

Abstract
Transposons are abundant components of eukaryotic genomes, and play important role in genome evolution. The knowledge about these elements should contribute to the understanding of their impact on the host genomes. The hAT transposon superfamily is one of the best characterized superfamilies in diverse organisms, nevertheless, a detailed study of these elements was never carried in sugarcane. To address this question we analyzed 32 cDNAs similar to that of hAT superfamily of transposons previously identified in the sugarcane transcriptome. Our results revealed that these hAT-like transposases cluster in one highly homogeneous and other more heterogeneous lineage. We present evidences that support the hypothesis that the highly homogeneous group is a domesticated transposase while the remainder of the lineages are composed of transposon units. The first is common to grasses, clusters significantly with domesticated transposases from Arabidopsis, rice and sorghum and is expressed in different tissues of two sugarcane cultivars analyzed. In contrast, the more heterogeneous group represents at least two transposon lineages. We recovered five genomic versions of one lineage, characterizing a novel transposon family with conserved DDE motif, named SChAT. These results indicate the presence of at least three distinct lineages of hAT-like transposase paralogues in sugarcane genome, including a novel transposon family described in Saccharum and a domesticated transposase. Taken together, these findings permit to follow the diversification of some hAT transposase paralogues in sugarcane, aggregating knowledge about the co-evolution of transposons and their host genomes.
Electronic supplementary material
The online version of this article (doi:10.1007/s00438-011-0670-8) contains supplementary material, which is available to authorized users.
Keywords: Sugarcane, Transposon, hAT transposase, Domesticated transposase
Introduction
The activity of transposable elements (TE) and their inherent ability to change location within chromosomes results in mutational activity mediating structural and functional changes in genomes. Gene disruption upon insertion, gene capture or modified gene expression patterns are examples of TE-genome interplay (Wessler 2006; Lisch and Bennetzen 2011). These changes provide genetic variability and are thought to influence genome evolution and species formation (McClintock 1984). The recruitment of transposase functional domains to create new host genes exemplifies the importance of these elements as active players in the evolution of the genomes (Britten 2004; Volff 2006; Casola et al. 2007; Feschotte 2008).
The simplest autonomous transposons are DNA sequences that encode a transposase protein responsible for their mobilization in the genome and are flanked by terminal inverted repeats (TIRs). The transposase has a DDE catalytic domain that recognizes the TIRs and promotes the excision and insertion of the element in a different site of the genome (Yuan and Wessler 2011). A non-autonomous element can be mobilized in trans through the recognition of its TIRs by a functional transposase encoded by an autonomous element. The Ac/Ds family comprises autonomous and non-autonomous elements and was originally described as “controlling elements of the gene” by Barbara McClintock in maize (McClintock 1946). Ac is one of the defining members of the hAT superfamily of transposons, together with Tam3 from Antirrhinum majus (Hehl et al. 1991), and hobo from Drosophila melanogaster (Streck et al. 1986; Calvi et al. 1991). hAT transposases exhibit conserved domains that have been identified in plants, fungi and animals as well as in humans (Kempken and Windhofer 2001). The most characteristic domains are the zinc finger at the amino terminal region of the protein, and two other conserved domains at the carboxy terminal region, one involved in dimerization (Essers et al. 2000) and other of whose function is still unknown.
The mechanism through which a transposase is incorporated by the genome to the formation of a new gene is called domestication. The hAT-like domesticated transposase DAYSLEEPER was described as a factor that binds to the Kubox1 motif present in the upstream region of some genes, including the DNA repair gene Ku70, and it is essential for the development of Arabidopsis thaliana (Bundock and Hooykaas 2005). Other plant genomes have similar transposase-derived genes such as wheat, barley and rice (Muehlbauer et al. 2006) with detectable expression, but no function has yet been assigned.
Sugarcane is one of the most economically important worldwide crops, providing raw material to the sugar and ethanol industry. Commercial sugarcane cultivars (2n = 100–130) are the recent hybrids with a highly polyploid and aneuploid genome (D’Hont et al. 1996; Grivet et al. 2004). Plants can support large numbers of highly heterogeneous TE families, thought to be due to the elevated frequency of polyploidy (Kraitshtein et al. 2010). In this context, sugarcane is proving to be an interesting model to study TE evolution and expression. Exploring TE diversification and distribution represents an interesting opportunity to unravel new mobile elements and potentially new gene functions.
Previous studies described the expression of 162 fully sequenced sugarcane cDNAs similar to several TE families (Araujo et al. 2005). Of these, 32 were similar to the hAT superfamily of transposons (Rubin et al. 2001). The present work describes the diversity, structure and expression of sugarcane hAT transposase paralogues. Our results reveal three distinct lineages of hAT-like transposase paralogues in sugarcane genome. One lineage presents characteristics that suggest domestication. We also recovered genomic copies of other lineage, identifying a novel transposon family present in Saccharum, named SChAT. Analyses of TE content in sugarcane should contribute to the understanding of their impact on the evolution and functioning of polyploid genomes.
Materials and methods
Sequence analyses: clustering of protein sequences
Nucleotide sequences of sugarcane hAT-like cDNA clones previously sequenced and annotated in our lab were analyzed. The annotation criterion to characterize it as hAT-like was a cutoff of −50 in a BLASTX against the transposase of the element Ac. These cDNAs came from several sugarcane hybrid cultivars (Table S1 in the supplementary material). These sequences were translated to amino acids using the program BioEdit Sequence Alignment Editor (Hall 1999). hAT-like sequences from sorghum, rice, maize, tomato, tobacco, grape, arabidopsis and populus were also retrieved from GenBank through BLASTX against Ac transposase aminoacid sequence, and introduced into the alignment. Accession numbers are as follows: Sorghum sequences (1–4): XP_002463929.1, XP_002442948.1, XP_002444199.1 and XP_002455637.1. Rice sequences (1–6): AC134929.2, AK067574, AK102230, AK105085, AK121128 and BAC10751. Maize sequence AY111033. Tomato sequences (Lycop1–3): AK319242.1, BT014568.1 and AC234415.1. Tobacco sequences (1–3): X97569.1, BT014568.1 and AK319242.1. Grape sequences (Vitis1–3): AM487463.2, AM486739.1 and XP_002275062.1. Arabidopsis sequences (Ath1–5): NM_106651, AL137079, AC009322, AC011717 and AAD24567. Populus sequences (1–4): XP_002325202.1, XP_002327524.1, XP_002332734.1 and XP_002331299.1. hobo transposase (P12258), Ac transposase (CAA29005), Tam3 transposase (CAA38906), DAYSLEEPER (AY728267.1). Two conserved motifs were aligned, one containing residues 555–594 of Ac transposase, and the dimerization domain (PFAM acession number PF05699) containing residues 682–719 of Ac transposase. The alignment was carried out using the program ClustalX 1.81 (Thompson et al. 1997), with default multiple alignment parameters. The program MEGA4.0 was used to generate the corresponding trees (Tamura et al. 2007), using the distance method and neighbor joining algorithm, with default parameters. Bootstrap test of 1,000 replicates was made. The GenBank accession numbers of genomic copies SCHAT-G1, SCHAT-G2, SCHAT1, SCHAT2 and SCHAT3 are HM067360, HM067361, HM067362, HM067363 and HM067364, respectively.
