

Abstract
Protein kinase D (PKD) is activated within cells by stimulation of multiple G protein coupled receptors (GPCR). Earlier studies demonstrated a role for PKC to mediate rapid activation loop phosphorylation-dependent PKD activation. Subsequently, a novel PKC-independent pathway in response to Gαq-coupled GPCR stimulation was identified. Here, we examined further the specificity and PKC-dependence of PKD activation using COS-7 cells cotransfected with different Gq-family Gα and stimulated with aluminum fluoride (AlF4−). PKD activation was measured by kinase assays, and Western blot analysis of activation loop sites Ser744, a prominent and rapid PKC transphosphorylation site, and Ser748, a site autophosphorylated in the absence of PKC signaling. Treatment with AlF4− potently induced PKD activation and Ser744 and Ser748 phosphorylation, in the presence of cotransfected Gαq, Gα11, Gα14 or Gα15. These treatments achieved PKD activation loop phosphorylation similar to the maximal levels obtained by stimulation with the phorbol ester, PDBu. Preincubation with the PKC inhibitor GF1 potently blocked Gα11-, Gα14-, and Gα15-mediated enhancement of Ser748 phosphorylation induced by AlF4−, and largely abolished Ser744 phosphorylation. In contrast, Ser748 phosphorylation was almost completely intact, and Ser744 phosphorylation was significantly activated in cells cotransfected with Gαq. Importantly, the differential Ser748 phosphorylation was also promoted by treatment of Swiss 3T3 cells with Pasteurella multocida toxin, a selective activator of Gαq but not Gα11. Taken together, our results suggest that Gαq, but not the closely related Gα11, promotes PKD activation in response to GPCR ligands in a unique manner leading to PKD autophosphorylation at Ser748.
1. Introduction
A large number (~1000) of G protein-coupled receptors (GPCR1) utilize only 15 individual heterotrimeric G protein alpha subunits (Gα) to transmit their signals to effector proteins and ion channels [1, 2]. Liganded GPCR physiologically activate Gα residing at the cytoplasmic face of the plasma membrane by reducing their intrinsic GTPase activity, concomitant with exchange of bound GDP for GTP [3, 4]. The ubiquitously expressed Gαq and Gα11 belong to the Gq family of Gα that also includes isoforms with more restricted expression, i.e., Gα14 expressed in kidney, liver, lung, pancreas and spleen and Gα15/16 that represents similar, hematopoetic cell-specific genes expressed in mice and humans, respectively [4]. Consistent with the very similar length and amino acid composition (359-aa each, with 90% identity) in human Gαq and Gα11, these proteins are functionally indistinguishable to stimulate PLCβ isoform activity [4–7]. PLCs hydrolyze membrane phosphoinositide 4, 5 bisphosphate to generate two key second messengers, inositol 1, 4, 5 trisphosphate that mediates Ca2+ signals [8], and diacylglycerol (DAG) that, in turn, stimulates membrane binding and activation of distinct effectors, including the classical and novel isoforms of PKC [9, 10]. Furthermore, Gαq and Gα11 also appear to couple with similar GPCRs, contributing to the view that, rather than conferring specificity, they represent highly redundant signaling components. Indeed, whereas Gαq−/− and Gα11−/− gene knockout mice undergo normal development, double knockouts are embryonically lethal due to cardiomyocyte hypoplasia [11]. However, Gαq−/− mice exhibit impaired motor coordination [12] and craniofacial defects not compensated by a normal Gα11 allele [11], and additional reports identified discrete roles for Gαq and Gα11 in regulating ion channel activity in neuronal cells [13, 14]. Thus, while data support a high degree of redundant signaling function between these two closely related Gα, evidence also exists for the existence of specific Gαq-dependent signaling pathways. However, the differential effectors and/or signal transducing modes of these important molecules have not been identified.
Protein kinase D (PKD), a novel protein kinase family belonging to the CAMK group, comprises the mammalian isoforms PKD1, PKD2 and PKD3 [15, 16]. These protein kinases, like PKCs also respond to rises in DAG by binding to membrane surfaces, due to the presence of PKC homology 1 (C1) domains located in the N-terminal portion of the molecule [17, 18]. Previous work from this laboratory led to a model of PKD family activation in response to GPCR stimulation, in which PKCs and PKDs are recruited to the membrane surface by PLC-induced DAG production and then PKCs act as direct upstream activators of PKDs [16]. A central feature of this model is the conversion of PKD to an activated form that persists when isolated from cell extracts, due to specific phosphorylation of two distinct sites, (Ser744 and Ser748 in murine PKD1) within the activation loop (also referred as activation segment or T loop) of the kinase catalytic domain [16, 19–22]. Many reports demonstrated PKD1 activation through multiple GPCRs in a number of distinct cell systems and in response to a variety of agonists [21, 23–31]. Previous studies indicated that members of the Gq, Gi and G12/13 families are capable of coupling to distinct GPCR-elicited signal transduction pathways that promote rapid PKD activation within cells [20, 21, 23–32]. Whereas Gα12/13 appear to provide alternate accessory pathways to augment PKD activation, our early studies established the members of the Gq family as central mediators of the rapid induction of PKC-mediated PKD phosphorylation [20, 31, 32]. study
More recent studies revealed that Gq-coupled receptor agonists not only induce rapid, PKC-dependent PKD activation but also a PKC-independent phase characterized by a distinct pattern of PKD activation loop phosphorylation [33, 34]. The PKC-independent phase of PKD activation, characterized by Ser748 autophosphorylation, is critical for mitogenic signaling induced by Gq-coupled GPCR agonists [34, 35]. However, it is not yet clear whether individual Gq family Gα are capable of stimulating PKD activation during each of the distinct phases, or whether the distinct pathways reflect the involvement of the same or different effectors. In fact, the precise contribution(s) of the multiple members of the Gq family to biphasic PKD activation remains unknown.
On the basis of these considerations, we aimed to define the role of different members of the Gq family in promoting PKD activation. Specifically, we determined whether PKD activation mediated by Gαq can be extended to other members of this protein family (i.e. Gα11, Gα14 and Gα15). To analyze the role of each member of the Gq family in PKD activation, we measured their capacity to promote PKD plasma membrane translocation and induce PKC-dependent or -independent PKD T loop phosphorylation at Ser744 and/or Ser748. Surprisingly, we found that stimulation of Gαq but not of Gα11, Gα14 or Gα15 induced sequential PKC-dependent and PKC-independent phases of PKD activation loop phosphorylation. Our results reveal a novel dimension in Gq signaling, namely the non-redundant ability of Gαq to induce PKC-independent PKD activation loop phosphorylation.
