

Abstract
A series of 1,3-disubstituted ureas possessing a piperidyl moiety has been synthesized to investigate their structure-activity relationships as inhibitors of the human and murine soluble epoxide hydrolase (sEH). Oral administration of thirteen 1-aryl-3-(1-acylpiperidin-4-yl)urea inhibitors in mice revealed substantial improvements in pharmacokinetic parameters over previously reported 1-adamantyl-urea based inhibitors. For example, 1-(1-(cyclopropanecarbonyl)piperidin-4-yl)-3-(4-(trifluoromethoxy)phenyl)urea (52) showed a 7-fold increase in potency, a 65-fold increase in Cmax† and a 3300 fold increase in AUC over its adamantane analogue 1-(1-adamantyl)-3-(1-propionylpiperidin-4-yl)urea (2). This novel sEH inhibitor showed a 1000 fold increase in potency when compared to morphine by reducing hyperalgesia as measured by mechanical withdrawl threshold using the in vivo carrageenan induced inflammatory pain model.
Introduction
The soluble epoxide hydrolase (sEH, E.C. 3.3.2.3) converts epoxides to the corresponding diols by the catalytic addition of a water molecule. The enzyme is implicated in several disease states for its ability to metabolize fatty acid epoxides such as epoxyeicosatrienoic acids (EETs) and leukotoxin, important endogenous signaling lipids, to less active dihydroxyeicosatrienoic acids (DHETs)1 and toxic, pro-inflammatory leukotoxin diols,2 respectively. sEH inhibitors are of growing interest for therapeutic use because they have been shown to increase the in vivo concentration of EETs and other fatty acid epoxides resulting in anti-inflammatory,3 anti-hypertensive,4 neuroprotective,5 and cardioprotective effects.6-8 Several reviews have been published concerning the mechanism of action and diverse biological roles of EETs and the sEH inhibitors that stabilize them.9-16 Of particular note Marino (2009)17 recently reviewed the chemistry of sEH inhibitors and Shen (2010)18 summarized the patent literature in the sEH field.
The prototypical inhibitors dicyclohexyl urea and 12-(3-adamantane-1-yl-ureido)dodecanoic acid (AUDA), while potent in vitro, suffer from poor physical properties and poor in vivo stability. Amides, carbamates, and other pharmacophores17,18 have been explored as alternative pharmacophores in an attempt to improve physical properties and show structure activity relationships similar to ureas, but the disubstituted ureas remain the most studied class of inhibitors due to their high potency,19-23 and promising pharmacokinetics.24,25 Although earlier studies found that trisubstitued ureas had reduced potency,9,26 with proper substituents piperidine based trisubstituted ureas have been found to be potent inhibitors of the enzyme.27-30 In the last year several other promising pharmacophores have been reported.17,18,21,28,31 We previously reported inhibitors incorporating a polar moiety, such as an N-acyl piperidine or cyclohexyloxybenzoic acid to one side of the urea, which yielded a substantial improvement in water solubility and oral bioavailability while retaining excellent potency.24,25,32,33 The adamantyl moiety retained in many of these potent second-generation sEH inhibitors provided sensitive characteristic mass spectral fragmentation. However, this moiety is prone to rapid metabolism, often leading to low drug concentrations in the blood and short in vivo half-life.
Replacement of the adamantyl group with a phenyl ring has been explored in our earlier work and has yielded several highly potent inhibitors, warranting further investigation.25,33,34 Thus, in this study, we further investigated urea based sEH inhibitors by optimizing the 1-aryl-3-(1-acylpiperidin-4-yl)urea core structure. Based on the reported 2-fold increase in potency of the N-propionylpiperidine over the N-acetylpiperidine in a group of adamantylureas,25,32 a series of inhibitors conserving the N-propionyl piperidine moiety was synthesized to probe the SAR of the aryl group. Additional inhibitors conserving 4-trifluoromethoxyphenyl as the aryl group and varying in N-acylpiperidine or N-sulfonylpiperidine substitution were synthesized to examine the effects of polar and basic side chains on potency.
Inhibitors were first screened for potency in vitro against homogenous recombinant murine sEH and human sEH, and their octanol-water partition coefficients were determined to help direct our search for more ‘drug-like’ molecules. Pharmacokinetic screening by oral cassette dosing was then undertaken in mice for thirteen piperidines exhibiting good in vitro potency and desirable structural characteristics. In addition several bridging compounds were studied to relate this study to previous publications. The in vivo anti-inflammatory and analgesic bioactivity of one sEHi was evaluated using carrageenan induced inflammatory pain model.
Chemistry
Scheme 1 outlines the two general synthetic routes used to form the unsymmetrical 1,3-disubstituted urea pharmacophore. Aryl isocyanates were purchased or formed from their corresponding anilines by reaction with triphosgene in the presence of aqueous base.35 The heptafluoroisopropylanilines required for compounds 38 and 39 were prepared as described.36
Scheme 1.
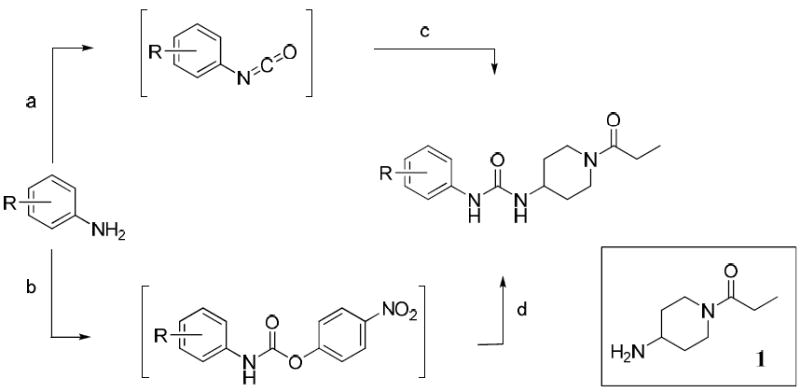
Synthesis of N-propionyl piperidine analogues
Reaction conditions: (a) triphosgene, DCM, sat. NaHCO3 or 1N NaOH, sat. NaCl, 0 °C, 10min; (b) 4-nitrophenyl chloroformate, Et3N, THF, 0 to 50 °C, 1-3h; (c) 1, THF, 0 °C to rt, 1-24h; (d) 1, DMF, 70 °C, 4h.
Amine 1 was prepared from 4-aminopiperidine by protection of the primary amine as its benzyl imine,37 reaction with propionyl chloride in the presence of triethylamine and subsequent deprotection. All isocyanates were reacted with amine 1 to give the desired (1-propionylpiperidin-4-yl)ureas 2–16, 18-21 and 24–40. Saponification of methyl ester 21 with methanolic NaOH afforded benzoic acid 22. Phenol 23 was prepared via 4-benzyloxyisocyanate to avoid formation of a carbamate side product.