Evolutionary analysis
The nucleotide cDNA sequences from the three lineages were aligned separately, as follows: domesticated lineage 074: TE016, TE017, TE035, TE048, TE074, TE096, TE124, TE132, TE203, TE207, TE217, TE223 and TE265. Lineage 191: TE191 and TE221. Lineage 257: TE001, TE154 and TE257. Sequence alignments were performed using ClustalW (Thompson et al. 1994) in the Bioedit program (Hall 1999) and corrected manually if necessary, removing the nucleotides relative to stop codons or frameshifts. The number of synonymous substitutions per synonymous site (dS) and non-synonymous substitutions per non-synonymous site (dN), the dN:dS ratios and codon-based Z test were calculated through the modified Nei-Gojobori method with the Jukes-Cantor correction, implemented in the MEGA4.0 software (Tamura et al. 2007). Time divergence between the two loci of the domesticated lineage was estimated using T = dS/2k, where dS is the estimated number of synonymous substitutions per synonymous site, and k is the average synonymous substitution rate, assuming a rate of 0.0065 mutations per site per MY, as previously reported to the Adh loci of grasses (Gaut et al. 1996).
Molecular hybridization
Probes were based on the nucleotide sequences of cDNA clones TE074, TE191 and TE257, and were 182, 187 and 146 bp in length, respectively. To avoid cross hybridization, primers were designed outside the conserved domains to amplify unique probes for each clone. PCR-synthesized probes were labeled with α32 dCTP using a Random Primers DNA Labeling System kit (Invitrogen # 18187-013). Molecular hybridizations were performed on 10 μg of XbaI-digested genomic DNA from the following species: potato (Solanum tuberosum), orchid (Catassetum fimbriatum), pineapple (Ananas comosus), rice (Oryza sativa), maize (Zea mays), two ancestral species of the genus Saccharum, Saccharum spontaneum (variety Mandalay) and Saccharum officinarum (variety Badila), and sugarcane hybrid cultivars SP-89-1115 and SP-80-3280. DNA was extracted using a modified version of the CTAB method (Porebski et al. 1997); 10 units of XbaI (New England Biolabs) were used for each 1 μg of genomic DNA, according to the manufacturer’s instructions. DNA was digested at 37°C for 16 h. The hydrolyzed DNA was electrophoresed (1.8 V/cm, 16 h) through a 0.8% agarose gel, in 0.5× TBE buffer. The gel was washed in 0.25 N HCl for 15 min, in 0.4 N NaOH for 15 min, and then transferred by capillary action to nylon membrane (Gene Screen Plus Hybridization Transfer Membrane–Life Science) in 0.4 N NaOH buffer for 16 h. After transfer, membranes were washed in 2× SSC solution for 15 min and dried at room temperature. Membranes were placed for pre-hybridization at 65°C under rotation for 4 h in a solution of 10% Dextran, 1% SDS, 6% NaCl in deionized water. After the addition of the probe the hybridization time was 16 h. Membranes were then washed twice with 0.5× SSC/0.1% SDS for 30 min at 65°C, and twice with 0.1× SSC/0.1% SDS for 30 min at 65°C. The membranes were exposed to X-Ray films (Kodak T-MAT G/RA Film) in cassettes at −70°C; films were developed according to the manufacturer’s instructions.
RNA expression analysis
The probes used on RNA blots were TE074 (182 bp) and TE191 (187 bp), i.e., the same probes as for DNA hybridization. A probe for the ubiquitin gene was used as a control for constitutive expression. RNA was extracted from the leaves and roots of sugarcane plants of the variety R570 grown in the greenhouse, from in vitro calli from the variety SP-89-1115. Six-months-old plants grown under field conditions were used for total RNA extraction form apical meristem, stem, mature and immature leaves. RNA was extracted using the lithium chloride method, adapted from Sambrook et al. (1989). To samples containing 10 μg of RNA, MOPS was added to a final concentration of 1×, formaldehyde 6.5% and formamide 48%. Samples were incubated at 60°C for 15 min, and buffer (bromophenol blue 0.25%, xylene cyanol 0.25%, EDTA 1 mM and glycerol 50%) was added subsequently to 16% and ethidium bromide to 0.35%. Samples were electrophoresed in denaturing agarose gels (1.5% agarose, 6.7% formaldehyde and MOPS 1×) at 80 V for 3 h. After electrophoresis, the gel was washed three times in deionized water for 10 min, and once in 10× SSC for 40 min, followed by capillary transfer to nylon membrane (Gene Screen Plus Hybridization Transfer Membrane–Life Science), in 10× SSC solution for 18 h. After transfer, the membrane was rinsed quickly in 10× SSC, baked at 80°C for 2 h and exposed to UV light for 1 min to fix the RNA. The membrane was placed in a hybridization bottle for pre-hybridization at 65°C under rotation for 4 h in a solution of 0.5 M phosphate buffer, 7% SDS and 1 mM EDTA in deionized water. Hybridization with probes occurred at 65°C under rotation for 40 h and the membrane was then washed with 2× SSC for 5 min at room temperature, twice with 2× SSC/SDS 0.5% for 30 min at 65°C, and finally with 2× SSC for 5 min at room temperature. X-Ray films (Kodak T-MAT G/RA Film) were exposed to the membranes in cassettes at −70°C, and developed according to the manufacturer’s instructions. Signals on the films were quantified by densitometric analysis. Signal intensity was determined using the “Kodak Digital Science 1D” program. After subtraction of background noise, data were normalized to the ubiquitin hybridization signal.
TIR and subsequent genomic copy recovery
A two-step approach was used to access the genomic copies. First, an inverse PCR (iPCR) was performed using 10 μg of genomic DNA from cultivar SP80-3280 digested with EcoRI (New England Biolabs) and ligated to form circular molecules using T4 DNA Ligase (Fermentas), according to manufacturer’s instructions. These circularized fragments of genomic DNA enabled outward amplification from primers designed on the outermost portion of TE221. A nested primers strategy was used in the iPCR. In the first round of amplification 100 ng of circularized genomic DNA was used as template, and the reactions were submitted to the following thermal cycle: 2 min at 94°C, 35 rounds of 30 s at 94°C, 30 s at 57°C and 1 min at 72°C, and a final extension time of 72°C for 10 min. In the second round 1 μl of the first round PCR, with a final volume of 50 μl, was used as template to the second amplification, which was submitted to the same thermal cycle. The primers used are presented in Table 1. The product of the iPCR was cloned in the pGEM-T-Easy, according to manufacturer’s instructions, and sequenced using primers of the vector. The TIRs of the element were identified in these sequenced fragments amplified in the iPCR. New primers were designed on the sequences of the TIRs, and a regular PCR was performed. This amplification was made using “Elongase Enzyme Mix” (Invitrogen), according to the manufacturer’s instructions and utilizing 80 ng of genomic DNA of the hybrid sugarcane variety SP-80-3280 as template, in a final volume of 50 μl. The reaction was submitted to the following thermal cycle: 2 min at 94°C, 35 rounds of 30 s at 94°C, 30 s at 55°C and 4 min at 68°C, and a final extension time of 68°C for 10 min. PCR products were resolved in low-melting agarose gels, the bands of interest were excised and purified through “GFX PCR DNA and Gel Purification Kit” (Amersham), cloned in the vector “pGEM-T-Easy” (Promega) and sequenced in both directions with the primers anchored on the vector. The sequences obtained, SChAT-G1 and SChAT-G2, were analyzed using BLASTN, BLASTX and ORF Finder on Genbank (http://www.ncbi.nlm.nih.gov) and TIGR (http://www.tigr.org).
Table 1.
