:2–13. doi: 10.1016/j.neures.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Neumann H, Schweigreiter R, Yamashita T, Rosenkranz K, Wekerle H, Barde YA. Tumor necrosis factor inhibits neurite outgrowth and branching of hippocampal neurons by a rho-dependent mechanism. J Neurosci. 2002;22:854–862. doi: 10.1523/JNEUROSCI.22-03-00854.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning H, Wang H, Zhao L, Zhang C, Li XY, Chen YH, Xu DX. Maternally-administered lipopolysaccharide (LPS) increases tumor necrosis factor alpha in fetal liver and fetal brain: its suppression by low-dose LPS pretreatment. Toxicol Lett. 2008;176:13–19. doi: 10.1016/j.toxlet.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Nouel D, Burt M, Zhang Y, Harvey L, Boksa P. Prenatal exposure to bacterial endotoxin reduces the number of GAD67- and reelin-immunoreactive neurons in the hippocampus of rat offspring. Eur Neuropsychopharmacol. 2011 doi: 10.1016/j.euroneuro.2011.08.001. [DOI] [PubMed] [Google Scholar]
- Paintlia MK, Paintlia AS, Singh AK, Singh I. Attenuation of lipopolysaccharide-induced inflammatory response and phospholipids metabolism at the feto-maternal interface by N-acetyl cysteine. Pediatr Res. 2008;64:334–339. doi: 10.1203/PDR.0b013e318181e07c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palsson-McDermott EM, O'Neill LA. Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4. Immunology. 2004;113:153–162. doi: 10.1111/j.1365-2567.2004.01976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panganiban G, Rubenstein JL. Developmental functions of the Distal-less/Dlx homeobox genes. Development. 2002;129:4371–4386. doi: 10.1242/dev.129.19.4371. [DOI] [PubMed] [Google Scholar]
- Patterson PH. Maternal infection: window on neuroimmune interactions in fetal brain development and mental illness. Curr Opin Neurobiol. 2002;12:115–118. doi: 10.1016/s0959-4388(02)00299-4. [DOI] [PubMed] [Google Scholar]
- Patterson PH, Nawa H. Neuronal differentiation factors/cytokines and synaptic plasticity. Cell. 1993;72 Suppl:123–137. doi: 10.1016/s0092-8674(05)80032-7. [DOI] [PubMed] [Google Scholar]
- Phillips TM, Wellner EF. Measurement of naproxen in human plasma by chip-based immunoaffinity capillary electrophoresis. Biomed Chromatogr. 2006;20:662–667. doi: 10.1002/bmc.673. [DOI] [PubMed] [Google Scholar]
- Phillips TM, Wellner EF. Analysis of inflammatory biomarkers from tissue biopsies by chip-based immunoaffinity CE. Electrophoresis. 2007;28:3041–3048. doi: 10.1002/elps.200700193. [DOI] [PubMed] [Google Scholar]
- Pletnikov MV, Rubin SA, Vasudevan K, Moran TH, Carbone KM. Developmental brain injury associated with abnormal play behavior in neonatally Borna disease virus-infected Lewis rats: a model of autism. Behav Brain Res. 1999;100:43–50. doi: 10.1016/s0166-4328(98)00111-9. [DOI] [PubMed] [Google Scholar]
- Qiu Z, Sweeney DD, Netzeband JG, Gruol DL. Chronic interleukin-6 alters NMDA receptor-mediated membrane responses and enhances neurotoxicity in developing CNS neurons. J Neurosci. 1998;18:10445–10456. doi: 10.1523/JNEUROSCI.18-24-10445.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigato C, Buckinx R, Le-Corronc H, Rigo JM, Legendre P. Pattern of invasion of the embryonic mouse spinal cord by microglial cells at the time of the onset of functional neuronal networks. Glia. 2011;59:675–695. doi: 10.1002/glia.21140. [DOI] [PubMed] [Google Scholar]
- Rossignol E. Genetics and function of neocortical GABAergic interneurons in neurodevelopmental disorders. Neural Plast. 2011;2011:649325. doi: 10.1155/2011/649325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell NJ, Hopkins SJ. Cytokines and the nervous system II: Actions and mechanisms of action. Trends Neurosci. 1995;18:130–136. doi: 10.1016/0166-2236(95)93890-a. [DOI] [PubMed] [Google Scholar]