Compounds 17 and 31 were prepared by conversion of the corresponding aniline to the intermediate 4-nitrophenyl carbamate, which was then reacted with amine 1 to give the desired urea. Intermediate 41 (Scheme 2) was prepared by the reaction of 4-trifluoromethoxyphenyl isocyanate with 1-BOC-4-aminopiperidine. BOC de-protection gave piperidine 42, which was converted to N-acyl compounds 47–50 and 52 by an EDCI mediated coupling reaction with the respective carboxylic acid.37 Acetylpiperazine 51 was prepared by de-benzylation of 50 and subsequent N-acetylation. Trifluoroacetyl compound 53 was prepared by the reaction of intermediate 42 with ethyl trifluoroacetate. Trihydroxybenzoyl compound 54 was prepared by coupling of 42 with tris O-benzyl protected gallic acid (44) followed by hydrogenolysis.38,39 Intermediate 42 was also converted to N-sulfonyl compounds 55–59 by reaction with the respective sulfonyl chlorides.
Scheme 2.
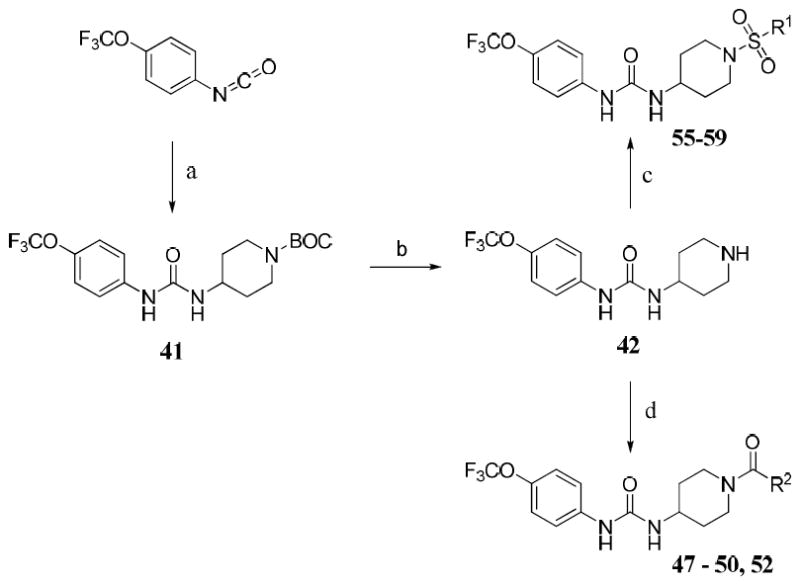
Synthesis of N-acyl and N-sulfonyl piperidine analogues
Reagents and conditions: (a) 1-BOC-4-aminopiperidine, THF, 0 °C to rt, 12h; (b) 1N HCl in MeOH, reflux, 3h; (c) R1SO2Cl, Et3N, THF, 0 °C to rt, 12h; (d) EDCI, R2COOH, DMAP, DCM, rt, 12-24h.
Results & Discussion
Effects of Phenyl Substitution on Potency
In Tables 1-4 a partial structure is shown at the top and the R groups are detailed in the tables. Full structures are shown in the supplementary materials along with detailed synthetic methods. Previous studies have shown that the steric bulk of groups adjacent to the urea is positively correlated with inhibitor potency,26 as was again observed for compounds 2–8 (Table 1). This effect is more directly attributed to the hydrophobicity of these groups, as indicated by the positive correlation between potency (-logIC50) and logP values (r = 0.67/0.87, human/murine) for simple side chains in 2-8. Although replacing the cyclohexyl ring (4) with a more compact phenyl ring (7) caused an 11-fold drop in potency against the human enzyme, substitution of the phenyl ring allowed access to electronically and sterically diverse structures (Tables 2 and 3). Compound 6 provides a bridging structure to recent literature compounds.27 Inclusion of a pyridine on the left side of the molecule as in compound 9 resulted in a dramatic reduction in potency in the piperidine series, although good potency of such pyridines are reported in the patent literature,17,18 and other reports.22,27-30 Inclusion of such polar groups are attractive to improve physical properties, pharmacokinetic properties, and ease of formulation.
Table 1.
Alkyl, carbocycle and unsubstituted aryl analogues
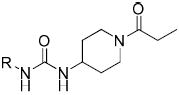 | ||||
|---|---|---|---|---|
| IC50(nM)a | ||||
| Compd | R | Human | Murine | logP (±0.5)b |
| 2 |
|
2.8 | 1.2 | 3.1 |
| 3 |
|
3.9 | 0.9 | 2.3 |
| 4 |
|
12 | 3.5 | 1.8 |
| 5 |
|
3.2 | 0.4 | 3.5 |
| 6 |
|
2.7 | 7.4 | 1.8 |
| 7 |
|
130 | 49 | 1.3 |
| 8 |
|
3.0 | 4.2 | 2.4 |
| 9 |
|
3,800 | >5,000 | ndc |
IC50 values were determined with a fluorescent assay using homogenous recombinant murine and human enzymes (see methods).
Confidence refers to logP value. See supplementary material for full explaination.
The HPLC method used is limited to logP values greater than zero.
Table 4.
N-Acyl and N-sulfonyl piperidine analogues
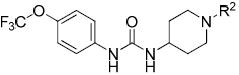 | ||||
|---|---|---|---|---|
| IC50 (nM) | ||||
| Compd | R2 | Human | Murine | logP (±0.5) |
| 47 |
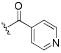
|
0.7 | 1.3 | 2.4 |
| 48 |
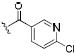
|
0.6 | 0.7 | 2.9 |
| 49 |
|
3.1 | 5.0 | 2.6 |
| 50 |
|
1.5 | 18 | 3.8 |
| 51 |
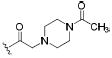
|
0.5 | 1.2 | 2.4 |
| 52 |
|
0.4 | 0.4 | 2.7 |
| 53 |
|
0.4 | 0.4 | 3.1 |
| 54 |
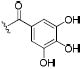
|
0.5 | 2.7 | 2.0 |
| 55 |
|
2.9 | 2.0 | 2.2 |
| 56 |
|
0.4 | 0.7 | 2.6 |
| 57 |
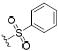
|
1.8 | 0.4 | 3.1 |
| 58 |
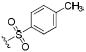
|
0.4 | 0.4 | 3.5 |
| 59 |
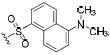
|
0.8 | nda | 4.3 |
nd = Not determined
Table 2.
Substituted phenyl analogues
|
| ||||
|---|---|---|---|---|
| IC50 (nM) | ||||
| Compd | R | Human | Murine | logP (±0.5) |
| 10 |
|
1,700 | >5,000 | 1.6 |
| 11 |
|
40 | 8.7 | 1.8 |
| 12 |
|
43 | 55 | 1.8 |
| 13 |
|
8.3 | 1.3 | 2.3 |
| 14 |
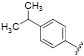
|
2.8 | 3.3 | 2.8 |
| 15 |
|
87 | 8.7 | 1.0 |
| 16 |
|
3.5 | 0.4 | 2.8 |
| 17 |
|
61 | 100 | 1.1 |
| 18 |
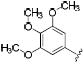
|
>5,000 | >5,000 | 0.8 |
| 19 |
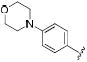
|
2,000 | 650 | 0.2 |
| 20 |
|
38 | 97 | 1.7 |
| 21 |
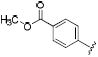
|
140 | 64 | 1.6 |
| 22 |
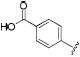
|
330 | 1,000 | 0.4 |
| 23 |
|
406 | 1,400 | 0.0 |
Table 3.