2. Materials and Methods
2.1. Cell culture and transfections
COS-7 cells were cultured in medium comprising DMEM (high glucose) supplemented with 10% FBS, in a humidified cell culture incubator supplied with 10% CO2 in air. Cells were sub-cultured prior to confluence every 3–4 days using fresh medium. The mammalian expression constructs pcDNA3-PKD [36], GFP-PKD [37] and pPKD-RFP [38] for wild type PKD, and chimeric fusions of either GFP or RFP with PKD, respectively, have been described previously. Constructs encoding wild type Gαq, Gα11, Gα14 and Gα15, and mutant Gαq-Q209L and Gα11-Q209L in pcDNA3.1 were obtained from the Missouri S & T cDNA Resource center (Rolla, MO). All constructs were propagated in an appropriate bacterial host and purified by standard molecular biology techniques. For transfections, cells were sub-cultured into 35-mm or 60-mm dishes at 5 × 104 cells/ml to achieve 50–60% confluency. The day after passaging, DNA complexes were prepared with 1–2 μg DNA and 2.5 μl Bio-T (Bioland Scientific, LLC, Cerritos, CA) per 35-mm dish in fresh DMEM, according to the manufacturer’s instructions, and introduced to the cell culture conditioned medium. Following 4–5 h to take up complexes, medium was removed and replaced with fresh complete medium. After 24–48 h, culture medium was replaced with fresh DMEM and cells were preincubated with or without inhibitors and then stimulated with 10 μM AlF4− (10 μM AlCl3, 10 mM NaF) or PDBu or left unstimulated, as required for each type of experiment.
Stock cultures of Swiss 3T3-PKD.GFP cells, which overexpress wild type PKD and control Swiss 3T3-GFP cells were generated as previously described [24, 30]. The cells were maintained at 37°C in DMEM, supplemented with 10% FBS in a humidified atmosphere containing 10% CO2 and 90% air. For experimental purposes, cells were plated in 35 mm dishes at 1 × 105 cells/dish and grown in DMEM containing 10% FBS for 7–9 days until they became confluent and quiescent [39].
2.2. In vitro kinase assays
Following treatment with inhibitors and/or stimuli, cells were lysed in a lysis buffer containing 50 mM Tris-HCl, pH 7.5, 2 mM EGTA, 2 mM EDTA, 1 mM dithiothreitol, 100 μg/ml leupeptin, 1 mM 4-(2-aminoethyl)-benzenesulfonyl fluoride, hydrochloride (AEBSF), and 1% Triton X-100, and PKD was immunoprecipitated using a PKD-specific antibody (C-20, Santa Cruz Biotechnologies) from the cleared cell lysates as described previously (17). Immunoprecipitates were washed at 4°C, twice with lysis buffer and twice with kinase buffer comprising 10 mM MgCl2 and 1 mM DTT in 30 mM Tris-HCl, pH 7.4 prior to in vitro kinase or syntide-2 assays. For in vitro kinase assays, γ-32P-ATP (1 μCi/assay)/cold ATP (200 μM) in kinase buffer (10 μl) were added to washed immunoprecipitates, with mixing, and transferred to a 30°C water bath for 10 min. Proteins in the samples were separated by SDS-PAGE, then gels were dried and autoradiographed to detect the autophosphorylated PKD. Syntide-2 assays were initiated by adding 10 μl of a mixture containing γ-32P-ATP (1 μCi/assay), syntide-2 (final concentration, 2 mg/ml) in ATP (200 μM) to washed immunoprecipitates and transferring to a 30°C water bath. Reactions were terminated by addition of 75 mM phosphoric acid, and subsequently spotted to P81 filter paper. Papers were washed twice for 5 min in 75 mM phosphoric acid, dehydrated and fixed with an acetone rinse, dried with heat under vacuum for 10 min using a gel-drier between two sheets of 3MM filter paper, and radioactivity associated with 32P-labeled peptide measured by Cerenkov radiation in a scintillation counter.
2.3. Western blot analysis
For analysis of PKD phosphorylation in immunoprecipitates, cells were transfected and/or stimulated as for in vitro kinase assays, then PKD was immunoprecipitated, washed, extracted, separated on SDS-PAGE gels and blotted to PVDF membranes. For all other Western blot analysis of cellular protein expression and phosphorylation, cells transfected, preincubated and treated with stimuli were washed briefly with ice-cold PBS and then lysed directly in 2×SDS-PAGE sample buffer. Samples were scraped together, collected in microfuge tubes and stored at −20°C. In preparation for SDS-PAGE, viscous DNA in the samples was sheared by passage through a 25-guage syringe/needle three times. Proteins were separated on 9% SDS-PAGE gels under reducing conditions, then blotted to PVDF membranes as described previously [23]. Membranes were probed with antibodies to detect PKD phosphorylated at Ser744 (Cell Signaling #2054) at 1:1000, Ser748 (Millipore 07-882) at 1:1000, total PKD (sc-638 and sc-639) at 1:500, total Gαq, Gα11, Gα14 and Gα15 (Santa Cruz Gq/11 antibody sc-392) at 1:500, by blocking (1 h) and subsequent overnight incubation in PBS containing 0.1% Tween-20 (PBS-T) supplemented with 5% BSA. Membranes were then incubated with HRP-conjugated anti-rabbit secondary antibody at 1:2500 in PBS-T for 1 h, washed in fresh PBS-T (twice for a total of 20 min) and the immunoreactive bands detected using ECL reagent (GE Healthcare) and a FUJI LAS-4000 Mini Luminescent Image Analyzer. Band intensities were quantified using Multiguage V 3.0 software.
2.4. Fluorescence microscopy
COS-7 cells were transfected using Bio-T, with or without Gαq or Gα11 together with GFP-PKD, PKD-RFP or other constructs in combinations indicated in the text. Cells were subjected to treatments, using fresh medium at 37°C as described previously and as indicated in Figures, then imaged as for fluorescein or rhodamine epifluorescence using a Axioskop II microscope (Carl Zeiss) with Achroplan 100X water-immersion objective, equipped with a SPOT Pursuit Digital CCD Camera and SPOT II imaging software (Diagnostic Instruments, Inc). Images were prepared for display in Figures using Adobe Photoshop with identical settings.