Primers used in the iPCR assay
| Nested PCR | Name | Sequence |
|---|---|---|
| External primer | IPPTE221-1F | 5′ GACCAGAAGGCCTGCAAC 3′ |
| IPPTE221-1R | 5′ GACTAATGACTGTAACTTATCAACG 3′ | |
| Internal primer | IPPTE221-2F | 5′ GCTCAAAGGGATTATCATTCC 3′ |
| IPPTE221-2R | 5′ GCATCTTCAGTCTTGGCTCA 3′ |
External and internal nested PCR primers are indicated
Recover of SChAT elements from sequenced BACs
BACs were chosen by hybridization of lineage-specific probes (TE074 and TE191) against a commercial BAC library (Clemson University Genomics Institute) according to the manufacturer’s instructions. Positive clones, BACs SHCRBa167_G07 (containing SChAT2), SHCRBa160_F01 (containing SChAT1) and SHCRBa185_O22 (containing SChAT3) were sequenced using GS-FLX pyrosequencer (Roche), according to the manufacturer’s instructions. The SChAT family elements were characterized and compared with Ac transposase and the SChAT-G1 element. Two conserved motifs were aligned, one containing the residues 140–196, i.e., the Zinc finger DNA ligation domain of Ac transposase (Pfam accession number PF05699), and the second motif was the dimerization domain, containing the residues 682–719 of Ac transposase (Pfam acession number PF05699). The alignment was carried out using the multiple alignment program ClustalX (MUSCLE tool), with default parameters.
Results
Uncovering hAT-like transposases in sugarcane
A local sugarcane mobilome database of expressed TEs had previously been constructed, based on the full-length sequencing of 162 cDNAs related to TEs (Rossi et al. 2001; Araujo et al. 2005). Thirty-two cDNAs were identified as similar to the hAT superfamily of transposons and were analyzed further despite frameshifts and in-frame stop codons being detected. The diversity of the sequences was evaluated through alignment of two conserved domains characteristic of hAT superfamily transposases (Fig. 1a). These domains are present in the predicted sequence of 21 sugarcane clones, which were aligned at the protein level with related sequences recovered from sorghum, rice, maize, arabidopsis, tomato, tobacco, grape and populus, and the reference sequences from Ac and Tam3 of the hAT superfamily. The resulting analysis revealed a subdivision of the sugarcane sequences into distinct lineages. A total of 13 clones cluster in a particular lineage, that ranges from 86 to 100% nucleotide identity and which are almost identical at the amino acid level in the region of the conserved domains. This set of sequences, comprising TE016, TE017, TE035, TE048, TE074, TE096, TE124, TE132, TE203, TE207, TE217, TE223 and TE265, was named arbitrarily lineage 074, based on the cDNA chosen to design the probe used for further hybridization assays (Fig. 1a). Lineage 074 clusters with the domesticated transposase DAYSLEEPER from arabidopsis (Bundock and Hooykaas 2005) in a branch supported by a bootstrap of 88%. This branch also displays a clear division between monocot and eudicot sequences (Fig. 1b). Lineage 074 sequences exhibit 89% amino acid similarity with DAYSLEEPER, along a stretch of 75 amino acids concatenated from two protein domains. The remaining sequences are heterogeneous lineages and cluster with the original plant transposable elements Ac and Tam3 (Fig. 1b). Two of these lineages were named lineage 191 and lineage 257, according to the cDNA used as probe for further hybridizations. An ellipse in Fig. 1b highlights these cDNAs.
Fig. 1.
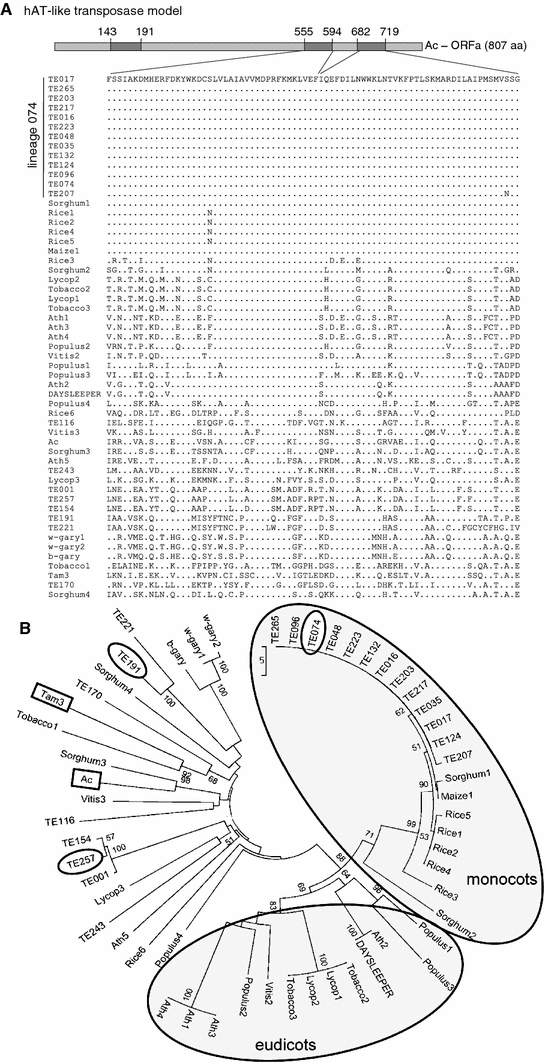
Multiple-alignment of conserved hAT superfamily transposase domains. a Alignment of a conserved domain that encompasses amino acids 555–594 of Ac transposase (Pfam PB001541), and the dimerization domain (Pfam PF05699) encompassing amino acids 682–719. The alignment comprises amino acid sequences of the transposons Activator and Tam3, sugarcane cDNAs, the monocots Zea mays (Maize1), Sorghum bicolor (Sorghum1-4), Oryza sativa (Rice1-6), and the dicotyledoneous Arabidopsis thaliana (Ath1-5), Nicotiana tabacum (Tobacco1-3), Vitis vinifera (Vitis1-4), Solanum lycopersicon (Lycop1-3) and Populus trichocarpa (Populus1-4) and DAYSLEEPER. The conserved lineage 074 is indicated with a bar. b Distance tree generated from aligned hAT-related sequences. The tree refers to the concatenated sequences from both alignments presented in a. Boxes highlight the founding elements of the hAT superfamily, Ac and Tam3, and the cDNAs used as probes, by small ellipses. Large ellipses define monocots and eudicots. Bootstrap values are indicated in the branches as numbers
Since different lineages were identified in sugarcane cDNAs, a genomic hybridization assay was performed to check the relative distribution and abundance between Saccharum species and hybrid cultivars, as well as among different grasses and other angiosperms (Fig. 2). Lineages 074, 191 and 257 were chosen to genomic hybridization assay. Under stringent conditions, probes from any of these lineages did not hybridize with distantly related monocots that neither belong to the Poaceae family, pineapple (Bromeliaceae) or orchid (Orchidaceae), nor to potato (Solanaceae; a dicot genome). Lineage 074 hybridized to all grasses analyzed (sugarcane modern cultivars, S. officinarum, S. spontaneum, maize and rice) with a low copy number profile. The 146 bp probe of TE257 hybridized with all Saccharum genomes, while the 187 bp probe of TE191 hybridized exclusively with sugarcane hybrid genomes and with S. officinarum. The TE257 probe is located between the nucleotides 1377 and 1523 of the cDNA, and the TE191 probe between the nucleotides 1074 and 1261, both non-conserved regions of the cDNAs outside of the conserved domains to avoid cross hybridization. We also used the whole nucleotide sequence of TE074 as a query sequence in BLASTN to search through three fully sequenced grass genomes: rice (International Rice Genome Sequencing Project 2005), sorghum (Paterson et al. 2009) and maize (Schnable et al. 2009). We found three copies in sorghum (chromosomes 1, 2 and 8), five copies in rice (chromosomes 1, 3 and 3 copies in the chromosome 5), and six copies in maize (chromosomes 1, 5 and 7 and 3 copies in the chromosome 10), indicating a low copy number in grasses and showing to be localized in more than one genomic locus.