Halophenyl urea analogues
|
| ||||
|---|---|---|---|---|
| IC50 (nM) | ||||
| Compd | R | Human | Murine | logP (±0.5) |
| 24 |
|
79 | 110 | 1.4 |
| 25 |
|
10 | 23 | 2.2 |
| 26 |
|
3.6 | 15 | 2.4 |
| 27 |
|
7.2 | 1.4 | 2.5 |
| 28 |
|
39 | 20 | 1.7 |
| 29 |
|
300 | 780 | 1.6 |
| 30 |
|
21 | 6.6 | 2.2 |
| 31 |
|
1100 | 2900 | 2.0 |
| 32 |
|
3.4 | 0.6 | 2.9 |
| 33 |
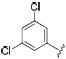
|
0.4 | 1.0 | 3.3 |
| 34 |
|
>5,000 | >5,000 | 1.3 |
| 35 |
|
4.1 | 2.3 | 3.0 |
| 36 |
|
0.7 | 6.5 | 2.4 |
| 37 |
|
17 | 8.8 | 2.4 |
| 38 |
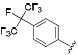
|
0.4 | 0.7 | 3.5 |
| 39 |
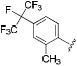
|
17 | 28 | 3.8 |
| 40 |
|
3.7 | 2.8 | 2.5 |
Compared to the unsubstituted phenyl compound 7, the inhibition potencies increased dramatically when small non-polar meta or para (11, 12) substituents were added. Their presence at the ortho position (10) has a clear negative effect on potency. Increasing the size of the hydrophobic para substituent in compounds 12-16 yielded a 3 to 46-fold increase in potency over 7. However, the presence of polar para substituents (17-23) resulted in less potent inhibitors. The phenol 23, a likely metabolite of 15, was a poor inhibitor, presumably due to unfavorable electronic character and polarity. Compound 20 was far less potent than anticipated due to the high polarity of the nitro functionality, despite having a favorable electron deficient urea. Methyl ester and corresponding carboxylic acid compounds 21 and 22, respectively, showed similarly diminished potency. The poor performance of highly polar substituents led us to investigate halophenyl analogues (Table 3). Halogens can increase polarity as a result of their inherent electronegativity, and can also serve to block metabolism at particularly reactive sites and reduce metabolism of the aromatic group by decreasing its electron density.
Thus, compounds 24-27 were synthesized to slow metabolic oxidation of the aromatic ring by cytochrome P450 enzymes (CYPs). These compounds also revealed a slight electronic effect on potency, which was less clear in previous studies.34,40 The observed increase in potency (-log IC50) was correlated with electron withdrawing characteristics according to classical Hammett substituent constants (r = 0.82) and the 1H-NMR chemical shifts of the urea N-H adjacent to the phenyl ring (r = 0.77).41 This effect, in the absence of confounding steric effects, was well revealed in comparing para versus meta fluorination. meta-Fluorophenyl (28, σ = 0.337, 1H-NMR δ 8.57) showed a 2-fold and 5-fold lower IC50 against the human and murine enzymes, respectively, than para-fluorophenyl (24, σ = 0.062, 1H-NMR δ 8.36). Along these lines the 3,5-dichloro substituents yielded a particularly potent sEH inhibitor (33, σ = 0.746, 1H-NMR δ = 8.74). An electron withdrawing group presumably strengthens hydrogen bonding interaction of the urea hydrogen with Asp334 at the catalytic site of the human enzyme – or Asp333 of the murine enzyme – by inductive withdrawal of the nitrogen lone electron pair, further polarizing the urea N-H bond. Fine optimization of 3,4 and 3,5 disubstituted electron withdrawing groups should yield additional potent compounds. The fluorinated para-isopropyl derivative 38 showed an increase in activity over the corresponding isopropyl phenyl derivative 14. As observed previously,40 and for the ortho-tolyl compound 10, ortho mono- or di-halogenation in compounds 29, 31 and 34 drastically decreased potency. However, this effect may be mitigable by the addition of a large hydrophobic para substituent, such as perfluoroisopropyl in compound 39. It is difficult to discern between hydrophobic and electronic contributions to inhibitor potency in vitro. Experimental logP values and calculated molar volumes (data not shown) are highly predictive of the relative potencies of the carbocyclic, alkylphenyl and phenyl ether analogues. However, these criteria do not fully account for the high potency observed for halophenyl compounds, highlighting an electronic contribution to inhibitor potency.
Comparison of Piperidine N-Substituents
The 4-trifluoromethoxyphenyl moiety was used as a metabolically stable replacement for the adamantyl ring of our earlier generation of inhibitors,25,33,40 and was thus conserved in order to investigate N-substitution of the piperidine moiety. N-Acyl and N-sulfonyl substitution of the 1-(piperidin-4-yl)-3-(4-(trifluoromethoxy)phenyl)urea core structure (intermediate 42) yielded multiple highly potent inhibitors (Table 4).
Compounds 47-51 demonstrated the feasibility of introducing a basic nitrogen to allow formulation of the inhibitor as a salt. Analogues 55 and 56 indicate that the sulfonamide group is a good isosteric replacement for the amide, yielding comparably potent inhibitors. These two functional groups may provide valuable pharmacokinetic and pharmacodynamic differences. While bulky substituents on the phenyl moiety improve potency, bulky N-substitution on the piperidine (50, 51, 54, 57-59) did not substantially improve potency over such compounds as 40 or 56. Although bulky N-substitution leads to some increases in potency, previous studies in canines showed that large N-substituents decreased blood levels dramatically following oral administration.25
Small N-acyl or N-sulfonyl substituents may improve hydrophobic interaction with the enzyme while minimizing the need for conformational change upon entry into the catalytic tunnel. The presence of a small hydrophobic group (52, 53) improved potency approximately 9-fold over compound 40 and reached the LOQ of our in vitro assay. Although highly potent, the hydrolytic instability of the trifluoroacetamide 53 makes it unsuited for in vivo use. Cyclopropanecarboxamide 52, however, is a reasonable modification that improves metabolic stability over propionamide 40 without significantly increasing the molecular weight or logP value. Proper substitution on the phenyl ring is crucial for attaining good potency. A varying degree of polarity, bulk and basicity is extremely well tolerated by the target enzyme if attached to the piperidine nitrogen, away from the urea pharmacophore.
Pharmacokinetic Screening in Mice
Carbocycle and phenyl substituted ureas significantly improved pharmacokinetics in comparison to the earlier inhibitors AUDA,34 AEPU (1-[1-acetypiperidin-4-yl]-3-adamantanylurea)24,32 and 1-(1-adamantyl)-3-(1-propionylpiperidin-4-yl)urea compound 232 (Table 5 and supporting information). Our results suggest that the adamantane ring is generally less favorable, in terms of ADME, than other groups. The results also demonstrate that molecules can be fine tuned to optimize ADME and physical properties while retaining high inhibitory potency on the enzyme.
Table 5.