3. Results
3.1. PKD catalytic activation and activation loop phosphorylation at Ser744 and Ser748 in response to Gαq and Gα11 stimulation
AlF4− is known to known to activate Gα proteins by mimicking the γ-phosphate of GTP in the presence of GDP [40]. Here, we used COS-7 cells transfected with PKD and Gα proteins as a model system to examine PKD activation in response to stimulation by members of the Gq family. PKD activity was determined by in vitro kinase assays of autophosphorylation (Fig 1A) and phosphorylation of an exogenous peptide substrate, syntide-2 (Fig. 1B). As in previous studies, PKD isolated from unstimulated cells exhibited low basal activity, and a positive control for full PKD activation was achieved by stimulation for 10 min with 200 nM PDBu. Due to the relatively low levels of endogenously expressed Gq/11 family Gα, PKD was not significantly activated by AlF4− stimulation in PKD-only transfected COS-7 cells. AlF4− stimulation over a longer time course (75 min) also failed to induce significant PKD activation in PKD-only transfected cells (Supplementary Fig. 1). In contrast, in cells cotransfected with PKD and Gαq, AlF4− stimulation for 10 min potently stimulated PKD catalytic activation (Fig. 1A and 1B).
Fig. 1. AlF4− stimulation induces PKD activation in COS-7 cells cotransfected with either Gαq or Gα11.

COS-7 cells were transfected with PKD, alone or together with G protein alpha subunits (Gαq or Gα11). After 48 h, cells were either left unstimulated or stimulated with phorbol 11,12 dibutyrate (PDBu) to stimulate maximal PKD activation, or 10 μM AlF4− for 10 min. Cells were then lysed, and PKD was immunoprecipitated using C-20 antiserum and collected on protein A-agarose beads for analysis by in vitro kinase assays or Western blot. A, PKD activity, as measured by autophosphorylation (IVK). Representative autoradiograms are shown above and data of three experiments is presented below as the mean +/- SEM. B, PKD activity measured by phosphorylation of an exogenous substrate, syntide-2. PKD catalytic activity was robustly stimulated by AlF4− only in cells cotransfected with Gαq or Gα11. Results shown are the mean c.p.m. +/− SEM n=3. C, Western blot analysis. PKD activation via Gαq or Gα11 in the presence of PDBu or AlF4− was accompanied by phosphorylation at activation loop sites Ser744 and Ser748 as detected with phosphor-specific antibodies.
In a previous study, significant PKD activity was stimulated in response to lysophosphatidic acid-responsive GPCR in embryonic fibroblasts from mice expressing Gαq and Gα11, but not in those from Gαq/Gα11 double knock-out mice (20). However, the precise contribution of Gα11 was not examined. Here, we demonstrate that cotransfection of PKD with Gα11 also gave rise to potent stimulation of PKD activity when cells were stimulated with AlF4− (Fig. 1A and 1B). PKD activation induced through Gαq or Gα11 was comparable to that achieved by stimulation with PDBu. Importantly, control cells co-transfected with Gαq or Gα11 but not stimulated with AlF4− did not exhibit elevated basal PKD activity (Fig. 1).
Next, we determined whether AlF4− induces PKD phosphorylation at the activation loop residues Ser744 and Ser748 using specific phosphospecific antibodies for monitoring phosphorylation at each site. As shown in Fig 1C, AlF4− specifically induced PKD Ser744 and Ser748 phosphorylation only in cells cotransfected with either Gαq or Gα11 but not in cells transfected only with PKD (Fig. 1C). We verified that PDBu stimulation induced maximal PKD phosphorylation at both activation loop sites in cells transfected with PKD or cotransfected with PKD and Gαq or Gα11 (Fig. 1C). These data imply that enhanced PKD activation loop phosphorylation (Fig. 1C) is responsible for the stimulation of PKD activity induced via stimulation of Gαq or Gα11 (Fig. 1A and B). Since Gα11-mediated PKD activation and PKD Ser748 phosphorylation had not previously been demonstrated, the results in Fig. 1 extend earlier studies and verify that our model system is useful for determining mechanisms of PKD activation in response to Gq family Gα, in the absence of receptor stimulation.
3.2. Gαq and Gα11 mediate plasma membrane translocation of PKD in response to AlF4− stimulation
Fluorescent chimeric fusion proteins, i.e., GFP-PKD and PKD-RFP were previously used to demonstrate dynamic re-localization of PKD [38, 41–44]. Here, we examined re-localization of these fusion proteins in response to acute activation of Gαq or Gα11, to gain insight into the role of Gα in the PKD activation process. Since this has not been characterized previously in COS-7 cells, we verified that treatment with PDBu, co-transfection with Gαq and treatment with AlF4− or cotransfection with Gαq-QL did not alter the diffuse distribution of transiently transfected GFP in these cells (Supplementary Fig. 2A and data not shown). As shown in Supplementary Fig 2B, GFP-PKD was localized in the cytoplasm, and also in the nuclei in a fraction of cells (20–25%), consistent with regulated nucleocytoplasmic shuttling [42]. Treatment with AlF4− in cells without ectopic expression of Gq did not induce GFP-PKD relocalization. These data are in line with those in Fig. 1 demonstrating that PKD transfected alone is not activated by cell treatment with AlF4−. We verified that cell stimulation with PDBu (200 nM for 10 min) induced dramatic relocalization of GFP-PKD to the cell membrane surface.
To determine whether Gq family Gα induce GFP-PKD redistribution in response to AlF4−, we next examined the localization of GFP-PKD cotransfected with Gαq or Gα11. Consistent with a requirement for stimulation of the Gα to elicit PKD plasma membrane relocalization, GFP-PKD remained cytosolic/nuclear localized in unstimulated COS-7 cells co-transfected with either Gαq (Fig 2A, top panel) or Gα11 (Fig 2A, middle panel). In contrast, GFP-PKD dramatically shifted to a prominent plasma membrane localization that entirely obscured the cytoplasmic compartment in the Gαq or Gα11-cotransfected cells stimulated with AlF4− (Fig. 2A). In addition, GFP-PKD fluorescence concentrated at the periphery of many of these cells (Fig 2A). In contrast, AlF4− stimulation failed to induce plasma membrane translocation of a PKD deletion mutant, GFP-PKD-ΔCys2, lacking the second C1 domain, i.e., C1b (Fig. 2A, bottom panel).
Fig. 2. AlF4−-activated Gαq or Gα11 induces GFP-PKD plasma membrane translocation.