Fig. 2.
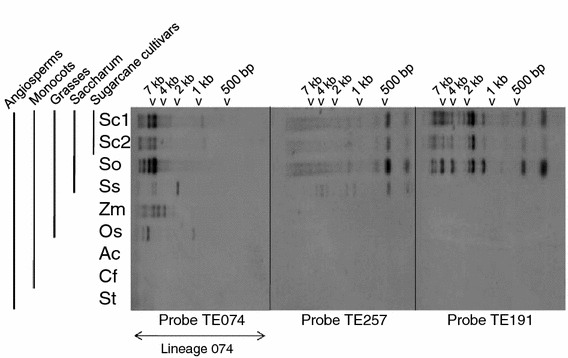
Molecular hybridization of TE074, TE191 and TE257 probes with blots of angiosperm genomic DNA digested with the endonuclease XbaI. Species used were: sugarcane hybrid variety SP-89-1115 (Sc1); sugarcane hybrid variety SP-80-3280 (Sc2); Saccharum officinarum variety Badila (So); Saccharum spontaneum variety Mandalay (Ss); Zea mays (Zm); Oryza sativa (Os); Ananas comosus (Ac); Catassetum fimbriatum (Cf) and Solanum tuberosum (St). Molecular weight markers are indicated along the top of each blot
The expression of the lineage 074 was examined by hybridizing total RNA prepared from leaves and roots of greenhouse grown R570 plants and in vitro calli from SP89-1115 cultivar (Table 2) as well as RNA from apical meristem, stem, mature leaf and young leaf of field grown plants from R570 and SP80-3280 cultivars (Fig. 3). Expression was detected in all tissues and the developmental stages analyzed, indicating a wide distributed expression. In addition to meristems, callus, leaves and roots were also analyzed and a sevenfold higher expression was observed in callus compared to the latter tissues. By contrast, probing the membranes with TE191 revealed no expression (data not shown).
Table 2.
Relative expression levels of a TE074-related element in different tissues
| Probe | Absolute value (a) | Background (b) | a − b | Normalization to ubiquitin |
|---|---|---|---|---|
| TE074 | ||||
| Calli | 234.7109 | 181.0375 | 53.6734 | 1.8253 |
| Roots | 185.5706 | 181.0375 | 4.5331 | 0.1734 |
| Leaves | 188.0968 | 181.0375 | 7.0593 | 0.2424 |
| Ubiquitin | ||||
| Calli | 220.2342 | 190.8299 | 29.4043 | – |
| Roots | 216.9712 | 190.8299 | 26.1413 | – |
| Leaves | 219.9508 | 190.8299 | 29.1209 | – |
Values were obtained from a semi-quantitative Northern blot hybridization assay. Total RNA from leaves, roots and calli of sugarcane hybrid cultivar was hybridized with a probe corresponding to TE074 and a probe from the constitutive gene ubiquitin. The intensity of the bands (a) and the background (b) were measured by densitometry. Values are normalized dividing the TE074 value by its corresponding ubiquitin value
Fig. 3.
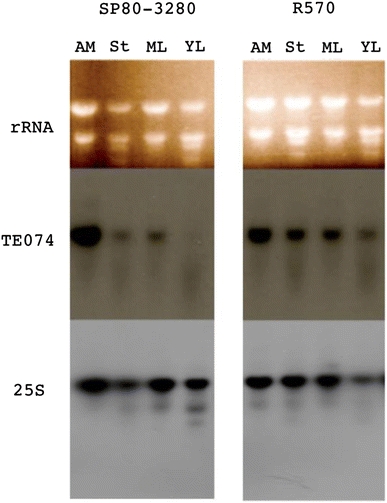
TE074 expression pattern analysis. Six-month-old plant tissues of sugarcane cultivars R570 and SP80-3280 were harvested and total RNA extracted. A northern blot assay was carried out using apical meristem (AM), stem (ST), mature leaf (ML) and immature leaf (YL). P-32 labeled probes of TE074 clone and 25S were used for the hybridization assay
Selective constraints were evaluated to explore the hypothesis that lineage 074 contains sequences from a domesticated transposase in opposition to the other two transposon lineages identified. It is expected that a functional gene, as proposed to the domesticated lineage, should be under a stronger selective pressure to maintain its coding sequence, while the transposon lineages are more tolerant to mutations (Feschotte and Pritham 2007). The synonymous (dS) and non-synonymous (dN) substitution rates, as well as dN:dS values, were calculated for cDNA sequences from the lineage 074 and the other two lineages, 191 and 257. Assuming that synonymous substitutions are under neutral evolution, dN:dS = 1 will represent neutral evolution, dN:dS <1 will correspond to purifying selection, and dN:dS >1, positive Darwinian selection.
Among thirteen lineage 074 cDNAs compared, nine presented dN:dS values under unity when compared with all the other cDNAs from the same lineage. For the lineage 074 subgroup formed by cDNAs TE048, TE096, TE207 and TE265, dN:dS ratios ranged from 0.223 to 0.256, with Z test P values ranging from 0.000 to 0.047. This means that these sequences are under purifying selection. Although the Z test P values were above the cutoff 0.05 to the other cDNAs from lineage 074, the dN:dS values were under unity to the majority of the sequences. In only three cases this ratio was above unity (Fig. S2; supplementary material). These results suggest a purifying selection to the majority of the lineage 074 sequences, corroborating a characteristic of a functional gene. Nevertheless, the purifying selection over the other lineage 074 sequences is more relaxed than the one over the subgroup formed by the cDNAs TE048, TE096, TE207 and TE265. Among the cDNAs from lineages 191 and 257 analyzed, no statistically validated data were obtained.
To examine the diversity among lineage 074 cDNA clones, a nucleotide sequence alignment was performed. Although these sequences are very similar at the amino acid level, nucleotide alignment revealed their remarkable subdivision into two highly conserved groups (Fig. 4). TE048, TE096, TE207 and TE265 diverge from the other group, suggesting that these sequences could be transcribed from two distinct loci. The separation time between these two hypothetical loci was calculated, and range from 32.23 to 35.6 MYA.
Fig. 4.