Pharmacokinetic Screening Resultsa
| Compound | Cmax (nM) | Tmax (min) | t1/2 (min) | AUCt (×104nM·min) |
|---|---|---|---|---|
| AUDAb | 14 | 80 | 126 | 0.3 |
| 2 | 138 | 45 | 78 | 1.9 |
| 3 | 2,770 | 60 | 50 | 35 |
| 4 | 4,600 | 50 | 56 | 58 |
| 12 | 5,570 | 30 | 72 | 92 |
| 13 | 4,810 | 30 | 56 | 74 |
| 14 | 5,860 | 50 | 58 | 95 |
| 15 | 13,000 | 70 | 82 | 283 |
| 24 | 8,410 | 83 | 378 | 467 |
| 25 | 18,300 | 188 | 381 | 1,360 |
| 27 | 3,790 | 440 | 881 | 375 |
| 35 | 5,940 | 230 | 814 | 516 |
| 40 | 17400 | 220 | 980 | 1400 |
| 52 | 8900 | 190 | 1180 | 985 |
Values are from single oral cassette dosing at 5 mg/kg in 120-150 μl of 20% PEG400 v/v in oleic ester rich triglyceride
Full pharmacokinetic profiles are shown in Figures S1 – S3 and plots of murine AUC as a function of the IC50 on the human enzyme and the murine enzyme are shown if Figures S4 and S5 in the supplementary material.
AUDA gave biphasic kinetics
Replacing the adamantane with cycloalkanes in compounds 2-4 resulted in substantially higher blood levels. For example, substitution of the adamantyl ring (2) with a cyclohexyl ring (4) resulted in a 33-fold and 81-fold increase in Cmax and AUC, respectively while the cycloheptyl ring (3) increased exposure while retaining potency on the recombinant enzymes (Figures S4, S5). Likewise, the 4-alkylphenyl compounds (12-14) showed improved PK profiles similar to that of compound 4. Interestingly, homologation of the para-alkyl group altered potency in vitro, with 14 exhibiting the lowest IC50 against both enzymes, but did not significantly alter PK. The 4-methoxyphenyl (15) surprisingly showed a further increase in Cmax and AUC by approximately 2-fold and 3- fold, respectively, over compounds 3 and 12-14, suggesting that an ether substituent helps to improve absorption and favorably alters metabolism. Compounds 2, 3, 4 and 12-15 were largely cleared in approximately 8 hours.
The 4-halophenyl compounds 24, 25 and 27 are more persistent than the former materials and have drastically increased Cmax and AUC, supporting our hypothesis that metabolism of the 1-aryl-3-(1-propionylpiperidin-4-yl)urea core structure occurs predominantly at the phenyl moiety. To bridge these data to the anti-cancer drug Sorafenib,42 which is also a potent sEH inhibitor,43 compound 35 was tested.
The Sorafenib-like compound 35 exemplifies excellent stability with two electron withdrawing groups on the phenyl ring.43 Interestingly, the chlorophenyl compound 25 showed significantly better PK than the other mono-halogenated analogues tested. While the lower Cmax and AUC of 4-iodophenyl compound 27 is likely a consequence of its poor solubility, the large difference between 4-fluorophenyl (24) and 4-chlorophenyl (25) suggests that the improved ADME of 25 is a result of favorable polarity and electronics.
Pain reduction in mice
Compound 52 was selected for biological studies as showing both high potency and good pharmacokinetics in mice (Figures S1 - S5). Several studies have demonstrated a dramatic reduction of inflammation by sEH inhibitors both alone and when combined with aspirin and non-steroidal anti-inflammatory drugs.3 Thus compound 52 was tested in an inflammation driven pain model (Figure 1).
Figure 1.
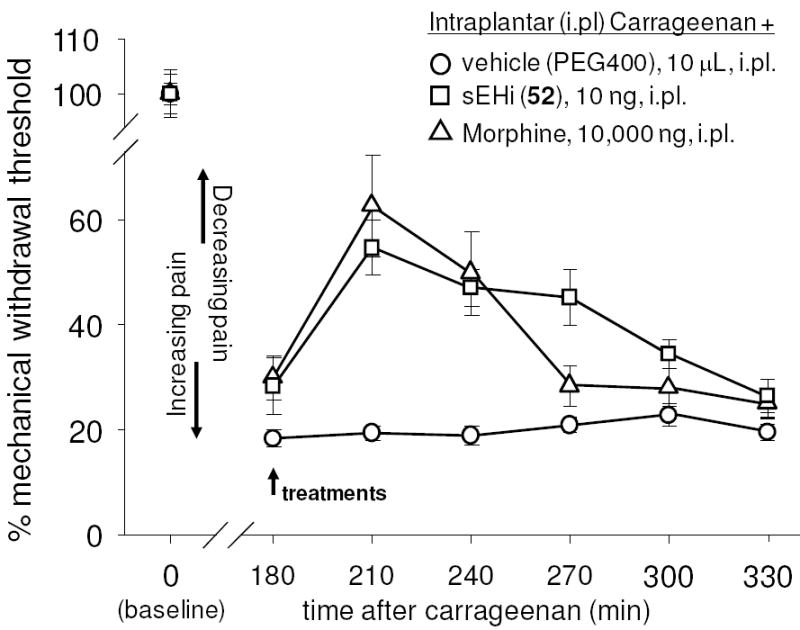
The soluble epoxide hydrolase inhibitor 52 (sEHi) reduces local inflammatory pain at a far lower dose than morphine. The inflammatory agent carrageenan was administered at time 0 resulting in a stable hyperalgesic response for the duration of the experiment. Zero time points represent normal response to pain. Treatment at 180 minutes post carrageenan with either morphine or compound 52 reversed symptoms. Standard deviation represents the average of 6 animals with regard to mechanical withdrawal threshold.
For this study local inflammatory pain was induced by a single, intraplantar injection of carrageenan in rats. The animals’ mechanical withdrawal thresholds, expressed as a percent of control, were measured before (baseline) and 3 hr after carrageenan administration. At this time vehicle or compounds were administered into the plantar surface of the inflamed paw, indicated by an arrow, in a volume of 10 μL. Mechanical withdrawal thresholds were then monitored (n=6 per group). Intraplantar carrageenan led to significant pain observed as a dramatic decrease in mechanical withdrawal threshold of animals. This state was sustained over the course of the experiment. The sEHi (10 ng/paw) and morphine (10,000 ng /paw) significantly reversed carrageenan induced pain in a time dependent manner although the sEHi was more stable than morphine (sEHi vs morphine, ANOVA followed by Dunnett’s 2-sided t-test, p > 0.3) 270 minutes post treatment even though the dose of the sEH inhibitor was 1,000 lower than the morphine dose.