A, COS-7 cells were cotransfected with either GFP-PKD (upper and middle panels), or GFP-PKD-ΔCys2 (lower panel), together with either Gαq (upper and lower panels) or Gα11 (middle panel), as indicated. After 48 h, the cells were imaged before and after stimulation with 10 μM AlF4− for 45 min. B, ALF4−-activated wild-type Gαq or constitutively active Gαq-QL mutant potently induces plasma membrane translocation of a distinct fusion protein with RFP attached to the C-terminal position of PKD (PKD-RFP), but not of the RFP protein. COS-7 cells were transfected with either RFP (lower panel, left) or PKD-RFP either alone (upper panel), together with Gαq (middle panel) or constitutively active mutant Gαq-QL (lower panel), and either left unstimulated or stimulated with 10 μM AlF4− as indicated, prior to imaging.
We also investigated Gα-dependent translocation of a distinct fusion protein with red fluorescent protein from Discosoma sp. (RFP) attached to the C-terminal position of PKD. Thus, similar to GFP-PKD, PKD-RFP transfected alone in COS-7 cells was predominantly cytosolic and redistributed to the plasma membrane in response to PDBu (data not shown) but not AlF4− stimulation (Fig. 2B, top panel). In contrast, PKD-RFP was potently relocalized to plasma membrane in response to AlF4− in cells co-transfected with either Gαq (Fig. 2B, middle panel) or Gα11 (data not shown). Moreover, PKD-RFP, but not the fluorescent protein lacking PKD, exhibited constitutive plasma membrane localization in cells cotransfected with constitutively active mutant forms of the Gα, i.e., Gαq-QL (Fig. 2B, bottom panel) or Gα11-QL (data not shown). Taken together, the results in Fig. 2 reinforce the notion that plasma membrane translocation, predominantly mediated by the C1b of PKD, is necessary for Gα-induced PKD activation.
3.3. Specific PKC-independent enhancement of activation loop Ser748 phosphorylation by Gαq
Many agonists of Gq-coupled receptors induce biphasic PKD activation in a variety of cell types, characterized by rapid PKC-dependent followed by PKC-independent PKD activation [33–35]. The hallmark of the PKC-independent phase is PKD autophosphorylation at Ser748. It is not known whether members of the Gq family can elicit both phases of PKD activation. To determine whether Gα mediate biphasic PKD activation in response to AlF4−, we measured PKD phosphorylation at its activation loop in cells cotransfected with PKD and Gαq, Gα11, Gα14 or Gα15, over a longer time course. Since the PKC-dependent and PKC-independent phases characteristic of GPCR-induced PKD activation exhibit distinct patterns of PKD activation loop phosphorylation [33, 34], we analyzed phosphorylation of Ser744 and Ser748, in lysates of cells treated with or without the broad-spectrum PKC inhibitor, GF109203X (GFI).
We initially verified that AlF4− stimulation of cells transfected with only PKD over a 75-min time course induced only very low levels of Ser744 or Ser748 phosphorylation (Supplementary Fig. 1). These data indicated that, in agreement with the very weak stimulation of PKD activity obtained in Fig. 1, endogenous Gq levels in COS-7 cells are not sufficient to mediate significant PKD activation loop phosphorylation in response to AlF4−. In contrast, AlF4− stimulation induced a dramatic time-dependent increase in both Ser744 and Ser748 phosphorylation in PKD + Gαq transfected cells in the absence of GFI (Fig. 3). However, these cells unexpectedly exhibited distinct kinetic patterns of phosphorylation of the two sites analyzed. In particular, Ser748 phosphorylation responded rapidly (within 10 min) to its peak level when stimulated with AlF4− in the absence of GFI (Fig. 3). Even more surprisingly, Ser748 phosphorylation in these cells was strikingly stimulated by AlF4− treatment even when preincubated with GFI, indicating a selective role of Gαq to promote PKC-independent Ser748 phosphorylation (Fig. 3). Thus, despite basal levels of Ser748 being essentially abolished during preincubation of these cells with GFI, AlF4− stimulation nevertheless elicited strong Ser748 phosphorylation over time, reaching ~75% of the maximal levels obtained with PDBu stimulation. In fact, during 25–45 min of stimulation, the levels of Ser748 phosphorylation obtained either with or without preincubation with GFI were indistinguishable. Moreover, Ser744 phosphorylation in the presence of GFI also appeared to be induced to a significant degree in cells with exogenous Gαq (Fig. 3). These results suggest that, whereas a PKC-dependent pathway mediates the elevated basal PKD Ser748 phosphorylation in Gαq-cotransfected cells, Gαq also mediates a distinct, PKC-independent mechanism that promotes phosphorylation of this residue upon AlF4− stimulation.
Fig. 3. Time-dependent activation loop phosphorylation of PKD at Ser744 and Ser748 in response to Gαq stimulation.

COS-7 cells were transfected with PKD and Gαq. After 48 h, cells were preincubated with or without GFI (3.5 μM), then stimulated with 10 μM AlF4−. Cells were lysed at the times indicated and samples analyzed by Western blot using antibodies to detect PKD activation loop phosphorylation. Blots were reprobed to verify equal expression and loading of total PKD protein. Small panels above the Figure indicate protein expression after transfection of PKD with or without Gαq. Graphs below, depicting data from measurements of Ser744 (left) and Ser748 phosphorylation (right), represent the mean ± SEM, n=4 in the absence, or n = 3 in the presence of GFI.
3.4. The pattern of PKD activation loop phosphorylation by stimulation of Gα11 with AlF4- is distinct from that of Gαq
PKD activation loop phosphorylation in COS cells transfected with PKD and Gα11 and stimulated with AlF4− are shown in Fig. 4. In these cells, like those expressing ectopic Gαq, AlF4− induced striking increases in both Ser744 and Ser748 phosphorylation over time in the absence of GFI. However, a pattern distinct from that obtained by Gαq stimulation also emerged. In particular, the kinetics of increase in Ser744 and Ser748 phosphorylation were concordant, both rising linearly for approximately 20 min and leveling off thereafter (Fig. 4). In addition, the responses of Ser744 and Ser748 phosphorylation to AlF4− stimulation were also similar to one another when measured in cells treated with the PKC inhibitor. Thus, in cells cotransfected with PKD and Gα11 and exposed to GFI, AlF4− stimulation failed to increase either Ser744 or Ser748 above approximately 30% of maximal values obtained with PDBu stimulation (Fig. 4). These data therefore indicate that, whereas Gα11 was capable of promoting robust PKC-dependent PKD activation loop phosphorylation in response to AlF4− stimulation, this Gα isoform, in contrast to Gαq, did not induce any detectable PKC-independent stimulation of Ser748 phosphorylation.
Fig. 4. Time-dependent activation loop phosphorylation of PKD at Ser744 and Ser748 in response to Gα11 stimulation.