- Rubenstein JL. Three hypotheses for developmental defects that may underlie some forms of autism spectrum disorder. Curr Opin Neurol. 2010;23:118–123. doi: 10.1097/WCO.0b013e328336eb13. [DOI] [PubMed] [Google Scholar]
- Schlotz W, Phillips DI. Fetal origins of mental health: evidence and mechanisms. Brain Behav Immun. 2009;23:905–916. doi: 10.1016/j.bbi.2009.02.001. [DOI] [PubMed] [Google Scholar]
- Sgado P, Dunleavy M, Genovesi S, Provenzano G, Bozzi Y. The role of GABAergic system in neurodevelopmental disorders: a focus on autism and epilepsy. Int J Physiol Pathophysiol Pharmacol. 2011;3:223–235. [PMC free article] [PubMed] [Google Scholar]
- Shi L, Fatemi SH, Sidwell RW, Patterson PH. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J Neurosci. 2003;23:297–302. doi: 10.1523/JNEUROSCI.23-01-00297.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. 2007;27:10695–10702. doi: 10.1523/JNEUROSCI.2178-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugitani Y, Nakai S, Minowa O, Nishi M, Jishage K, Kawano H, Mori K, Ogawa M, Noda T. Brn-1 and Brn-2 share crucial roles in the production and positioning of mouse neocortical neurons. Genes Dev. 2002;16:1760–1765. doi: 10.1101/gad.978002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson Ray M, Weickert CS, Wyatt E, Webster MJ. Decreased BDNF, trkB-TK+ and GAD67 mRNA expression in the hippocampus of individuals with schizophrenia and mood disorders. J Psychiatry Neurosci. 2011;36:195–203. doi: 10.1503/jpn.100048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissir F, Bar I, Jossin Y, De Backer O, Goffinet AM. Protocadherin Celsr3 is crucial in axonal tract development. Nat Neurosci. 2005;8:451–457. doi: 10.1038/nn1428. [DOI] [PubMed] [Google Scholar]
- Tran TS, Kolodkin AL, Bharadwaj R. Semaphorin regulation of cellular morphology. Annu Rev Cell Dev Biol. 2007;23:263–292. doi: 10.1146/annurev.cellbio.22.010605.093554. [DOI] [PubMed] [Google Scholar]
- Tucker NR, Middleton RC, Le QP, Shelden EA. HSF1 is essential for the resistance of zebrafish eye and brain tissues to hypoxia/reperfusion injury. PLoS One. 2011;6:e22268. doi: 10.1371/journal.pone.0022268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urakubo A, Jarskog LF, Lieberman JA, Gilmore JH. Prenatal exposure to maternal infection alters cytokine expression in the placenta, amniotic fluid, and fetal brain. Schizophr Res. 2001;47:27–36. doi: 10.1016/s0920-9964(00)00032-3. [DOI] [PubMed] [Google Scholar]
- Walker BR, Diefenbach KS, Parikh TN. Inhibition within the nucleus tractus solitarius (NTS) ameliorates environmental exploration deficits due to cerebellum lesions in an animal model for autism. Behav Brain Res. 2007;176:109–120. doi: 10.1016/j.bbr.2006.08.008. [DOI] [PubMed] [Google Scholar]
- Wan G, Zhaorigetu S, Liu Z, Kaini R, Jiang Z, Hu CA. Apolipoprotein L1, a novel Bcl-2 homology domain 3-only lipid-binding protein, induces autophagic cell death. J Biol Chem. 2008;283:21540–21549. doi: 10.1074/jbc.M800214200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Meng XH, Ning H, Zhao XF, Wang Q, Liu P, Zhang H, Zhang C, Chen GH, Xu DX. Age- and gender-dependent impairments of neurobehaviors in mice whose mothers were exposed to lipopolysaccharide during pregnancy. Toxicol Lett. 2010a;192:245–251. doi: 10.1016/j.toxlet.2009.10.030. [DOI] [PubMed] [Google Scholar]
- Wang X, Mori T, Sumii T, Lo EH. Hemoglobin-induced cytotoxicity in rat cerebral cortical neurons: caspase activation and oxidative stress. Stroke. 2002;33:1882–1888. doi: 10.1161/01.str.0000020121.41527.5d. [DOI] [PubMed] [Google Scholar]