Conclusion
The sEH inhibitors are promising pharmacological leads based on their successful use in multiple models of human disease including prevention and reversal of cardiac hypertrophy,16,44 and a dramatic reduction of inflammation and pain when either used alone or with non-sterodial anti-inflammatory drugs.3,45 As discussed in the introduction there are several excellent lead structures under investigation as sEH inhibitors.17,18 Disubstituted urea sEH inhibitors with substituted piperidines as the secondary pharmacophore were selected as the focus of this study because they are small, drug like, conformationally restricted structures that are synthetically straight forward.25,32 Earlier compounds in this series showed attractive pharmacokinetic properties.24,25 Although many criteria are used for the selection of an investigational new drug candidate or a probe for investigating the arachidonic acid cascade in experimental animals, an estimate of exposure indicated by blood levels (Table 5, Figures S1 – S3) and enzyme inhibition as an indication of potency (Tables 1-4 and S2) are important considerations as are physical properties. When the potential efficacy of compounds is expressed as a function of exposure in murine blood and potency on the recombinant human enzyme, compound 52 represents an improvement of over 104 from the bridging compound AUDA reported 10 years ago (Figure S4).9,19 It is important to look at a compound’s potency in the model species being used and a similar trend with a 105 improvement is seen when murine potency data are used (Figure S5). Evaluation of potency in the model species used is critical. For example there is a good correlation between data on the recombinant mouse and rat enzymes, while there is not a perfect correlation between IC50s on human and murine enzymes (r2 = 0.77; rho = 0.80; Figure S6), and a poor correlation among canine, rodent and human enzymes particularly for the piperidine containing sEH inhibitors.25
Figure S3 compares the blood pharmacokinetic profile of compound 52 with the closely related bridging compound APAU previously published from this laboratory.32 The activity of these acyl pyperidine compounds is similar on the human and murine recombinant enzymes but compound 52 is about 30 times more potent on the human than the murine enzyme.25,32 The data in Figure S3 indicate a very short halflife and small AUC for APAU. When the AUC/IC50 is used as a metric there is a 5,500 fold improvement with compound 52 over the earlier APAU.
Thus multiple compounds in this series and particularly compounds such as 40 and 52 should be reasonable compounds to use in murine models of disease. Both inhibitors have similar potencies on the rodent and human enzyme and reasonable pharmacokinetics. In particular the data in Figure 1 demonstrate the efficacy of compound 52 in a pain model. More broadly, these data demonstrate rational ways to fine tune inhibitors in this series as potential therapeutics. It is possible that combinations of these and related structural units will yield highly potent inhibitors and with pharmacokinetics fine tuned for use in various species.
Experimental Section
General
All reagents and solvents were purchased from commercial suppliers and were used without further purification. All reactions were performed under an inert atmosphere of dry nitrogen. Flash chromatography was performed on silica gel using a dry loading technique, where necessary for poorly soluble products, and elution with the appropriate solvent system. Melting points were determined using an OptiMelt melting point apparatus and are uncorrected. 1H-NMR spectra were collected using a Bruker Avance 500MHz spectrometer or Varian Mercury 300MHz spectrometer. Accurate masses were measured using a Micromass LCT ESI-TOF-MS equipped with a Waters 2795 HPLC. logP and purity analyses were performed using a Hewlett Packard 1100 HPLC equipped with a diode array detector. A Phenomenex Luna 150mm × 4.6mm, 5μ, C-18 column was used for all HPLC analyses. For purity analysis, final products were dissolved in MeOH:H2O (3:1, v/v) at 10μg/mL, and 100μL injections were analyzed in triplicate by HPLC-UV with detection at 210nm, 230nm, 254nm and 290nm. HPLC conditions were the same as those for logP determination (see below). Purity was judged as the percent of total peak area for each wavelength. The lowest observed purity is reported. Compounds were also judged to be pure based on thin layer chromatography visualized with short wave UV and stained with basic potassium permanganate. Compounds were ≥95% pure by HPLC-UV, except where specifically noted (see Supplementary Information). All compounds were evaluated by LC-MS and NMR specifically to insure there was no symmetrical urea impurity present since these compounds can be very active.
logP Determination
Octanol-water partition coefficients were determined by an HPLC method following OECD guideline 117. The accepted error for this method is ±0.5 log units of shake flask values (OEDC guideline 107). Isocratic MeOH:H2O (3:1, v/v), 50 mM ammonium acetate in MeOH:H2O (3:1, v/v) adjusted to pH 9.0, and MeOH:H2O (3:1, v/v) adjusted to pH 3.0 with H3PO4 were used for neutral, basic and acidic analytes, respectively, with a flow rate of 0.75 mL/min. The HPLC method was validated using compounds 24 and 54, which were found to have log P values of 1.9 and 2.3, respectively, using the shake flask method (OECD guideline 107).25 Many alogrithms for calculation of logP values experience difficulty with urea compounds. For example the r2 for the correlation for the CLogP values and shake flask values (OECD guidelines 107 and 122) was <0.3,25 while the correlation with the HPLC method used herein was ~0.7 (Figure S7).
Method A – Synthesis of Aryl and Alkyl Isocyanates
The aniline or amine (1mmol) was added to an ice cold, stirred biphasic mixture of DCM (10mL) and saturated sodium bicarbonate (10mL), or 1N NaOH (3mL) in brine (7mL) where noted. Stirring was stopped momentarily, triphosgene (0.37 eq.) in DCM (1mL) added via syringe to the lower DCM layer and stirring continued for 10 minutes. The DCM layer was removed and filtered through a bed of magnesium sulfate. The filtrate was evaporated to afford the corresponding isocyanate, which was used without further purification.
Method B – Synthesis of Ureas via Isocyanate
The isocyanate (1mmol) was dissolved or suspended in dry THF (3-5mL) and cooled in an ice bath. The amine (1mmol) was dissolved in dry THF (1mL) and slowly added to the reaction. Stirring was continued for 1 to 24 hours at rt. The reaction was quenched with dilute HCl (or water where the BOC group was present) and extracted into ethyl acetate. The combined organic phase was dried, evaporated and purified.
Method C – Synthesis of Ureas via 4-Nitrophenylcarbamate
To an ice cold solution of 4-nitrophenyl chloroformate (1eq) in dry THF was added Et3N (1.3eq) and the appropriate aniline (1eq) dissolved in dry THF. The reaction was allowed to warm to rt, stirred for 30 minutes and then filtered. The filtrate was evaporated and dissolved in DMF. Amine 1 was added and the reaction warmed to 50°C for 1-3 hours. The mixture was cooled to rt, diluted with ethyl acetate, and the organic phase washed with 1N NaOH until the wash was free of yellow p-nitrophenol. The organic phase was dried, evaporated and purified.
Method D – Synthesis of N-Acyl Piperidine Analogues
To a solution of 41 (1eq) in DCM was added the corresponding carboxylic acid (1.1eq), DMAP (1eq) and EDCI (1.1eq). The reaction was stirred for 12-24 hours at rt, and neutral products worked up by partition with EtOAc and 1N HCl (basic products by partition with saturated sodium bicarbonate and EtOAc) and the organic phase was dried, evaporated and purified.
Method E – Synthesis of N-Sulfonyl Piperidine Analogues
To a solution of 41 (152mg, 0.5mmol) in dry THF (5mL) was added Et3N (1.3eq) and the corresponding sulfonyl chloride (1eq) in dry THF (1mL). The reaction was stirred for 12 hours, quenched with 1N HCl and filtered to collect the resulting precipitate, which was further purified.