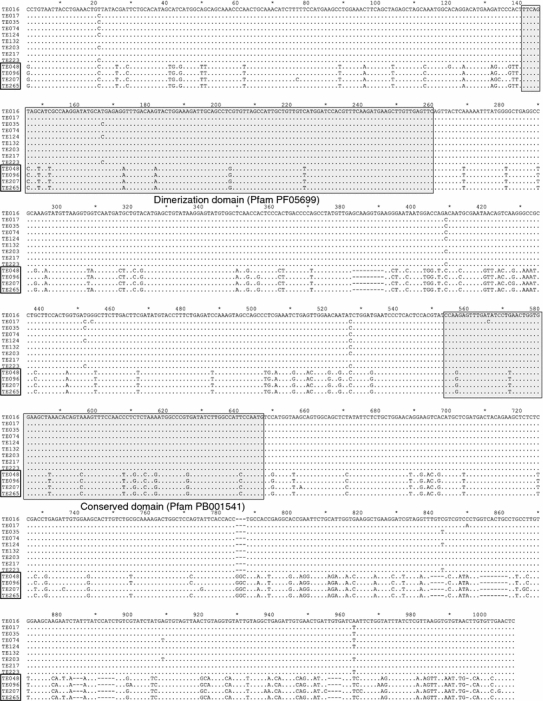
Multiple alignment of a 1,008 bp nucleotide segment shared by the lineage 074 cDNAs. Alignment was performed with ClustalX using default parameters. Shaded boxes indicate conserved domains of the hAT transposase superfamily in the nucleotide sequence as presented. Boxed names on the left highlight a subgroup within lineage 074
SChAT, a novel family of transposons present in the sugarcane genome
Two independent strategies were used to recover full-length copies of hAT transposons from the sugarcane genome: amplification from genomic DNA and searches in sugarcane BACs sequenced in our laboratory. Inverse PCR (iPCR) experiments enabled us to access genomic sequences adjacent to transposons related to lineage 191. Primers were designed on both subterminal regions of the cDNA TE221, directed toward the ends. TE221 was chosen because its sequence is the longer among lineage 191 cDNAs. This approach leads to two possibilities of amplification: the first is the amplification of two extremities of one copy of the element, including untranscribed regions and flanking genomic regions. The second possibility is amplification of the extremities of two identical or very similar tandem copies of the element instead of two flanks of the same copy. Two bands, with sizes of 1,062 and 755 bp, were amplified by iPCR (Fig. S1a; supplementary material). Analysis of these sequenced fragments revealed the absence of the restriction site for the endonuclease EcoRI, which was used to cleave the genomic DNA. This information indicates that the terminal sequences obtained, including the putative TIRs, correspond to copies sitting in tandem in the genome. The pair of TIRs isolated is composed of 21 reverse complementary base pairs, with 15 matches among them. No target site duplication was found. Primers were designed based on these TIR sequences, directed inwards to amplify a full-length copy. Amplifications making use of these primers allowed the recovery of two genomic versions of the sugarcane hAT element from the genome of cultivar SP80-3280. These elements, 4,616 bp and 3,838 bp in length, were named SChAT-G1 and SChAT-G2, respectively (Fig. S1b; supplementary material). SChAT-G2 is a deleted copy of SChAT-G1 with a deletion of 777 bp in the 3′ portion of SChAT-G1 comprising positions 2,822–3,624 (Fig. S1c; supplementary material).
Searching sugarcane BACs revealed three copies of the SChAT family, named SChAT1, SChAT2 and SChAT3. Figure 5a presents a schematic representation of the coding region structure among the elements comprising the SChAT family. The cDNAs TE191, TE221 and TE257 are also included as well as the Ac element from maize. All these elements have a dimerization domain in their carboxy terminal portion and a zinc finger domain in the amino terminal region, except SChAT3, which is truncated. SChAT2 has two frameshifts in its predicted coding sequence, represented by the black vertical bars in Fig. 5a. SChAT-G1 and SChAT1 are larger elements and both present the potential to be autonomous elements. SChAT-G1 shares 51% similarity with the Ac transposase, on a segment covering 77% of the full Ac sequence. Nevertheless, there are 2,540 nucleotides downstream of the putative stop codon of SChAT-G1 predicted transposase that present similarity to the SNF2 helicase domain. In addition, there is probably a deletion in the 5′ untranscribed region of SChAT-G1, between the putative TATA box and the putative start codon, which are separated by only two nucleotides. SChAT1 exhibits 51% protein similarity along a region covering 77% of the full Ac transposase. ORF analysis indicates putative start and stop codons located at positions 1,049 and 2,870 of its nucleotide sequence, respectively.
Fig. 5.
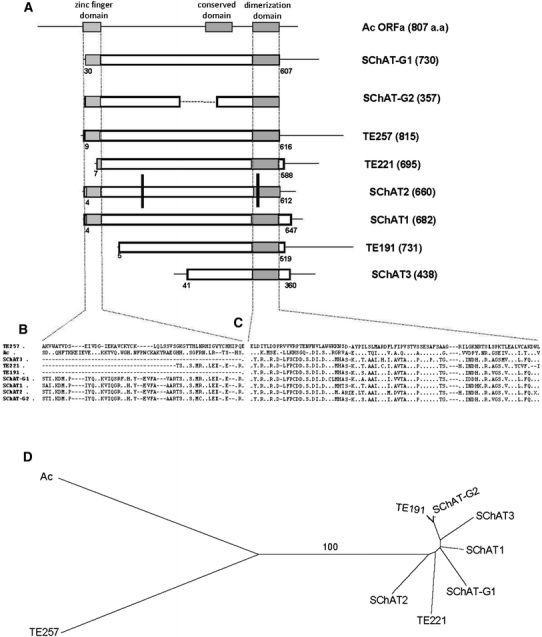
Comparison of hAT transposons recovered from sugarcane genome and the Ac transposase. a Schematic representation comparing the amino acid sequence of the functional transposase of the Ac element from maize, the sugarcane cDNAs TE191, TE221 and TE257, and the predicted amino acid sequences of the transposase of the five SChAT elements found in sugarcane: SChAT1, SChAT2, SChAT3, SChAT-G1 and SChAT-G2. The numbers in brackets in each line correspond to the size of the predicted ORF. The three hAT superfamily domains are highlighted in gray boxes. Black bars indicate the presence of frameshift mutations on the copy of SChAT2. b Detailed alignment of the zinc finger domain. c Detailed alignment of the hAT dimerization domain. d Distance tree based on the concatenated sequences aligned in b and c. Bootstrap value above 50% is presented
SChAT1, SChAT2, SChAT3, SChAT-G1 and SChAT-G2 are the first DNA transposons transcriptionally active isolated from sugarcane genome. The detailed alignments of the zinc finger and the dimerization domains are presented in Fig. 5b, c. A phylogenetic tree constructed based on the alignment of the concatenated sequences of these domains reveals that TE191, TE221 and the SChAT elements cluster in a branch supported by 100% bootstrap, while TE257 is more distantly related (Fig. 5d). These results suggest the presence of at least two distinct hAT-like transposon lineages inhabiting the sugarcane genome, one called lineage 191 related to TE191 cDNA, and a second related to TE257 cDNA, that were not recovered by amplification or by searches in BAC sequences in the present study.
To strengthen the observation of recovery of a potentially active version of SChAT 191 lineage and the existence of a domesticated variant, the identification of the DDE motif described in Muehlbauer et al. (2006) was made. Figure 6 displays the alignment of four lineage 074 cDNAs, w-gary1, w-gary2, b-gary, the two recovered genomic versions SChAT1 and SChAT2 along with the Ac transposase. All the six domains (A–F) previously described in Muehlbauer et al. (2006) were identified. Arrows in the figure highlight the predicted DDE motif necessary for the enzymatic cleavage activity. Only the canonical Ac transposase and SChAT1 possesses the expected amino acid triad at the correct position. The domesticated SChAT versions found in sugarcane present a GDG motif in the corresponding positions. Another conserved motif of hAT transposases is a CxxH downstream of the second D residue of the DDE motif, required to the catalytic activity of the enzyme (Zhou et al. 2004). Three of the four lineage 074 cDNAs analyzed presented the correct motif: TE096, TE048 and TE124 as well as SChAT1, SChAT2 and the Ac transposase. In summary, we recovered five genomic copies of a novel hAT superfamily of transposons in sugarcane, named SChAT. It was the first family of transposons isolated from sugarcane genome. Among these copies there is one potential autonomous element, presenting a full-length ORF and the intact motifs characteristic of the superfamily.
