

Abstract
Sodium hydride is a common reagent for substrate activation in nucleophilic substitution reactions. Sodium hydride can behave both as a base and as a source of hydride. This dual ability in the presence of an electrophile such as benzyl bromide results in the formation of byproducts when dimethylformamide or acetonitrile are used as solvents for these reactions. The structural nature of these byproducts is revealed in this report.
Sodium hydride is a commonly used base for deprotonation of alcohols, phenols, amides, ketones, esters and other functional groups for the promotion of their nucleophilic substitution.1 Typically, sodium hydride and the reagents are mixed in polar aprotic solvents such as DMSO, DMF or acetonitrile for these SN2-type reactions. Whereas the literature is replete with examples of the use of these solvents in such reactions, our chance discovery of reactivity of sodium hydride with two of these commonly used solvents, DMF and acetonitrile, indicates that certain undesired side reactions involving these two solvents might be common, but unrecognized. As is disclosed in this report, the root cause of these complicating side-reactions is the dual role that sodium hydride exhibits as a base and as a source of hydride.
The type of byproducts that will be disclosed in this report are generally difficult to detect with common visualization methods and are easy to overlook. Using benzyl bromide as the electrophile helped us to isolate the byproducts by UV detection and the structures were ultimately assigned by X-ray analysis of single crystals.
During benzylation of compound 1 in the presence of NaH and benzyl bromide in DMF, we identified an unusual solid byproduct (Figure 1A). The 1H NMR spectrum of this byproduct was simple (Figure 1B), and was without resonances originating from the glucal substrate. Assignment of structure was done by X-ray analysis. This compound crystallized in the monoclinic space group P21/c. Once in hand, the X-ray structure surprised us, as it showed that the solid, compound 3, was a derivative of dimethylamine (Figure 1C). The origin of the dimethylamine moiety was clearly DMF, which served as the solvent for the reaction. We note that DMF was freshly distilled prior to use in the reaction and that there was no trace of dimethylamine in DMF as discerned by a negative ninhydrin assay.
Figure 1.
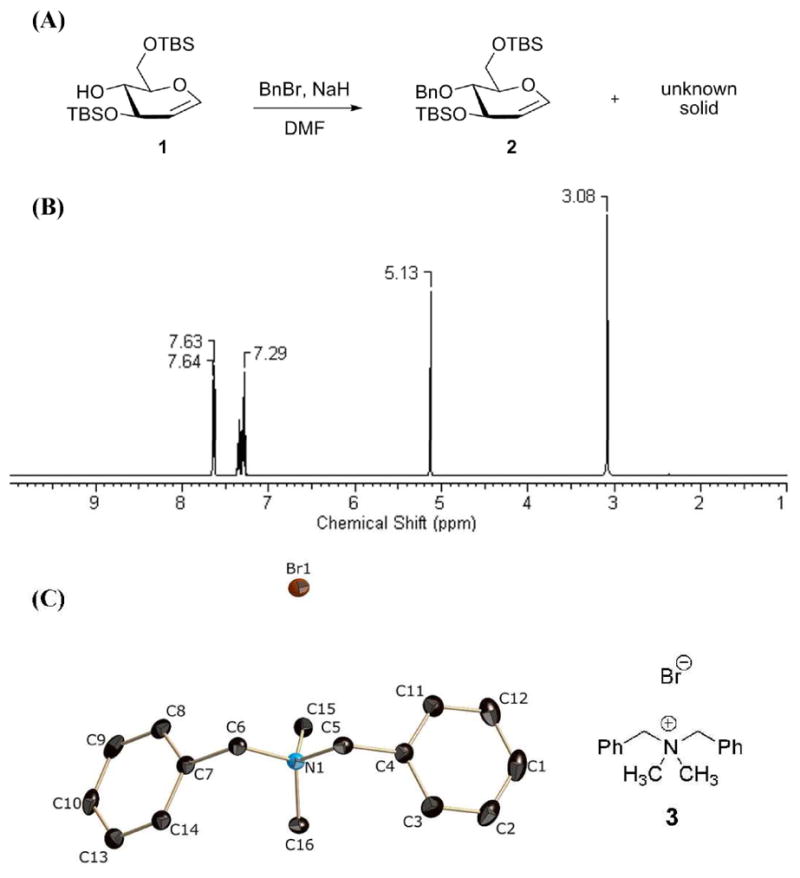
(A) Benzylation of compound 1. (B) The 1H NMR spectrum of the unknown solid in CDCl3. (C) The ORTEP diagram of the X-ray structure of compound 3 is shown at 50% probability level. Hydrogen atoms are omitted for clarity.