Enzyme Purification
Recombinant murine and human sEH were produced in a polyhedron positive baculovirus expression system, and they were purified by affinity chromatography as previously reported.46-48
IC50 Assay Conditions
IC50 values were determined using a sensitive fluorescent based assay.49 Cyano(2-methoxynaphthalen-6-yl)methyl trans-(3-phenyl-oxyran-2-yl) methyl carbonate (CMNPC) was used as the fluorescent substrate. Human sEH (1nM) or murine sEH (1nM) was incubated with the inhibitor for 5 min in pH 7.0 Bis-Tris/HCl buffer (25mM) containing 0.1mg/mL of BSA at 30°C prior to substrate introduction ([S] = 5μM). Activity was determined by monitoring the appearance of 6-methoxy-2-naphthaldehyde over 10 minutes by fluorescence detection with an excitation wavelength of 330 nm and an emission wavelength of 465 nm. Reported IC50 values are the average of three replicates with at least two datum points above and at least two below the IC50. The fluorescent assay as performed here has a standard error between 10 and 20%, suggesting that differences of two-fold or greater are significant.49
Pharmacokinetic (PK) Studies
Male CFW mice (7 week old, 24-30 g) were purchased from Charles River Laboratories. All the experiments were performed according to protocols approved by the Animal Use and Care Committee of University of California, Davis. Inhibitors (1 mg each) were dissolved in 1 mL of oleic ester-rich triglyceride containing 20% polyethylene glycol (average molecular weight: 400) to give a clear solution for oral administration. Since many of these compounds are high melting and relatively water insoluble, it is important that they are in true solution to study their pharmacokinetics. The 20% PEG 400 in oleic ester rich triglyceride gave true solutions for all compounds reported. To avoid ill defined levels of linoleate esters (18:2) in the vehicle we used the synthetic triglyceride of oleic esters (18:1) or triolein for vehicle. Cassette 1 contained compounds 2, 24, 25, and 27, cassette 2, compounds 12, 13, and 14, cassette 3 compounds 3, 4 and AUDA, cassette 4 compounds 2, 15, and 35 and cassette 5 compounds 40 and 52. Each cassette was orally administered to 3 or 4 mice at a dose of 5 mg/kg in 120-150μl of vehicle depending on animal weight. Blood (10 μL) was collected from the tail vein using a pipette tip rinsed with 7.5% EDTA(K3) at 0, 0.5, 1, 1.5, 2, 4, 6, 8, 24 hours after oral dosing with the inhibitor. The blood samples were prepared according to the methods detailed in our previous study.24 Blood samples were analyzed using an Agilent 1200 Series HPLC equipped with a 4.6 mm X 150 mm Inertsil ODS-4 3 μm column (GL Science Inc., Japan) held at 40 °C and coupled with an Applied Biosystems 4000 QTRAP hybrid, triple-quadrupole mass spectrometer. The instrument was equipped with a linear ion trap and a Turbo V ion source and was operated in positive ion MRM mode (see Table S1). The solvent system consisted of water/acetic acid (999/1 v/v, solvent A) and acetonitrile/acetic acid (999/1 v/v; solvent B). The gradient was begun at 30% solvent B and was linearly increased to 100% solvent B in 5 min. This was maintained for 3 min, then returned to 30% solvent B in 2 min. The flow rate was 0.4 mL/min. The injection volume was 10 μL and the samples were kept at 4 °C in the auto sampler. Optimized conditions for mass spectrometry are in Table S1.
For clarity standard deviation is not included in Figure S1. There is less than 5% variation in compound levels in replicate blood samples from the same mice. Thus the standard deviation shown in Figure S2A-F represents variation among mice treated with the same compound. The PK parameters of individual mice were calculated by fitting the time dependent curve of blood inhibitor concentration (Figure S2) to a non-compartmental analysis with the WinNonlin software (Pharsight, Mountain View, CA). Parameters determined include the time of maximum concentration (Tmax), maximum concentration (Cmax), half-life (t1/2), and area under the concentration–time curve to terminal time (AUCt). In separate studies to determine dose linearity of selected compounds, pharmacokinetic parameters determined by cassette dosing were found to be predictive of results from dosing individual compounds.24,50 Figure S3 compares the pharmacokinetics of compound 52 with that of the bridging compound APAU.24,25,32 Graphs of exposure as a function of potency are shown in Figures S4 and 5.
Inflammatory pain model
This study was approved by UC Davis Animal Care and Use Committee. Male Sprague-Dawley rats weighing 250-300 gr were obtained from Charles River Inc., and maintained in UC Davis animal housing facilities with ad libitum water and food on a 12hr:12hr light-dark cycle. Behavioral nociceptive testing was conducted by assessing mechanical withdrawal threshold using an electronic von Frey anesthesiometer apparatus (IITC, Woodland Hills, CA).45 The controller was set to ‘maximum holding’ mode so that the highest applied force (in grams) upon withdrawal of the paw was displayed. Three measurements were taken at 1-2 min interstimulus intervals. Data were normalized to percentage values using the formula: mechanical withdrawal threshold (gr) × 100)/ mechanical withdrawal threshold (gr) before carrageenan. Compound 52 was tested using the intraplantar carrageenan elicited inflammatory pain model. Following baseline measurements, carrageenan (50 µL, 1% solution of carrageenan) was administered into the plantar area of one hind paw. Three hour following this, post-carrageenan responses were determined. Immediately after, the vehicle (10 μL PEG400), the sEH inhibitor or morphine sulphate (10 μg in 10 μL saline) were administered into the same paw by intraplantar injection in a volume of 10 µL. Responses following compound administration were monitored over the course of 2.5 hours.
Supplementary Material
Acknowledgments
We thank Dr. Joszef Lango for assistance with accurate mass determination. This work was supported in part by NIEHS R01 ES002710, NIEHS/Superfund Basic Research Program P42 ES004699 and NIH/NHLBI R01 HL059699. BDH is a George and Judy Senior fellow of the American Asthma Foundation.
Footnotes
Abbreviations: sEH, soluble epoxide hydrolase; EETs, epoxyeicosatrienoic acids; DHETs, dihydroxyeicosatrienoic acids; AUDA, 12-(3-adamantane-1-yl-ureido)dodecanoic acid; SAR, structure-activity relationship; T1/2, half-life; Cmax, maximum concentration; IC50, half maximal inhibitory concentration; AUC, area under the curve; LOQ, limit of quantitation.