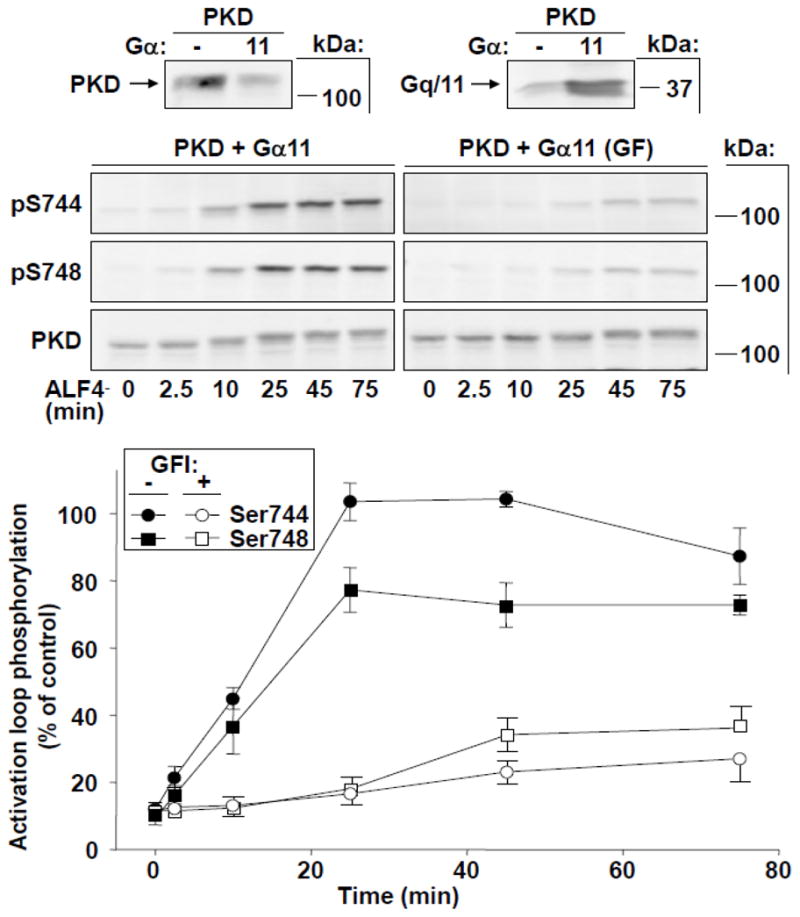
COS-7 cells were transfected with PKD and wild-type Gα11. After 48 h, cells were preincubated with or without GFI (3.5 μM), then stimulated with 10 μM AlF4− to activate Gα11. Cells were lysed at the times indicated and samples analyzed by Western blot using antibodies to detect PKD activation loop phosphorylation. Small panels above the Figure indicate protein expression after transfection of PKD with or without Gα11. Lower panel: Combined data from measurements of Ser744 and Ser748 phosphorylation represent the mean ± SEM, n=4 in the absence, or n = 3 in the presence of GFI.
3.5. Time-dependent induction of PKD activation loop phosphorylation in response to Gα14 and Gα15 signaling stimulated by AlF4−
Whereas all previous studies have primarily focused on Gαq and Gα11 isoforms, several cell types express other Gq family Gα, including Gα14 and Gα15/16. In its 355 aa, human Gα14 is 82% identical to either Gαq or Gα11. At 374 aa, Gα16 is longer and more divergent, sharing a 362-aa region of homology with Gαq that is only 56% identical [4]. It was not known whether stimulation of Gα14 and Gα15 also mediates PKD activation. As shown in Fig. 5A and B, stimulation with AlF4− elicited robust phosphorylation of both Ser744 and Ser748 in cells cotransfected with PKD and either Gα14 or Gα15. In these cells, Ser744 and Ser748 phosphorylation increased gradually for the first 20–25 min, reached a peak and/or leveled off, and remained at near these peak values for at least 75 min. Interestingly, Ser744 and Ser748 phosphorylation in response to AlF4− stimulation in these cells, like that found with Gα11, was predominantly PKC-dependent. Specifically, we detected only minimal phosphorylation of either Ser744 or Ser748 at all times in the presence of GFI. Thus, the pattern of PKD activation loop phosphorylation induced by AlF4− stimulation of Gα14 or Gα15, like Gα11, is mediated exclusively through a PKC-dependent pathway.
Fig. 5. Time-dependent activation loop phosphorylation of PKD at Ser744 and Ser748 in response to Gα14 or Gα15 stimulation.
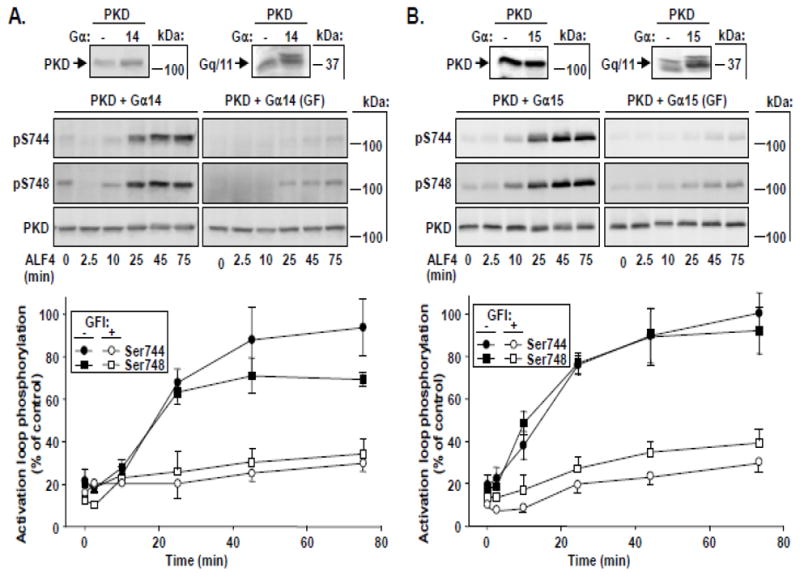
COS-7 cells were transfected with PKD and wild-type Gα14 (A) or Gα15 (B). After 48 h, cells were preincubated with or without GFI (3.5 μM), then stimulated with ALF4− to activate the respective Gα. Cells were lysed at the times indicated and samples analyzed by Western blot using antibodies to detect PKD activation loop phosphorylation. Small panels above A and B indicate protein expression after transfection of PKD with or without Gα14 (A) or Ga15 (B). Combined data in the lower panels represent the mean +/− SEM, n=4 in the absence, or n= 3 in the presence of GFI.