A search of the literature revealed that when DMF was treated with BnBr in the presence of K2CO3 as the base at 80 °C for 2 days, compound 3 was obtained as a major product.2 The process is presumably slow, because it is a hydrolytic event. In the observation from our lab, hydride reduction of DMF could result in the formation of sodium (dimethylamino)methanolate (4a), followed by dimethylamine (4b; Scheme 1). Subsequent benzylation, either step-wise on 4b or concerted on 4a yields 3 as a stable bromide salt. Compound 3 was obtained in 5–12% yield during benzylation of 1. Consumption of benzyl bromide by the side reaction lowered the yield of the desired product and gave a complex reaction mixture, which made purification more difficult.
Scheme 1.
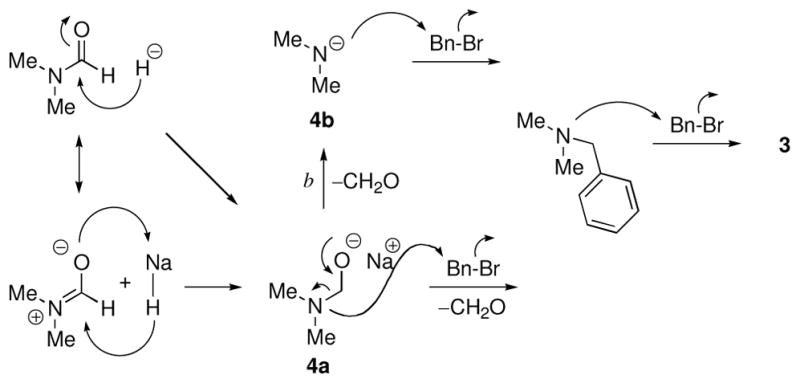
Possible mechanisms for the formation of compound 3.
A disproportionation reaction with DMF, resulting in dimethylamine and carbon monoxide, has also been reported.3 In this reaction, when a mixture of DMF and methyl bromide (in the absence of base) was heated at 80 °C for 6 days in a sealed tube, the formation of tetramethylammonium bromide and carbon monoxide were noted. To explore whether this disproportionation reaction, and not the involvement of NaH, was responsible for the formation of 3, we carried out two additional experiments. In one, DMF-d6 and benzyl bromide (and no NaH) were allowed to react. In the second, the mixture of DMF-d6, benzyl bromide, and NaH were allowed to incubate. These reactions were monitored by 1H NMR at room temperature over a period of 16 h. We did not detect any change in 1H NMR of the first reaction. The 1H NMR spectrum of the second reaction showed the disappearance of the methylene resonance (4.7 ppm) of benzyl bromide and the appearance of a new signal (5.2 ppm) from the resultant deuterated variant of compound 3. We hasten to add that there was no evidence of carbon monoxide formation in either reaction, as judged by a negative phosphomolybdic acid-palladium chloride test.
We identified another example of a side-reaction caused by entry of solvent into reaction with NaH. The O-benzylation of glucosamine derivative 5 (Figure 2A), using NaH as base and acetonitrile as solvent, gave a low yield of the product. Moreover, the isolated product (6) was contaminated by an impurity of unknown structure. The 1H NMR spectrum of the isolated impurity is shown as Figure 2B. Further analysis by NMR spectroscopy (H-H COSY, Dept and H-C HETCOR) revealed that the 3H doublet at δ 1.2 was a methyl coupled to the 1H multiplet at δ 3.2. The methylene resonance at δ 2.5 was diastereotopic based on the splitting pattern, while the two methylenes at δ 3.6 originated from two benzyl groups. The structure of this unknown again was solved by X-ray analysis. Crystals were grown by overnight diffusion of Et2O into a CH2Cl2 solution of the compound at room temperature. The structure revealed the overall reaction of two molecules of acetonitrile with two molecules of BnBr (Figure 2C). Compound 7 crystallized in the centrosymmetric space group P1̄. We note that the bond lengths between N1 and C7, N1 and C9, and N1 and C12 are 1.467 ± 0.001, 1.471 ± 0.002, and 1.465 ± 0.002 Å, respectively. These bond lengths confirm that these are all single bonds.
Figure 2.