Supporting Information Available. Blood PK profiles, mass spectrometer parameters, figures of exposure/potency ratios for selected compounds, correlation between IC50’s for human and murine sEH, correlation between experimental and ClogP values, conditions and fragmentation patterns for mass spectrometric analysis and cumulative table of results and properties for all inhibitors are presented. In addition synthetic details and analytical data are presented for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Spector AA, Fang X, Snyder GD, Weintraub NL. Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog Lipid Res. 2004;43:55–90. doi: 10.1016/s0163-7827(03)00049-3. [DOI] [PubMed] [Google Scholar]
- 2.Moghaddam MF, Grant DF, Cheek JM, Greene JF, Williamson KC, Hammock BD. Bioactivation of leukotoxins to their toxic diols by epoxide hydrolase. Nat Med. 1997;3:562–566. doi: 10.1038/nm0597-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schmelzer KR, Kubala L, Newman JW, Kim IH, Eiserich JP, Hammock BD. Soluble epoxide hydrolase is a therapeutic target for acute inflammation. Proc Natl Acad Sci USA. 2005;102:9772–9777. doi: 10.1073/pnas.0503279102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu ZG, Xu FY, Huse LM, Morisseau C, Draper AJ, Newman JW, Parker C, Graham L, Engler MM, Hammock BD, Zeldin DC, Kroetz DL. Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circ Res. 2000;87:992–998. doi: 10.1161/01.res.87.11.992. [DOI] [PubMed] [Google Scholar]
- 5.Iliff JJ, Alkayed NJ. Soluble Epoxide Hydrolase Inhibition: Targeting Multiple Mechanisms of Ischemic Brain Injury with a Single Agent. Future Neurol. 2009;4:179–199. doi: 10.2217/14796708.4.2.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yousif MH, Benter IF, Roman RJ. Cytochrome P450 metabolites of arachidonic acid play a role in the enhanced cardiac dysfunction in diabetic rats following ischaemic reperfusion injury. Auton Autacoid Pharmacol. 2009;29:33–41. doi: 10.1111/j.1474-8673.2009.00429.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Katragadda D, Batchu SN, Cho WJ, Chaudhary KR, Falck JR, Seubert JM. Epoxyeicosatrienoic acids limit damage to mitochondrial function following stress in cardiac cells. J Mol Cell Cardiol. 2009;46:867–875. doi: 10.1016/j.yjmcc.2009.02.028. [DOI] [PubMed] [Google Scholar]
- 8.Xu D, Li N, He Y, Timofeyev V, Lu L, Tsai HJ, Kim IH, Tuteja D, Mateo RKP, Singapuri A, Davis BB, Low R, Hammock BD, Chiamvimonvat N. Prevention and reversal of cardiac hypertrophy by soluble epoxide hydrolase inhibitors. Proc Natl Acad Sci USA. 2006;103:18733–18738. doi: 10.1073/pnas.0609158103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morisseau C, Hammock BD. Epoxide hydrolases: Mechanisms, inhibitor designs, and biological roles. Annu Rev Pharmacol Toxicol. 2005;45:311–333. doi: 10.1146/annurev.pharmtox.45.120403.095920. [DOI] [PubMed] [Google Scholar]
- 10.Imig JD. Epoxide hydrolase and epoxygenase metabolites as therapeutic targets for renal diseases. Am J Physiol Renal Physiol. 2005;289:496–503. doi: 10.1152/ajprenal.00350.2004. [DOI] [PubMed] [Google Scholar]
- 11.Chiamvimonvat N, Ho CM, Tsai HJ, Hammock BD. The soluble epoxide hydrolase as a pharmaceutical target for hypertension. J Cardiovasc Pharmacol. 2007;50:225–237. doi: 10.1097/FJC.0b013e3181506445. [DOI] [PubMed] [Google Scholar]
- 12.Imig JD, Hammock BD. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat Rev Drug Discov. 2009;8:794–805. doi: 10.1038/nrd2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Y-XJ, Ulu A, Zhang L-N, Hammock BD. Soluble epoxide hydrolase in atherosclerosis. Curr Atheroscler Rep. 2010;12:174–183. doi: 10.1007/s11883-010-0108-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gross GJ, Nithipatikom K. Soluble epoxide hydrolase: a new target for cardioprotection. Curr Opin Investig Drugs. 2009;10:253–258. [PMC free article] [PubMed] [Google Scholar]
- 15.Fang X. Soluble epoxide hydrolase: a novel target for the treatment of hypertension. Recent Pat Cardiovasc Drug Discov. 2006;1:67–72. doi: 10.2174/157489006775244227. [DOI] [PubMed] [Google Scholar]
- 16.Harris TR, Li N, Chiamvimonvat N, Hammock BD. The potential of soluble epoxide hydrolase inhibition in the treatment of cardiac hypertrophy. Congest Heart Fail. 2008;14:209–224. doi: 10.1111/j.1751-7133.2008.08430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marino JP., Jr Soluble epoxide hydrolase, a target with multiple opportunities for cardiovascular drug discovery. Curr Top Med Chem. 2009;9:452–463. doi: 10.2174/156802609788340805. [DOI] [PubMed] [Google Scholar]
- 18.Shen HC. Soluble epoxide hydrolase inhibitors: a patent review. Expert Opin Ther Pat. 2010 doi: 10.1517/13543776.2010.484804. in press. [DOI] [PubMed] [Google Scholar]
- 19.Morisseau C, Goodrow MH, Dowdy D, Zheng J, Greene JF, Sanborn JR, Hammock BD. Potent urea and carbamate inhibitors of soluble epoxide hydrolases. Proc Natl Acad Sci USA. 1999;96:8849–8854. doi: 10.1073/pnas.96.16.8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim IH, Heirtzler FR, Morisseau C, Nishi K, Tsai HJ, Hammock BD. Optimization of amide-based inhibitors of soluble epoxide hydrolase with improved water solubility. J Med Chem. 2005;48:3621–3629. doi: 10.1021/jm0500929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xie YL, Liu YD, Gong GL, Smith DH, Yan F, Rinderspacher A, Feng Y, Zhu ZX, Li XP, Deng SX, Branden L, Vidovic D, Chung C, Schurer S, Morisseau C, Hammock BD, Landry DW. Discovery of potent non-urea inhibitors of soluble epoxide hydrolase. Bioorg Med Chem Lett. 2009;19:2354–2359. doi: 10.1016/j.bmcl.2008.09.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eldrup AB, Soleymanzadeh F, Taylor SJ, Muegge I, Farrow NA, Joseph D, McKellop K, Man CC, Kukulka A, De Lombaert S. Structure-based optimization of arylamides as inhibitors of soluble epoxide hydrolase. J Med Chem. 2009;52:5880–5895. doi: 10.1021/jm9005302. [DOI] [PubMed] [Google Scholar]
- 23.Anandan SK, Webb HK, Do ZN, Gless RD. Unsymmetrical non-adamantyl N,N’-diaryl urea and amide inhibitors of soluble expoxide hydrolase. Bioorg Med Chem Lett. 2009;19:4259–4263. doi: 10.1016/j.bmcl.2009.05.102. [DOI] [PubMed] [Google Scholar]
- 24.Liu JY, Tsai HJ, Hwang SH, Jones PD, Morisseau C, Hammock BD. Pharmacokinetic optimization of four soluble epoxide hydrolase inhibitors for use in a murine model of inflammation. Br J Pharmacol. 2009;156:284–296. doi: 10.1111/j.1476-5381.2008.00009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsai HJ, Hwang SH, Morisseau C, Yang J, Jones PD, Kim IH, Hammock BD. Pharmacokinetic Screening of Soluble Epoxide Hydrolase Inhibitors in Dogs. Eur J Pharm Sci. 2010;40:222–238. doi: 10.1016/j.ejps.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McElroy NR, Jurs PC, Morisseau C, Hammock BD. QSAR and classification of murine and human soluble epoxide hydrolase inhibition by urea-like compounds. J Med Chem. 2003;46:1066–1080. doi: 10.1021/jm020269o. [DOI] [PubMed] [Google Scholar]