3.6 Pasteurella multocida toxin induces biphasic PKD1 activation in Swiss 3T3 cells
Next, we asked whether selective activation of endogenous Gαq can also induce PKD activation through PKC-dependent and PKC-independent pathways. For these experiments we exploited Swiss 3T3 fibroblasts, a cell line used extensively as a model system to elucidate mechanisms of mitogenic signaling [10, 45] and biphasic PKD regulation [30, 34]. Bacterial toxins have provided powerful tools to elucidate the function of α subunits of heterotrimeric G proteins. In this context, Pasteurella multocida toxin (PMT), an extremely potent mitogen for Swiss 3T3 cells and other fibroblasts [46, 47], strikingly stimulates phospholipase C-mediated hydrolysis of phosphatidylinositol 4,5-bisphosphate [48] through selective modification and constitutive activation Gαq but not via the closely related Gα11 [49–52]. Consequently, we determined whether treatment with PMT for vatious times induces biphasic PKD activation in Swiss 3T3-PKD.GFP cells. Cultures of these cells were treated with 10 ng/ml PMT for either 2 h, a time shortly its internalization or 6 h, a time sufficient for complete internalization and action of PMT [46]. As shown in Fig. 6, treatment of Swiss 3T3-PKD.GFP cells with PMT induced a dramatic time-dependent increase in the phosphorylation of both Ser744 and Ser748 in the activation loop of PKD. Prior exposure to GF1 markedly inhibited PKD activation loop phosphorylation in the cultures treated with PMT for 2h. In contrast, stimulation of PKD phosphorylation at the activation loop residues, particularly at Ser748, was largely insensitive to PKC inhibition after 6h of treatment with PMT. We confirmed that GF1 was active in suppressing PKC activity within Swiss 3T3-PKD.GFP cells either 2h or 6 h after PMT, as shown by the complete inhibition of phosphorylation of MARCKS, a well known direct substrate of PKCs.
Fig. 6. PKC-dependent and PKC-independent PKD activation loop phosphorylation in response to PMT in Swiss 3T3 PKD.GFP cells.

Cultures of these cells were incubated in the absence (−) or in the presence (+) of 3.5 μM GF1 for 1 h prior to stimulation of the cells with 10 ng/ml PMT for either 2 h or 6 h. The cultures were then lysed with 2×SDS–PAGE sample buffer. All samples were analyzed by SDS-PAGE and Western blot using antibodies to detect PKD activation loop phosphorylation (Ser744 and Ser748) and MARCKS phosphorylation (Ser152/156), a marker of PKC activity, as well as tubulin to verify equal gel loading. The results shown here are representative autoluminograms; similar results were obtained in three independent experiments. Graphs below, depicting data from measurements of Ser744 (left) and Ser748 phosphorylation (right), represent the mean ± SEM, n=3 either in the absence or presence of GFI.
4. Discussion
It is well established that maximal PKD activation by cellular stimuli requires phosphorylation of both activation loop residues, Ser744 and Ser748. Multiple lines of evidence indicate that co-localized, activated PKC rapidly catalyzes direct transphosphorylation of these PKD sites [16]. More recent studies identified distinct GPCR-induced PKC-dependent and –independent phases of PKD activation with distinct kinase-dependency of phosphorylation sites and kinetics [33–35]. These studies also indicated that, in cells with inhibited PKC activity, Ser748 becomes preferentially autophosphorylated over a longer time course of GPCR stimulation. However, the precise role of different members of the Gq family in mediating this latter pathway remained unknown.
Based on the results presented here, we conclude that stimulation of each Gq family Gα, namely Gαq, Gα11, Gα14 and Gα15, initiate rapid PKD activation through PKC-dependent phosphorylation of the T loop residues Ser744 and Ser748. However, our data also reveal an unexpected new dimension in Gq function, i.e., a distinct output of Gαq and Gα11, that identifies a divergence point in signaling emanating from these closely related but distinct Gα. Thus, whereas Gα11, Gα14 or Gα15 mediate early and late PKD activation only through a PKC-dependent pathway, Gαq uniquely confers a PKC-independent phase of PKD activation via Ser748 phosphorylation. We substantiated that these findings are not restricted to ectopically overexpressed Gα proteins in COS-7 cells, since we demonstrated that treatment of Swiss 3T3 cells with PMT, an intracellular acting toxin that selectively activates Gαq but not Gα11[49–52], induced biphasic PKD activation loop phosphorylation in these cells. Since these results likely reflect differential Gα participation in distinct phases of PKD activation, our findings bring us a step closer to understanding the mechanisms involved in these distinct phases.
Studies with fluorescent PKD fusion proteins indicate that both Gαq and Gα11 induce dramatic PKD plasma membrane relocalization, but only when the Gα is activated either mutationally or by acute cell stimulation, consistent with a role of PLC-mediated DAG production in this process. In line with a previous study [43], we also obtained evidence of direct Gα-PKD complex formation, from co-immunoprecipitation experiments, which was not dependent on G protein activation (our unpublished results). Thus, it appears that events subsequent to PKD membrane translocation determine whether the enzyme becomes activated, yet only Gαq but not the closely related Gα11 is capable of stimulating PKC-independent PKD autophosphorylation.
The unique expression patterns of each Gα of the Gq family suggests that these transducers have tissue-specific signaling functions [4]. We propose that the level of expression of the different Gα is an important molecular element that determines the influence of cell context on the mechanism of agonist-induced PKD activation. Thus, it could be hypothesized that in cells that express Gα14 and/or Gα15/16, induction of PKD activation by stimulation of GPCR that may couple to these Gq family Gα will proceed via a PKC-dependent pathway. As for the more ubiquitous Gα11 and Gαq, our data suggest that these two highly similar gene products permit distinct pathways of PKD activation in cells expressing one versus the other Gα that is either entirely or only partially PKC-dependent, respectively. This potential diversity in PKD activation implies that pathways induced by stimulation of GPCR that couple to Gαq in cells expressing this Gα may be partially independent of PKC. Evidence obtained recently indicates that such pathways are of potential importance for proliferation in fibroblast and intestinal epithelial cells [15, 34, 35]. Indeed, a PKC-independent signaling pathway for PKD activation may be selectively advantageous for the promotion of proliferation, since EGF receptor transactivation, a critical element driving proliferative signal transduction in response to GPCRs [10], is subject to negative feedback regulation mediated by PKC activity in these cells [53].
Our data indicating this novel and unique role of Gαq in promoting biphasic PKD activation has further implications in normal cellular function and certain disease states [15], including diabetes mellitus [54, 55], hypertrophic cardiac failure [56] and melanocytic neoplasms [57, 58], in which overexpression and/or mutational activation of Gαq and PKD have been implicated in disease causation.