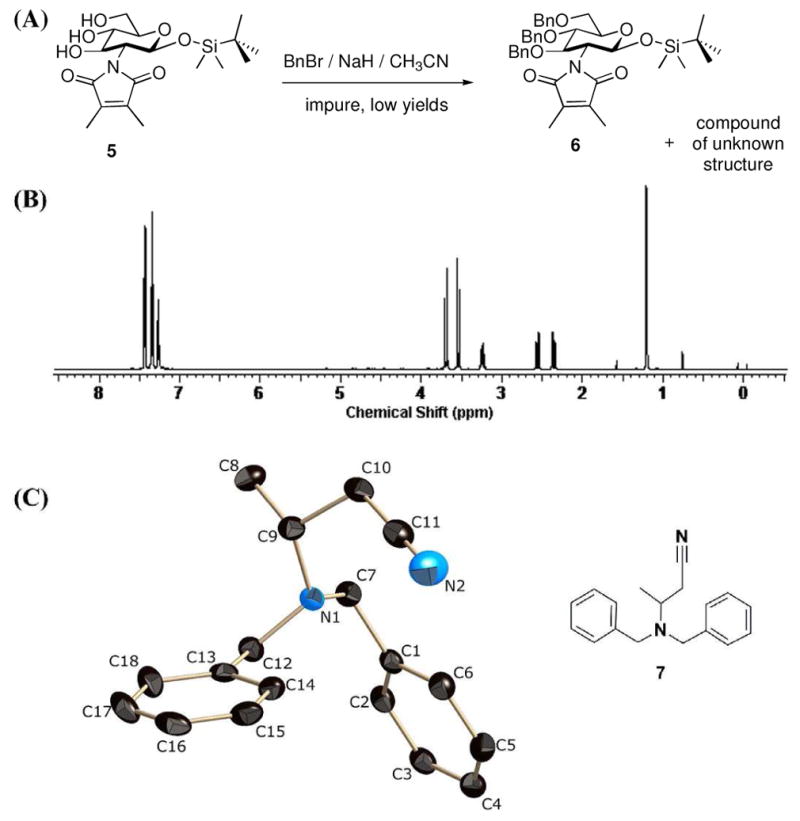
(A) Attempt at NaH-mediated benzylation of compound 5 in acetonitrile, (B) The 1H NMR spectrum of the compound of unknown structure in CDCl3 (C) The ORTEP diagram of the X-ray structure of the compound of unknown structure (compound 7) is shown at 50% probability level. Hydrogen atoms are omitted for clarity.
The structure for 7 implicates the existence of a sequential benzylation and reduction events. Since the pKa of alcohols (16–18) is substantially lower than the pKa of acetonitrile (25), in principle selective deprotonation of the alcohol by NaH (rather than reaction with acetonitrile) should prevail. However, when acetonitrile is used as solvent, the opportunity exists for competitive deprotonation of acetonitrile. It is this opportunity, along with a favorable entropic consideration (high concentration) for the solvent, which initiates the events leading to the formation of 7.
We set out to confirm the presumptive intermediates for 7 by examination of a reaction mixture of acetonitrile, benzyl bromide, and sodium hydride (Scheme 2A). Two sets of reaction conditions were examined. In one, NaH was added in several portions, to an acetonitrile solution of benzyl bromide. In the second, benzyl bromide was added in several portions, to a suspension of NaH in acetonitrile. The first reaction gave 3-phenylpropanenitrile (8) as the major product (55%), assigned by comparison of its 1H NMR spectrum with authentic material. The second reaction afforded a mixture of several compounds, including 8, which was removed from the mixture by vacuum distillation. Crystallization of the remaining mixture (from an EtOAc/hexanes cosolvent system) produced crystals of compound 7. The mother liquor was concentrated, and the crude material purified by preparative TLC to give yet another new compound. Its 1H NMR spectrum was similar to that of 7 and suggested the presence of structure arising from overall reaction of two molecules of acetonitrile with benzyl bromide. High-resolution MS confirmed the structure as that of 9. Compound 9 previously was synthesized by reaction of crotonitrile and benzylamine under reflux in ethanol.4
Scheme 2.

The proposed mechanism of NaH-mediated benzylation of acetonitrile.
Yields for compound 7, 8, and 9 were 4, 40, and 6%, respectively. Yields were calculated from the 1H NMR spectrum of the crude mixture for the second set of reaction conditions.