- 27.Shen HC, Ding FX, Deng Q, Xu S, Chen HS, Tong X, Tong V, Zhang X, Chen Y, Zhou G, Pai LY, Alonso-Galicia M, Zhang B, Roy S, Tata JR, Berger JP, Colletti SL. Discovery of 3,3-disubstituted piperidine-derived trisubstituted ureas as highly potent soluble epoxide hydrolase inhibitors. Bioorg Med Chem Lett. 2009;19:5314–5320. doi: 10.1016/j.bmcl.2009.07.138. [DOI] [PubMed] [Google Scholar]
- 28.Shen HC, Ding FX, Wang S, Deng Q, Zhang X, Chen Y, Zhou G, Xu S, Chen HS, Tong X, Tong V, Mitra K, Kumar S, Tsai C, Stevenson AS, Pai LY, Alonso-Galicia M, Chen X, Soisson SM, Roy S, Zhang B, Tata JR, Berger JP, Colletti SL. Discovery of a Highly Potent, Selective, and Bioavailable Soluble Epoxide Hydrolase Inhibitor with Excellent Ex Vivo Target Engagement. J Med Chem. 2009;52:5009–5012. doi: 10.1021/jm900725r. [DOI] [PubMed] [Google Scholar]
- 29.Kowalski JA, Swinamer AD, Muegge I, Eldrup AB, Kukulka A, Cywin CL, De Lombaert S. Rapid synthesis of an array of trisubstituted urea-based soluble epoxide hydrolase inhibitors facilitated by a novel solid-phase method. Bioorg Med Chem Lett. 2010;20:3703–3707. doi: 10.1016/j.bmcl.2010.04.078. [DOI] [PubMed] [Google Scholar]
- 30.Eldrup AB, Soleymanzadeh F, Farrow NA, Kukulka A, De Lombaert S. Optimization of piperidyl-ureas as inhibitors of soluble epoxide hydrolase. Bioorg Med Chem Lett. 2010;20:571–575. doi: 10.1016/j.bmcl.2009.11.091. [DOI] [PubMed] [Google Scholar]
- 31.Anandan SK, Do ZN, Webb HK, Patel DV, Gless RD. Non-urea functionality as the primary pharmacophore in soluble epoxide hydrolase inhibitors. Bioorg Med Chem Lett. 2009;19:1066–1070. doi: 10.1016/j.bmcl.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 32.Jones PD, Tsai HJ, Do ZN, Morisseau C, Hammock BD. Synthesis and SAR of conformationally restricted inhibitors of soluble epoxide hydrolase. Bioorg Med Chem Lett. 2006;16:5212–5216. doi: 10.1016/j.bmcl.2006.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hwang SH, Tsai HJ, Liu JY, Morisseau C, Hammock BD. Orally bioavailable potent soluble epoxide hydrolase inhibitors. J Med Chem. 2007;50:3825–3840. doi: 10.1021/jm070270t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morisseau C, Goodrow MH, Newman JW, Wheelock CE, Dowdy DL, Hammock BD. Structural refinement of inhibitors of urea-based soluble epoxide hydrolases. Biochem Pharmacol. 2002;63:1599–1608. doi: 10.1016/s0006-2952(02)00952-8. [DOI] [PubMed] [Google Scholar]
- 35.Nowick JS, Holmes DL, Noronha G, Smith EM, Nguyen TM, Huang SL. Synthesis of peptide isocyanates and isothiocyanates. J Org Chem. 1996;61:3929–3934. doi: 10.1021/jo960038n. [DOI] [PubMed] [Google Scholar]
- 36.Onishi Masanobu, Yoshiura Akihiko, Kohno Eiji, Tsubata Kenji. A process for producing perfluoroalkylaniline derivatives. 2000 June 7; EP 1006102. [Google Scholar]
- 37.Sonda S, Kawahara T, Murozono T, Sato N, Asano K, Haga K. Design and synthesis of orally active benzamide derivatives as potent serotonin 4 receptor agonist. Bioorg Med Chem. 2003;11:4225–4234. doi: 10.1016/s0968-0896(03)00412-7. [DOI] [PubMed] [Google Scholar]
- 38.Dhaon MK, Olsen RK, Ramasamy K. Esterification of N-protected alpha-amino-acids with alcohol/carbodiimide/4-(dimethylamino)-pyridine - racemization of aspartic and glutamic-acid derivatives. J Org Chem. 1982;47:1962–1965. [Google Scholar]
- 39.Ren Y, Himmeldirk K, Chen X. Synthesis and Structure-Activity Relationship Study of antidiabetic penta-O-galloyl-D-glucopyranose and its analogues. J Med Chem. 2006;49:2829–2837. doi: 10.1021/jm060087k. [DOI] [PubMed] [Google Scholar]
- 40.Hwang SH, Morisseau C, Do Z, Hammock BD. Solid-phase combinatorial approach for the optimization of soluble epoxide hydrolase inhibitors. Bioorg Med Chem Lett. 2006;16:5773–5777. doi: 10.1016/j.bmcl.2006.08.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Correlations were determined from plots of -logIC50 v. Hammet constants and 1H-NMR chemical shifts. Compounds 7, 12, 15, 24-28, 30, 32 and 33 were examined to minimize possible confounding effects on inhibitor potency, such as steric bulk, high polarity and ortho subtitution
- 42.Zhong HZ, Bowen JP. Molecular design and clinical development of VEGFR kinase inhibitors. Curr Top Med Chem. 2007;7:1379–1393. doi: 10.2174/156802607781696855. [DOI] [PubMed] [Google Scholar]
- 43.Liu J-Y, Park S-H, Morisseau C, Hwang SH, Hammock BD, Weiss RH. Sorafenib has soluble epoxide hydrolase inhibitory activity, which contributes to its effect profile in vivo. Mol Cancer Ther. 2009;8:2193–2203. doi: 10.1158/1535-7163.MCT-09-0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ai D, Pang W, Li N, Xu M, Jones PD, Yang J, Zhang Y, Chiamvimonvat N, Shyy JY-J, Hammock BD, Zhu Y. Soluble epoxide hydrolase plays an essential role in angiotensin II-induced cardiac hypertrophy. Proc Natl Acad Sci USA. 2009;106:564–569. doi: 10.1073/pnas.0811022106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Inceoglu B, Jinks SL, Ulu A, Hegedus CM, Georgi K, Schmelzer KR, Wagner K, Jones PD, Morisseau C, Hammock BD. Soluble epoxide hydrolase and epoxyeicosatrienoic acids modulate two distinct analgesic pathways. Proc Natl Acad Sci USA. 2008;105:18901–18906. doi: 10.1073/pnas.0809765105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beetham JK, Tian TG, Hammock BD. CDNA cloning and expression of a soluble epoxide hydrolase from human liver. Arch Biochem Biophys. 1993;305:197–201. doi: 10.1006/abbi.1993.1411. [DOI] [PubMed] [Google Scholar]
- 47.Grant DF, Storms DH, Hammock BD. Molecular-cloning and expression of murine liver soluble epoxide hydrolase. J Biol Chem. 1993;268:17628–17633. [PubMed] [Google Scholar]
- 48.Wixtrom RN, Silva MH, Hammock BD. Affinity purification of cytosolic epoxide hydrolase using derivatized epoxy-activated sepharose gels. Anal Biochem. 1988;169:71–80. doi: 10.1016/0003-2697(88)90256-4. [DOI] [PubMed] [Google Scholar]
- 49.Jones PD, Wolf NM, Morisseau C, Whetstone P, Hock B, Hammock BD. Fluorescent substrates for soluble epoxide hydrolase and application to inhibition studies. Anal Biochem. 2005;343:66–75. doi: 10.1016/j.ab.2005.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Watanabe T, Schulz D, Morisseau C, Hammock BD. High-throughput pharmacokinetic method: Cassette dosing in mice associated with minuscule serial bleedings and LC/MS/MS analysis. Anal Chim Acta. 2006;559:37–44. doi: 10.1016/j.aca.2005.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.