Fig. 6.
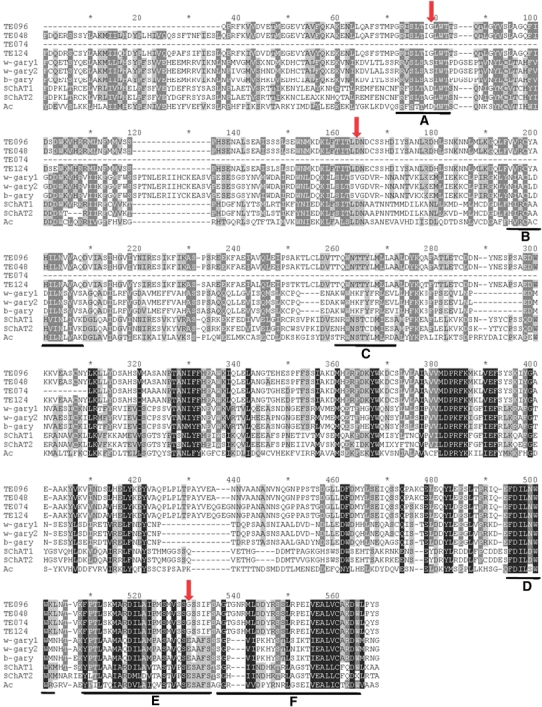
Alignment of the predicted aminoacid sequences of sugarcane cDNAs, gary transposases from barley and wheat, and the Ac transposase. Sugarcane sequences TE096, TE048, TE074 and TE124 refer to domesticated transposase, and SChAT1 and SChAT2 are transposons. The key DDE aminoacid motifs are indicated with arrows. The six regions (A–F) characteristics of the hAT superfamily are underlined
Discussion
The hAT superfamily of transposons presents remarkable historical significance for transposable elements studies because Ac, a belonging member of the superfamily, was the first transposable element observed in the early studies of Barbara McClintock. It is also one of the most studied superfamily of transposons. However, a deepened analysis of these elements was never made in sugarcane. In order to access the information about these elements present in sugarcane genome, we used a strategy based in the analysis of cDNAs. In the first part of this work we analyzed the diversity, genomic distribution, expression and selective constraints relative to a set of thirty-two cDNAs previously annotated as hAT-like by our group (Rossi et al. 2001; Araujo et al. 2005). The alignment of two conserved protein domains characteristic of hAT transposases present in the cDNAs with homologous of several species (see “Materials and methods”) resulted in the separation of distinct lineages among the sugarcane cDNAs (Fig. 1). Lineage 074 was particularly interesting, presenting a highly conserved aminoacid sequence among its sequences and clustering with the domesticated transposase DAYSLEEPER from Arabidopsis thaliana (Bundock and Hooykaas 2005). The branch containing these sequences is supported by 88% bootstrap (Fig. 1b). The clustering pattern suggests that all these transposases may be DAYSLEEPER homologous, also domesticated transposases. The division of this branch in monocot and eudicot suggests that the domestication of these transposases might occur before the separation between mono and eudicots.
Subsequently three lineages were investigated further regarding their genomic distribution: 074, 191 and 257. None of the lineages was present in the non-grasses genomes examined, potato (dicot), pineapple and orchid (monocots). While lineage 257 was present in all Saccharum genomes, lineage 191 was even more restrict and present only in the genomes of sugarcane hybrids and S. officinarum (Fig. 2). The contribution of S. spontaneum to chromosome content in the hybrids described to be about 15–25% (D’Hont et al. 1996). Thus it is reasonable that sequences which evolved after the divergence of ancestral Saccharum into S. spontaneum and S. officinarum might be present in S. officinarum and its hybrids, but not in S. spontaneum. On the other hand, lineage 074 was present in the genomes of all the grasses analyzed. Since all the probes were made carefully to avoid cross hybridization, this result reveals a higher degree of sequence conservation of lineage 074 when compared with the other two lineages. A search for lineage 074 homologous in maize (Schnable et al. 2009), rice (International Rice Genome Sequencing Project 2005) and sorghum (Paterson et al. 2009) genomes also found a low copy number: six, five and three copies, respectively. These results confirm a low copy number in grasses and the presence of more than one locus. Again, the results suggest that lineage 074 could be a domesticated transposase, since it is expected that a gene with cellular function should be more conserved in sequence and present in the genome as a lower copy number than transposons (Feschotte and Pritham 2007). In summary, the genomic hybridization results, in addition to the clustering pattern of TE074, TE191 and TE257 in the phylogenetic tree, indicate that hAT lineages display distinctly unique patterns, suggesting independent evolution of these paralogues, and the lineage 074 presents characteristics of a domesticated transposase.
We carried out the expression analysis of lineages 074 and 191 in four tissues of two sugarcane cultivars. Expression of lineage 074 was confirmed in all the samples analyzed. In opposition, no expression was detected to lineage 191 (Table 2; Fig. 3). Araujo et al. (2005) presented a global analysis of sugarcane cDNAs homologous to many TE families, including a macroarray in which they report the expression of several hAT-like clones in distinct tissues. Of the 13 clones from 074 lineage, several were expressed in tissues such as calli (TE017, TE048, TE096, TE203 and TE207), apical meristem (TE017, TE203 and TE207), leaf roll (TE017 and TE203), and mature and immature inflorescence (TE017 and TE203). Of the other lineages, only TE191 and TE221 were expressed in calli. Transcripts of lineage 074 are generally more abundant than those from the other lineages, indicating higher expression in the tissues investigated. These previous studies are in agreement with the expression results presented here, which demonstrate higher expression levels and distribution among diverse tissues in lineage 074, while no detectable expression was found to lineage 191. It has been extensively demonstrated that transposons are under strict transcriptional control mediated by their methylation status (Van Sluys et al. 1993; Scortecci et al. 1997; Bird 2007). These data support our hypothesis that lineage 074 refers to a domesticated transposase, presenting higher levels of expression, while the other lineages correspond to transposons, which are expected to be under transcriptional repression. The analysis of selective constraints also supports the hypothesis of lineage 074 as a domesticated transposase. An evaluation of dN:dS for the lineages 074, 191 and 257 revealed a purifying selection to the majority of the lineage 074 sequences, while to the other lineages no statistical supported data were obtained, may be due to the low number of sequences available to compare (Figure S2; supplementary material). The purifying selection acting over lineage 074 sustains a feature of a functional gene.