To further evaluate the mechanism, the products of the reaction of MeCN-d3 were examined. The 1H NMR spectra for 8-D and 9-D isolated from this reaction are shown in Figure 3. The formation of 8-D is readily explained by base-promoted benzylation of acetonitrile (Scheme 2B). The reaction of the acetonitrile carbanion (10) with a second acetonitrile molecule, and subsequent reaction with benzyl bromide, gives intermediate 14. Acetonitrile anion has two resonance structures (10 and 11). The conversion of 14 to 9-D or to 7-D requires reduction of the imine. The two-electron reduction presumably is by the hydride anion of NaH. The precedent for such chemistry by NaH is exemplified by carbonyl, halide, disulfide, disilane, silyl ether, azides, and isoquinoline reductions in the literature.5–11 Addition of hydride to imine 14 was confirmed by the presence of a singlet at δ 2.99 in the 1H NMR spectrum of 9-D (Figure 3B). The X-ray structure of compound 7 also confirmed an N1-C9 single bond (bond length of 1.471 ± 0.002 Å, Figure 2C). The conversion of 14 to 7-D might proceed in a stepwise or in a concerted manner. It is conceivable that the tautomer 13 would exist. The potential intermediate 13 could also serve as the recipient of the hydride, whereby it too will lead to the formation of 7-D and 9-D.
Figure 3.
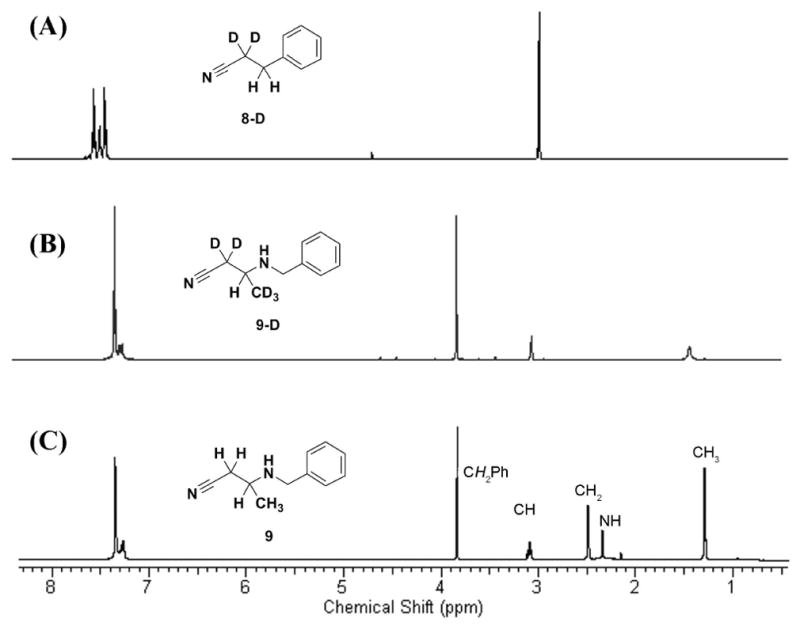
Comparison of 1H NMR spectra of (A) deuterated compound 8, of (B) deuterated compound 9 and of (C) compound 9 in CDCl3.
DMF and acetonitrile are commonly used solvents in alkylation reactions mediated by NaH. As reported herein, the use of these solvents with NaH entails complications, stemming from competing processes involving NaH both as a base and as a source of two electrons in competing reactions with the intended substrate or with the solvent. While many reactions in the literature have used these conditions with high product yields, there is no doubt that with a problematic substrate, the erosion of reagent stoichiometry as a result of these competing reactions with solvent could account for unacceptably low yields. Reactions of NaH are also performed in DMSO. Since explosions have been reported for the mixture of DMSO and NaH, we did not perform similar reactions in this solvent.12, 13
Recognition that these reactions do take place, as we have now documented, should influence how these reactions are carried out in the future. The presently unrecognized existence of these types of undesired side reactions is likely to be quite common.
EXPERIMENTAL SECTION
Compound 3
1H NMR (500 MHz, CDCl3), δ 3.08 (s, 6H, 2×CH3), 5.13 (s, 4H, 2×CH2Ph), 7.24 – 7.39 (m, 6H, Ar-H), 7.59 – 7.67 (m, 4H, Ar-H); 13C NMR (126 MHz, CDCl3), δ 47.8, 67.1, 127.3, 128.8, 130.3, 133.2; HRMS (FAB), calcd for C16H20N (M+), 226.1596, found 226.1599.