- Wang Y, Dye CA, Sohal V, Long JE, Estrada RC, Roztocil T, Lufkin T, Deisseroth K, Baraban SC, Rubenstein JL. Dlx5 and Dlx6 regulate the development of parvalbumin-expressing cortical interneurons. J Neurosci. 2010b;30:5334–5345. doi: 10.1523/JNEUROSCI.5963-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss MJ, Zhou S, Feng L, Gell DA, Mackay JP, Shi Y, Gow AJ. Role of alpha-hemoglobin-stabilizing protein in normal erythropoiesis and beta-thalassemia. Ann N Y Acad Sci. 2005;1054:103–117. doi: 10.1196/annals.1345.013. [DOI] [PubMed] [Google Scholar]
- Welch WJ. Mammalian stress response: cell physiology, structure/function of stress proteins, and implications for medicine and disease. Physiol Rev. 1992;72:1063–1081. doi: 10.1152/physrev.1992.72.4.1063. [DOI] [PubMed] [Google Scholar]
- Yang M, Crawley JN. Simple behavioral assessment of mouse olfaction. Curr Protoc Neurosci. 2009;Chapter 8(Unit 8):24. doi: 10.1002/0471142301.ns0824s48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying G, Wu S, Hou R, Huang W, Capecchi MR, Wu Q. The protocadherin gene Celsr3 is required for interneuron migration in the mouse forebrain. Mol Cell Biol. 2009;29:3045–3061. doi: 10.1128/MCB.00011-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Bausano B, Pike BR, Newcomb-Fernandez JK, Wang KK, Shohami E, Ringger NC, DeFord SM, Anderson DK, Hayes RL. TNF-alpha stimulates caspase-3 activation and apoptotic cell death in primary septo-hippocampal cultures. J Neurosci Res. 2001;64:121–131. doi: 10.1002/jnr.1059. [DOI] [PubMed] [Google Scholar]
- Zhaorigetu S, Wan G, Kaini R, Jiang Z, Hu CA. ApoL1, a BH3-only lipid-binding protein, induces autophagic cell death. Autophagy. 2008;4:1079–1082. doi: 10.4161/auto.7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Bar I, Achouri Y, Campbell K, De Backer O, Hebert JM, Jones K, Kessaris N, de Rouvroit CL, O'Leary D, Richardson WD, Goffinet AM, Tissir F. Early forebrain wiring: genetic dissection using conditional Celsr3 mutant mice. Science. 2008;320:946–949. doi: 10.1126/science.1155244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Analysis of microarray data shows goodness of fit within the two conditions and overall outcome for LPS versus saline. For the 4 h time-point samples, bioinformatic analysis shows A) Tukey box plot comparing the sample level distributions of Robust Multichip Average (RMA) normalized gene expression data, consisting of 29,215 gene probe measurements per distribution for 10 control samples (green) and 9 LPS samples (red) at 4 h post maternal LPS or saline injection (0.25 mg/kg); B) Covariance-based Principal Component Analysis (PCA) scatter-plot using RMA normalized gene expression data for 29,215 gene probes per sample; C) Pearson-based correlation Heat Map RMA using normalized gene expression data for 29,215 gene probes per sample; and D) RMA normalized gene expression based Heat Map depicting sample-to-sample relationships using gene probes (n = 3,285) selected as differentially changing between LPS and Control.
First tab: Final gene list representing all significantly up- and down-regulated genes that met the significance testing and fold change set at ≥ 1.3. Second tab: List of the top 10 most enriched annotated functions as determined by Ingenuity Pathways Analysis (IPA) processing of the up-regulated genes at 4 h post maternal LPS. The list contains the function, numbers of mapped and uniquely mapped genes, and gene symbols. Third tab: List of the top 10 most enriched annotated functions as determined by IPA processing of the down-regulated genes at 4 h post maternal LPS. The list contains the function, numbers of mapped and uniquely mapped genes, and gene symbols. It additionally contains the top 10 subcategories belonging to the overall enriched function “Nervous System Development & Function.”