A nucleotide sequence alignment was performed to analyze in detail the diversity among the lineage 074. The alignment revealed a well-defined subdivision of the sequences into two highly conserved groups (Fig. 4). TE048, TE096, TE207 and TE265 diverge from the other group. This pattern suggests that these cDNAs could be originated from two distinct loci. In addition to the split up of lineage 074 in two subgroups of sequences, it is known that duplication events have occurred during the Poaceae evolution (Paterson et al. 2004; Kim et al. 2009) and particularly in sugarcane (Ming et al. 1998). Homologues of TE074 were found in sorghum and rice in three chromosomes, and in maize in four chromosomes, denoting the existence of distinct loci. These data strongly suggest that the differences between the two subgroups of lineage 074 are due to its origin from two loci, and not a polymorphism between two alleles of the same locus. We calculated the separation time between the two hypothetical loci and the range obtained was from 32.23 to 35.6 MYA. Since the speciation of the Saccharum genus is estimated in 8–9 MYA (Jannoo et al. 2007; Wang et al. 2010) and the origin of the grasses is between 75 and 55 MYA (Kellogg 2001), thus it is reasonable to infer that the locus referent to lineage 074 was duplicated after the differentiation of the grasses, and significantly before the differentiation of Saccharum.
Based on the results presented, we propose the occurrence of a domestication event during the evolution process of the hAT-like transposases, before the divergence of mono and eudicots, approximately from 70 to 55 MYA (Kellogg 2001), and further demonstrate that these loci are expressed in sugarcane. Moreover, expression level is higher in undifferentiated tissues (calli and apical meristems) of different sugarcane cultivars in agreement with the aberrant phenotype of DAYSLEEPER arabidopsis mutants (Bundock and Hooykaas 2005). The results presented here corroborate the hypothesis that lineage 074 is a domesticated transposase.
The second part of this work presents a new transposon family in sugarcane. We recovered two genomic copies of lineage 191 elements from sugarcane genome using iPCR strategy. These copies were named SChAT-G1 and SChAT-G2. Subsequently, three copies of the same lineage were identified in sugarcane BACs: SChAT1, SChAT2 and SChAT3. From these, two elements presented the potential to be autonomous, SChAT-G1 and SChAT1. Both elements present the dimerization domain in the carboxy terminal portion and the zinc finger domain in the amino terminal region (Fig. 5a). SChAT-G1 and SChAT1 also share 51% protein similarity along a region covering 77% of the full Ac transposase. However SChAT-G1 presents 2,540 nucleotides downstream of the putative predicted stop codon that present similarity to the SNF2 helicase domain. Further analysis will be necessary to evaluate if this region represents an exon capture or if it has been functionally co-opted by the element. Further transcript analysis may contribute to solving this question. In addition, SChAT-G1 probably presents a deletion between the putative TATA box and the putative start codon, what probably makes it a non-functional element. SChAT1, on the other hand, exhibits all the six protein domains previously described in Muehlbauer et al. (2006), the DDE motif necessary for the enzymatic cleavage transposase activity, and the conserved motif CxxH downstream of the second D residue of the DDE motif, also required to the catalytic activity of the enzyme (Zhou et al. 2004) (Fig. 6). ORF analysis of SChAT1 indicates putative start and stop codons located at positions 1,049 and 2,870 of its nucleotide sequence, respectively, and the absence of premature stop codons or frameshifts. Although SChAT1 has characteristics indicative of an autonomous element, further studies in vitro are necessary to evaluate the functionality of the transposase encoded by this element.
With the aim to corroborate the correlation of the genomic copies isolated with lineage 191, on which the iPCR primers were designed, we carried out an alignment of the zinc finger and the dimerization domains (Fig. 5b–d). The clustering pattern obtained revealed that TE191, TE221 and the genomic SChAT elements cluster in a branch supported by 100% bootstrap, while TE257 and the maize Ac element represent independent branches. This result suggests the presence of at least two distinct hAT-like transposon lineages inhabiting the sugarcane genome, lineage 191 and lineage 257. No copy from lineage 257 was recovered by amplification or by searches in BAC sequences in the present study. Further analysis will be necessary to search for these elements as the sugarcane BAC sequence database expands. Five genomic copies recovered belong to lineage 191, supporting the existence of a true transposon lineage composed by the elements SChAT1, SChAT2, SChAT3, SChAT-G1 and SChAT-G2. To date these represent the first collection of DNA transposons recovered from the sugarcane genome. Nevertheless, transposition activity remains to be demonstrated.
Here we report a detailed analysis of hAT transposase paralogues in the sugarcane genome, using a collection of cDNAs as a starting point. We identified a putative domesticated transposase present in two loci, and at least two lineages of transposons. Previous studies have reported hAT-like elements transcriptionally active in rice (Jiao and Deng 2007) and grapevine (Benjak et al. 2008). Nevertheless, here we presented a combined approach to reveal these actively expressed hAT paralogues from sugarcane tissues. Our data suggest the presence of a domesticated hAT-like transposase in two loci of the sugarcane genome with clear transcriptional activity in several tissues of distinct sugarcane cultivars. This transposase is highly conserved among monocots and eudicots, suggesting early specialization. We also identified at least two lineages of transcriptionally active transposons restricted to Saccharum, including non-autonomous copies that are also expressed. For one of the lineages, we recovered five genomic copies including one potential autonomous element from a novel transposon family, SChAT. One of the isolated copies carries a SNF2-helicase domain. These findings provide the basis for further functional studies on the domesticated transposase gene as well as on the transposition mechanisms of the SChAT family, which will contribute to our understanding of the impact of hAT superfamily transposons on the sugarcane genome.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
We thank Dr. Magdalena Rossi and Paula Araújo for early discussion of the experiments, Dr. Josep Casacuberta for careful reading of the manuscript and suggestions, Dr. Nathalia de Setta for useful discussions about the evolutionary analysis, and Silvia Regina Blanco and Myna Nakabashi for technical assistance. This work had the following financial support: FAPESP and CNPq.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- Araujo PG, Rossi M, de Jesus EM, et al. Transcriptionally active transposable elements in recent hybrid sugarcane. Plant J. 2005;44(5):707–717. doi: 10.1111/j.1365-313X.2005.02579.x. [DOI] [PubMed] [Google Scholar]
- Benjak A, Forneck A, Casacuberta JM. Genome-wide analysis of the “Cut-and-Paste” transposons of grapevine. PLoS ONE. 2008;3(9):e3107. doi: 10.1371/journal.pone.0003107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2007;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- Britten RJ. Coding sequences of functioning human genes derived entirely from mobile element sequences. Proc Natl Acad Sci USA. 2004;101(48):16825–16830. doi: 10.1073/pnas.0406985101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bundock P, Hooykaas P. An Arabidopsis hAT-like transposase is essential for plant development. Nature. 2005;436(7048):282–284. doi: 10.1038/nature03667. [DOI] [PubMed] [Google Scholar]