Synthesis of Deuterated Compound 3
NaH (60% dispersion in oil, 20 mg, 0.5 mmol) was added to a vigorously stirred solution of benzyl bromide (70 μL, 0.6 mmol) in DMF-d6 (0.7 mL) in ice-water bath and was stirred for 16 h. The solution was mixed with hexanes and the top hexanes layer was removed. The DMF layer was analyzed by NMR. The sample was purified by recrystallization from chloroform.
Compound 3-D
1H NMR (500 MHz, DMF-d6) δ 5.24 (s, 4H), 7.43 – 7.52 (m, 6H), 7.73 – 7.80 (m, 4H); 13C NMR; 13C NMR (126 M Hz, DMF- d6) δ 46.5 (m), 65.8, 128.9, 130.3, 133.5, 139.5; HRMS (FAB), calcd for C16H14D6N (M+), 232.1972, found 232.1980.
Syntheses of Compounds 7, 8 and 9
Two methods were used.
Method A
NaH (60% dispersion on oil, 0.2 g, 5 mmol) was added to a vigorously stirred solution of benzyl bromide (0.7 mL, 6 mmol) in anhydrous acetonitrile (7 mL) in three portions over a period of 5 h in ice-water bath under a nitrogen atmosphere. The mixture was stirred overnight, then it was filtered through layers of silica gel and Celite and washed with acetonitrile. The combined filtrate was mixed with hexanes and top hexanes layer was removed. The acetonitrile layer was evaporated down to an oil, which was distilled (130 °C/20 mmHg) to yield compound 8 as a major product (55%). The 1H NMR spectrum of this sample matched that of the authentic 3-phenylpropanenitrile purchased from Sigma-Aldrich.
Method B
Benzyl bromide (0.7 mL, 6 mmol) was added to a vigorously stirred suspension of NaH (60% dispersion on oil, 0.2 g, 5 mmol) in anhydrous acetonitrile (7 mL) in three portions over a period of 5 h in ice-water bath under a nitrogen atmosphere. The mixture was stirred overnight and the resulting suspension was filtered through layers of silica gel and Celite and washed with acetonitrile. The combined filtrate was mixed with hexanes and top hexanes layer was removed. The bottom acetonitrile layer was evaporated under reduced pressure and the resultant oil was subjected to vacuum distillation to remove compound 8 from the crude reaction mixture. The remaining oil was dissolved in a mixture of ethyl acetate and hexanes (5 mL). Slow evaporation of solvent overnight gave colorless crystals of compound 7. 1H NMR (500 M Hz, CDCl3) δ 1.20 (d, J = 6.8 Hz, 3H), 2.36 (dd, J = 16.7, 6.8 Hz, 1H), 2.56 (dd, J = 16.7, 7.8 Hz, 1H), 3.24 (sxt, J = 6.9 Hz, 1H), 3.54 (d, J = 13.8 Hz, 2H), 3.69 (d, J = 13.8 Hz, 2H), 7.06 – 7.58 (m, 10H); 13C NMR (126 M Hz, CDCl3) δ 13.9, 21.9, 50.3, 53.3, 118.7, 127.1, 128.4, 128.6, 139.1; HRMS (FAB), calcd for C18H21N2 (M+H+), 265.1705, found 265.1703.
The mother liquor was evaporated and subjected to preparative thin-layer column chromatography to furnish compound 9. 1H NMR (500 MHz, CDCl3) δ 1.29 (d, J = 6.6 Hz, 3H), 2.34 (s, 1H), 2.48 (d, J = 5.6 Hz, 2H), 3.09 (m, 1H), 3.83 (s, 2H), 7.12 – 7.48 (m, 5H); 13C NMR (126 MHz, CDCl3) δ 20.2, 24.8, 49.3, 51.0, 117.8, 127.4, 128.1, 128.6, 139.4; HRMS (FAB), calcd for C11H15N2 (M+H+), 175.1235, found 175.1256.
Synthesis of Deuterated Compounds 8 and 9
CD3CN was subjected to method B as described above and the title compound was isolated by preparative thin-layer chromatography. Compound 8-D, 1H NMR (500 MHz, CDCl3) δ 2.95 (s, 2H), 7.22 – 7.43 (m, 5H); 13C NMR (126 M Hz, CDCl3) δ 18.6 (m), 31.1, 119.0, 127.0, 128.1, 128.2, 128.6, 137.9; HRMS (FAB), calcd for C9H7D2N (M+), 133.0861, found 133.0879.