Supplementary Material
Highlights.
The results presented indicate that stimulation of Gαq, but not of the closely related Gα11, promotes PKD activation in response to GPCR agonists leading to PKD autophosphorylation at the activation loop residue Ser-748.
Acknowledgments
This work was supported by the National Institutes of Health Grants R21DK 071783 (to RW) and DK 055003S1, DK 055003, DK 056930 and P30 DK41301 (to ER). We also acknowledge the help provided by the Morphology and Cell Imaging Core of the CURE: Digestive Diseases Research Center supported by P30 DK41301.
Footnotes
Abbreviations: PKC, protein kinase C; PKD, protein kinase D; GPCR, G protein-coupled receptor; Gα, heterotrimeric G protein alpha subunit; PLC, phospholipase C; CAMK, Ca2+/calmodulin-dependent protein kinase; AlF4−, aluminum fluoride; GFP, enhanced green fluorescent protein; RFP, red fluorescent protein; PDBu, phorbol 12,13 dibutyrate; DAG, diacylglycerol; PMT, Pasteurella multocida toxin; PBS, phosphate-buffered saline; PAGE, polyacrylamide gel electrophoresis; DMEM, Dulbecco’s modified Eagle medium; BSA, bovine serum albumin; AEBSF, 4-(2-aminoethyl)-benzenesulfonyl fluoride hydrochloride.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Offermanns S, Simon MI. Cancer Surveys. 1996;27:177–98. [PubMed] [Google Scholar]
- 2.Exton JH. Eur J Biochem. 1997;243:10–20. doi: 10.1111/j.1432-1033.1997.t01-1-00010.x. [DOI] [PubMed] [Google Scholar]
- 3.Wettschureck N, Offermanns S. Physiol Rev. 2005;85:1159–1204. doi: 10.1152/physrev.00003.2005. [DOI] [PubMed] [Google Scholar]
- 4.Hubbard KB, Hepler JR. Cell Signal. 2006;18:135–150. doi: 10.1016/j.cellsig.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 5.Xu X, Croy JT, Zeng W, Zhao L, Davignon I, Popov S, Yu K, Jiang H, Offermanns S, Muallem S, Wilkie TM. J Biol Chem. 1998;273:27275–27279. doi: 10.1074/jbc.273.42.27275. [DOI] [PubMed] [Google Scholar]
- 6.Exton JH. Annu Rev Pharmacol Toxicol. 1996;36:481–509. doi: 10.1146/annurev.pa.36.040196.002405. [DOI] [PubMed] [Google Scholar]
- 7.Rhee SG. Annu Rev Biochem. 2001;70:281–312. doi: 10.1146/annurev.biochem.70.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berridge MJ, Lipp P, Bootman MD. Nat Rev Mol Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- 9.Newton AC. Am J Physiol Endocrinol Metab. 2010;298:E395–402. doi: 10.1152/ajpendo.00477.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rozengurt E. J Cell Physiol. 2007;213:589–602. doi: 10.1002/jcp.21246. [DOI] [PubMed] [Google Scholar]
- 11.Offermanns S, Zhao LP, Gohla A, Sarosi I, Simon MI, Wilkie TM. Embo Journal. 1998;17:4304–4312. doi: 10.1093/emboj/17.15.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Offermanns S, Hashimoto K, Watanabe M, Sun W, Kurihara H, Thompson RF, Inoue Y, Kano M, Simon MI. Proc Nal Acad Sci USA. 1997;94:14089–94. doi: 10.1073/pnas.94.25.14089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haley JE, Abogadie FC, Delmas P, Dayrell M, Vallis Y, Milligan G, Caulfield MP, Brown DA, Buckley NJ. J Neurosci. 1998;18:4521–4531. doi: 10.1523/JNEUROSCI.18-12-04521.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haley JE, Delmas P, Offermanns S, Abogadie FC, Simon MI, Buckley NJ, Brown DA. J Neurosci. 2000;20:3973–3979. doi: 10.1523/JNEUROSCI.20-11-03973.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rozengurt E. Physiology. 2011;26:23–33. doi: 10.1152/physiol.00037.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rozengurt E, Rey O, Waldron RT. J Biol Chem. 2005;280:13205–13208. doi: 10.1074/jbc.R500002200. [DOI] [PubMed] [Google Scholar]
- 17.Valverde AM, Sinnett-Smith J, Van Lint J, Rozengurt E. Proc Natl Acad Sci USA. 1994;91:8572–8576. doi: 10.1073/pnas.91.18.8572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johannes FJ, Prestle J, Eis S, Oberhagemann P, Pfizenmaier K. J Biol Chem. 1994;269:6140–6148. [PubMed] [Google Scholar]
- 19.Iglesias T, Waldron RT, Rozengurt E. J Biol Chem. 1998;273:27662–27667. doi: 10.1074/jbc.273.42.27662. [DOI] [PubMed] [Google Scholar]
- 20.Yuan JZ, Slice L, Walsh JH, Rozengurt E. J Biol Chem. 2000;275:2157–2164. doi: 10.1074/jbc.275.3.2157. [DOI] [PubMed] [Google Scholar]
- 21.Waldron RT, Rey O, Iglesias T, Tugal T, Cantrell D, Rozengurt E. J Biol Chem. 2001;276:32606–32615. doi: 10.1074/jbc.M101648200. [DOI] [PubMed] [Google Scholar]
- 22.Waldron RT, Rozengurt E. J Biol Chem. 2003;278:154–63. doi: 10.1074/jbc.M208075200. [DOI] [PubMed] [Google Scholar]
- 23.Zugaza JL, Waldron RT, Sinnett-Smith J, Rozengurt E. J Biol Chem. 1997;272:23952–23960. doi: 10.1074/jbc.272.38.23952. [DOI] [PubMed] [Google Scholar]
- 24.Zhukova E, Sinnett-Smith J, Rozengurt E. J Biol Chem. 2001;276:40298–40305. doi: 10.1074/jbc.M106512200. [DOI] [PubMed] [Google Scholar]
- 25.Zhukova E, Sinnett-Smith J, Wong H, Chiu T, Rozengurt E. J Cell Physiol. 2001;189:291–305. doi: 10.1002/jcp.10018. [DOI] [PubMed] [Google Scholar]
- 26.Chiu T, Rozengurt E. Febs Letters. 2001;489:101–106. doi: 10.1016/s0014-5793(01)02076-2. [DOI] [PubMed] [Google Scholar]