- Calvi BR, Hong TJ, Findley SD, Gelbart WM. Evidence for a common evolutionary origin of inverted repeat transposons in Drosophila and plants: hobo, Activator and Tam3. Cell. 1991;66:465–471. doi: 10.1016/0092-8674(81)90010-6. [DOI] [PubMed] [Google Scholar]
- Casola C, Lawing AM, Betran E, Feschotte C. PIF-like transposons are common in Drosophila and have been repeatedly domesticated to generate new host genes. Mol Biol Evol. 2007;24(8):1872–1888. doi: 10.1093/molbev/msm116. [DOI] [PubMed] [Google Scholar]
- D’Hont A, Grivet L, Feldmann P, Rao S, Berding N, Glaszmann JC. Characterisation of the double genome structure of modern sugarcane cultivars (Saccharum spp.) by molecular cytogenetics. Mol Gen Genet. 1996;250:405–413. doi: 10.1007/BF02174028. [DOI] [PubMed] [Google Scholar]
- Essers L, Adolphs RT, Kunze R. A highly conserved domain of the maize Activator transposase is involved in dimerization. Plant Cell. 2000;12:211–223. doi: 10.1105/tpc.12.2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feschotte C. Transposable elements and the evolution of regulatory networks. Nat Rev Genet. 2008;9:397–405. doi: 10.1038/nrg2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feschotte C, Pritham EJ. DNA transposons and the evolution of eukaryotic genomes. Annu Rev Genet. 2007;41:331–368. doi: 10.1146/annurev.genet.40.110405.090448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaut BS, Morton BR, McCraig BC, Clegg MT. Substitution rate comparisons between grasses and palms: synonymous rate differences at the nuclear gene Adh parallel rate differences at the plastid gene rbcL. Proc Natl Acad Sci. 1996;93:10274–10279. doi: 10.1073/pnas.93.19.10274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivet L, Daniels C, Glaszmann JC, D′Hont A. A review of recent molecular genetics evidence for sugarcane evolution and domestication. Ethnobot Res Appl. 2004;2:9–17. [Google Scholar]
- Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser. 1999;41:95–98. [Google Scholar]
- Hehl R, Nacken WK, Krause A, Saedler H, Sommer H. Structural analysis of Tam-3, a transposable element from Antirrhinum majus, reveals homologies to the Ac element from Maize. Plant Mol Biol. 1991;16(2):369–371. doi: 10.1007/BF00020572. [DOI] [PubMed] [Google Scholar]
- International Rice Genome Sequencing Project The map-based sequence of the rice. Nature. 2005;436:793–800. doi: 10.1038/nature03895. [DOI] [PubMed] [Google Scholar]
- Jannoo N, Grivet L, Chantret N, Garsmeur O, Glaszmann JC, Arruda P, D’Hont A. Orthologous comparison in a gene-rich region among grasses reveals stability in the sugarcane polyploid genome. Plant J. 2007;50:574–585. doi: 10.1111/j.1365-313X.2007.03082.x. [DOI] [PubMed] [Google Scholar]
- Jiao Y, Deng XW. A genome-wide transcriptional activity survey of rice transposable element-related genes. Genome Biol. 2007;8:R28. doi: 10.1186/gb-2007-8-2-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg E. Evolutionary history of the grasses. Plant Physiol. 2001;125:1198–1205. doi: 10.1104/pp.125.3.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempken F, Windhofer F. The hAT family: a versatile transposon group common to plants, fungi, animals, and man. Chromosoma. 2001;110(1):1–9. doi: 10.1007/s004120000118. [DOI] [PubMed] [Google Scholar]
- Kim C, Tang H, Paterson AH. Duplication and divergence of grass genomes: interacting the Chloridoids. Tropical Plant Biol. 2009;2:51–62. doi: 10.1007/s12042-009-9028-3. [DOI] [Google Scholar]
- Kraitshtein Z, Yaakov B, Khasdan V, Kashkush K. Genetic and epigenetic dynamics of a retrotransposon after allopolyploidization of wheat. Genetics. 2010;186:801–812. doi: 10.1534/genetics.110.120790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisch D, Bennetzen JL. Transposable element origins of epigenetic gene regulation. Curr Opin Plant Biol. 2011;14:156–161. doi: 10.1016/j.pbi.2011.01.003. [DOI] [PubMed] [Google Scholar]
- McClintock B. Maize genetics. Year Book Carnegie Inst Washington. 1946;45:176–186. [PubMed] [Google Scholar]
- McClintock B. The significance of responses of the genome to challenge. Science. 1984;226:792–801. doi: 10.1126/science.15739260. [DOI] [PubMed] [Google Scholar]
- Ming R, Liu SC, Lin YR, da Silva J, Wilson W, Braga D, van Deynze A, et al. Detailed alignment of Saccharum and Sorghum chromosomes: comparative organization of closely related diploid and polyploid genomes. Genetics. 1998;150:1663–1682. doi: 10.1093/genetics/150.4.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muehlbauer GJ, Bhau BS, Syed NH, et al. A hAT superfamily transposase recruited by the cereal grass genome. Mol Genet Genomics. 2006;275(6):553–563. doi: 10.1007/s00438-006-0098-8. [DOI] [PubMed] [Google Scholar]
- Paterson AH, Bowers JE, Chapman BA. Ancient polyploidization predating divergence of the cereals, and its consequences for comparative genomics. Proc Natl Acad Sci. 2004;101(26):9903–9908. doi: 10.1073/pnas.0307901101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson AH, Bowers JE, Bruggmann R, et al. The Sorghum bicolor genome and the diversification of grasses. Nature. 2009;457(29):551–556. doi: 10.1038/nature07723. [DOI] [PubMed] [Google Scholar]
- Porebski S, Bailey LG, Baum BR. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol Biol Rep. 1997;15:8–15. doi: 10.1007/BF02772108. [DOI] [Google Scholar]
- Rossi M, Araújo PG, Van Sluys MA. Survey of transposable elements in sugarcane expressed sequence tags (ESTs) Genet and Mol Biology. 2001;24(1–4):147–154. [Google Scholar]
- Rubin E, Lithwick G, Levy AA. Structure and evolution of the hAT transposon superfamily. Genetics. 2001;158(3):949–957. doi: 10.1093/genetics/158.3.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- Schnable PS, Ware D, Fulton RS, et al. The B73 Maize Genome: complexity, diversity, and dynamics. Science. 2009;326:1112–1115. doi: 10.1126/science.1178534. [DOI] [PubMed] [Google Scholar]
- Scortecci KC, Dessaux Y, Petit A, Van Sluys MA. Somatic excision of the Ac transposable element in transgenic Arabidopsis thaliana after 5-azacytidine treatment. Plant Cell Phisiol. 1997;38(3):336–343. doi: 10.1093/oxfordjournals.pcp.a029171. [DOI] [PubMed] [Google Scholar]
- Streck RD, MacGaffey JE, Beckendorf SK. The structure of hobo transposable elements and their insertion sites. The EMBO Journal. 1986;5(13):3615–3623. doi: 10.1002/j.1460-2075.1986.tb04690.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 40. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improvingthe sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;24:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Sluys MA, Scortecci KC, Tempé J. DNA methylation associated to Ac element in Arabidopsis. Plant Physiol Biochem. 1993;31(6):805–813. [Google Scholar]
- Volff JN. Turning junk into gold: domestication of transposable elements and the creation of new genes in eukaryotes. Bioessays. 2006;28(9):913–922. doi: 10.1002/bies.20452. [DOI] [PubMed] [Google Scholar]
- Wang J, Roe B, Macmil S, Yu Q, Murray JE, Tang H, Chen C, Najar F, Wiley G, Bowers J, Van Sluys MA, Rokhsar DS, Hudson ME, Moose SP, Paterson AH, Ming R. Microcollinearity between autopolyploid sugarcane and diploid sorghum genomes. BMC Genomics. 2010;11:261. doi: 10.1186/1471-2164-11-261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessler SR. Transposable elements and the evolution of eukaryotic genomes. Proc Natl Acad Sci. 2006;103(47):17600–17601. doi: 10.1073/pnas.0607612103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Wessler SR. The catalytic domain of all eukaryotic cut-and-paste transposase superfamilies. Proc Natl Acad Sci. 2011;108(19):7884–7889. doi: 10.1073/pnas.1104208108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Mitra R, Atkinson PW, Burgess Hickman A, Dyda F, Craig NL. Transposition of hAT elements links transposable elements and V(D)J recombination. Nature. 2004;432:995–1001. doi: 10.1038/nature03157. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.