Compound 9-D
1H NMR (300 M Hz, CDCl3) δ 1.36 (br.s., 1H), 2.99 (s, 1H), 3.76 (s, 2H), 7.13 – 7.34 (m, 5H); 13C NMR (151 MHz, CDCl3) δ 19.6 (m), 24.4 (m), 48.9, 51.1, 118.0, 127.2, 127.9, 128.5, 139.6; HRMS (FAB), calcd for C11H10D5N2 (M+H+), 180.1549, found 180.1533.
Crystals Growth and Analysis
Repeated recrystallization from chloroform afforded colorless crystals of compound 3 suitable for X-ray diffraction analysis. For compound 7, crystals of suitable size for single-crystal X-ray diffraction analysis were obtained by diffusion of diethyl ether into the CH2Cl2 solution at room temperature overnight.
Crystals were examined under Infineum V8512 oil and placed on a MiTeGen mount, then transferred to the 100 K N2 stream of a Bruker SMART Apex CCD diffractometer. Unit cell parameters were determined from reflections with I > 10σ(I) harvested from three orthogonal sets of 30 0.5° ω scans. Data collection strategy was calculated using COSMO, included in the Apex2 suite of programs14 to maximize coverage of reciprocal space in a minimum amount of time. Average 4-fold redundancy of measurements was sought. Data were corrected for Lorentz and polarization effects, as well as for absorption.
Structure solution and refinement utilized the programs of the SHELXTL software package.15 Full details of the X-ray structure determinations are in the CIF files included as Supporting Information.
Previously, structure of compound 3 was determined by X-ray powder diffraction2 and later by X-ray single crystal diffraction with R value of 0.0637.16 Our data reported herein are of higher quality, with R value of 0.0215.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health.
Footnotes
Supporting Information Available: Compound characterization data, including copies of 1D NMR spectra (1H, 13C NMR, and Dept) and 2D NMR spectra (H-H COSY and H-C Hetcor) of compounds 3, 3-D, 7, 9, 8-D, 9-D and crystallographic information files (CIFs) of compounds 3 and 7. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Encyclopedia of Reagents for Organic Synthesis. Vol. 7. John Wiley & Sons; New York: 1995. pp. 4568–4571. [Google Scholar]
- 2.Ropponen J, Lahtinen M, Busi S, Nissinen M, Kolehmainen E, Rissanen K. New J Chem. 2004;28:1426–1430. [Google Scholar]
- 3.(a) Neumeyer JL, Cannon JG. J Org Chem. 1961;26:4681–4682. [Google Scholar]; (b) Armarego DD, Perrin WLF. Purification of Laboratory Chemicals. Butterworth Heinemann; 1997. p. 192. [Google Scholar]
- 4.Furukawa M, Okawara T, Terawaki Y. Chem Pharm Bull (Tokyo) 1977;25:1319–1325. [Google Scholar]
- 5.Caubere P. Angew Chem, Int Ed Engl. 1983;22:599–613. [Google Scholar]
- 6.Corriu RJP, Guerin C. J Chem Soc, Chem Commun. 1980:168–169. [Google Scholar]
- 7.Krull LH, Friedman M. Biochem Biophys Res Commun. 1967;29:373–377. doi: 10.1016/0006-291x(67)90465-2. [DOI] [PubMed] [Google Scholar]
- 8.Lee YJ, Closson WD. Tetrahedron Lett. 1974:381–384. [Google Scholar]
- 9.Natsume M, Kumadaki S, Kanda Y, Kiuchi K. Tetrahedron Lett. 1973:2335–2338. [Google Scholar]
- 10.Pi R, Friedl T, Schleyer PvR, Klusener P, Brandsma L. J Org Chem. 1987;52:4299–4303. [Google Scholar]
- 11.Shekhani MS, Khan KM, Mahmood K. Tetrahedron Lett. 1988;29:6161. [Google Scholar]
- 12.French FA. Chem Eng News. 1966;44:48. [Google Scholar]
- 13.Larsen RD, Davis P, Corley EG, Reider PJ, Lamanec TR, Grabowski EJJ. J Org Chem. 1990;55:299–304. [Google Scholar]
- 14.COSMO. Vol. 58 Bruker-AXS; Madison, WI: 2008. [Google Scholar]
- 15.Sheldrick GM. Acta Crystallogr A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 16.Busi S, Lahtinen M, Ropponen J, Valkonen J, Rissanen K. J Solid State Chem. 2004;177:3757–3767. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.