- 27.Chiu T, Rozengurt E. Am J Physiol Cell Physiol. 2001;280:C929–942. doi: 10.1152/ajpcell.2001.280.4.C929. [DOI] [PubMed] [Google Scholar]
- 28.Chiu T, Wu SS, Santiskulvong C, Tangkijvanich P, Yee HF, Jr, Rozengurt E. Am J Physiol Cell Physiol. 2002;282:C434–450. doi: 10.1152/ajpcell.00240.2001. [DOI] [PubMed] [Google Scholar]
- 29.Guha S, Rey O, Rozengurt E. Cancer Res. 2002;62:1632–1640. [PubMed] [Google Scholar]
- 30.Sinnett-Smith J, Zhukova E, Hsieh N, Jiang X, Rozengurt E. J Biol Chem. 2004;279:16883–93. doi: 10.1074/jbc.M313225200. [DOI] [PubMed] [Google Scholar]
- 31.Yuan J, Slice LW, Rozengurt E. J Biol Chem. 2001;276:38619–38627. doi: 10.1074/jbc.M105530200. [DOI] [PubMed] [Google Scholar]
- 32.Yuan J, Slice LW, Gu J, Rozengurt E. J Biol Chem. 2003;278:4882–4891. doi: 10.1074/jbc.M211175200. [DOI] [PubMed] [Google Scholar]
- 33.Jacamo R, Sinnett-Smith J, Rey O, Waldron RT, Rozengurt E. J Biol Chem. 2008;283:12877–12887. doi: 10.1074/jbc.M800442200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sinnett-Smith J, Jacamo R, Kui R, Wang YM, Young SH, Rey O, Waldron RT, Rozengurt E. J Biol Chem. 2009;284:13434–13445. doi: 10.1074/jbc.M806554200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sinnett-Smith J, Rozengurt N, Kui R, Huang C, Rozengurt E. J Biol Chem. 2011;286:511–20. doi: 10.1074/jbc.M110.167528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van Lint JV, Sinnett-Smith J, Rozengurt E. Journal of Biological Chemistry. 1995;270:1455–1461. doi: 10.1074/jbc.270.3.1455. [DOI] [PubMed] [Google Scholar]
- 37.Matthews S, Iglesias T, Cantrell D, Rozengurt E. Febs Letters. 1999;457:515–521. doi: 10.1016/s0014-5793(99)01090-x. [DOI] [PubMed] [Google Scholar]
- 38.Rey O, Young SH, Cantrell D, Rozengurt E. J Biol Chem. 2001;276:32616–32626. doi: 10.1074/jbc.M101649200. [DOI] [PubMed] [Google Scholar]
- 39.Seufferlein T, Withers DJ, Mann D, Rozengurt E. Molecular Biology of the Cell. 1996;7:1865–1875. doi: 10.1091/mbc.7.12.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nishimura A, Kitano K, Takasaki J, Taniguchi M, Mizuno N, Tago K, Hakoshima T, Itoh H. Proc Natl Acad Sci U S A. 2010;107:13666–13671. doi: 10.1073/pnas.1003553107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matthews SA, Iglesias T, Rozengurt E, Cantrell D. Embo Journal. 2000;19:2935–2945. doi: 10.1093/emboj/19.12.2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rey O, Sinnett-Smith J, Zhukova E, Rozengurt E. J Biol Chem. 2001;276:49228–49235. doi: 10.1074/jbc.M109395200. [DOI] [PubMed] [Google Scholar]
- 43.Oancea E, Bezzerides VJ, Greka A, Clapham DE. Dev Cell. 2003;4:561–574. doi: 10.1016/s1534-5807(03)00087-x. [DOI] [PubMed] [Google Scholar]
- 44.Rey O, Young SH, Yuan J, Slice L, Rozengurt E. J Biol Chem. 2005;280:22875–22882. doi: 10.1074/jbc.M503455200. [DOI] [PubMed] [Google Scholar]
- 45.Rozengurt E. Science. 1986;234:161–166. doi: 10.1126/science.3018928. [DOI] [PubMed] [Google Scholar]
- 46.Rozengurt E, Higgins T, Chanter N, Lax AJ, Staddon JM. Proc Natl Acad Sci USA. 1990;87:123–127. doi: 10.1073/pnas.87.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Higgins TE, Murphy AC, Staddon JM, Lax AJ, Rozengurt E. Proc Natl Acad Sci USA. 1992;89:4240–4244. doi: 10.1073/pnas.89.10.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Staddon JM, Barker CJ, Murphy AC, Chanter N, Lax AJ, Michell RH, Rozengurt E. J Biol Chem. 1991;266:4840–4847. [PubMed] [Google Scholar]
- 49.Murphy AC, Rozengurt E. J Biol Chem. 1992;267:25296–25303. [PubMed] [Google Scholar]
- 50.Wilson BA, Zhu X, Ho M, Lu L. J Biol Chem. 1997;272:1268–12675. doi: 10.1074/jbc.272.2.1268. [DOI] [PubMed] [Google Scholar]
- 51.Zywietz A, Gohla A, Schmelz M, Schultz G, Offermanns S. J Biol Chem. 2001;276:3840–3845. doi: 10.1074/jbc.M007819200. [DOI] [PubMed] [Google Scholar]
- 52.Orth JH, Preuss I, Fester I, Schlosser A, Wilson BA, Aktories K. Proc Natl Acad Sci U S A. 2009;106:7179–7184. doi: 10.1073/pnas.0900160106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Santiskulvong C, Rozengurt E. Cell Signal. 2007;19:1348–13457. doi: 10.1016/j.cellsig.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 54.Sumara G, Formentini I, Collins S, Sumara I, Windak R, Bodenmiller B, Ramracheya R, Caille D, Jiang H, Platt KA, Meda P, Aebersold R, Rorsman P, Ricci R. Cell. 2009;136:235–248. doi: 10.1016/j.cell.2008.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sassmann A, Gier B, Grone HJ, Drews G, Offermanns S, Wettschureck N. J Clin Invest. 2010;120:2184–2193. doi: 10.1172/JCI41541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dorn GW, II, Force T. J Clin Invest. 2005;115:527–537. doi: 10.1172/JCI24178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O/’Brien JM, Simpson EM, Barsh GS, Bastian BC. Nature. 2009;457:599–602. doi: 10.1038/nature07586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Patel M, Smyth E, Chapman PB, Wolchok JD, Schwartz GK, Abramson DH, Carvajal RD. Clin Cancer Res. 2011;17:2087–2100. doi: 10.1158/1078-0432.CCR-10-3169. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.