

Abstract
Alkyl β-D-xylopyranosides are highly surface active, biodegradable surfactants that can be prepared from hemicelluloses and are of interest for use as pharmaceuticals, detergents, agrochemicals and personal care products. To gain further insights into their structure-property and structure-activity relationships, the present study synthesized a series of hydrocarbon (-C6H13 to -C16H33) and fluorocarbon (-(CH2)2C6F13) alkyl β-D-xylopyranosides in four steps from D-xylose by acylation or benzoylation, bromination, Koenigs-Knorr reaction and hydrolysis, with the benzoyl protecting group giving better yields compared to the acyl group in the Koenigs-Knorr reaction. All alkyl β-D-xylopyranosides formed thermotropic liquid crystals. The phase transition of the solid crystalline phase to a liquid crystalline phase increased linearly with the length of the hydrophobic tail. The clearing points were near constant for alkyl β-D-xylopyranosides with a hydrophobic tail ≥ 8, but occurred at a significantly lower temperature for hexyl β-D-xylopyranoside. Short and long-chain alkyl β-D-xylopyranosides displayed no cytotoxicity at concentration below their aqueous solubility limit. Hydrocarbon and fluorocarbon alkyl β-D-xylopyranosides with intermediate chain length displayed some toxicity at millimolar concentrations due to apoptosis.
Keywords: Next-generation surfactants, Renewable precursors, Xylose, Structure-activity relationship, Structure-property relationship, Koenigs-Knorr reaction, Liquid crystals, Biocompatibility, Apoptosis, Necrosis
1. Introduction
Carbohydrate-based surfactants are becoming increasingly important as substitutes for poly(ethylene oxide)-based surfactants used in a broad range of industrial and consumer applications, such as pharmaceuticals, detergents, agrochemicals and personal care products.1 They can be synthesized from renewable raw materials using green chemistry processes, have excellent surface activity, and are readily biodegradable. Furthermore, carbohydrate-based surfactants are generally considered to be toxicologically safe. For example, many carbohydrate-based surfactants display little-to-no toxicity in cells in culture and have limited haemolytic activity.2 In particular starch, sugar beet and sugar cane are sources of glucose, sorbitol and sucrose, currently the most important carbohydrate moieties employed in the production of commercially available carbohydrate-based surfactants. In addition to these established sources of carbohydrate starting materials, hemicelluloses, the second most abundant natural polysaccharides after cellulose,3 are receiving increasing attention as sources for carbohydrate starting materials, such as xylose, for the synthesis of next-generation surfactants.
A range of chemical and enzymatic processes have been used to synthesize xylose-based surfactants. For example, simple alkyl xylopyranosides have been prepared by the acid-catalyzed reaction of D-xylose4 or xylan with alcohols.5 Enzymatic reactions that have been employed for the synthesis of alkyl xylopyranosides include, among others, xylanase-catalyzed transglycosylation reactions of xylan6 or Aspergillus niger β-xylosidase transxylosyl reactions of xylobiose2e, 7 with short chain alcohols. The yield of octyl xylopyranoside synthesized from xylan and octanol using acetone-dried cells of Aureobasidium pullulans as the xylanase source has been shown to increase in supercritical carbon dioxide or supercritical CHF3.8
While the physicochemical properties of the xylose-based surfactants synthesized by the various chemical and enzymatic approaches, including their surface activity and foaming properties, are well investigated and comparable to other carbohydrate surfactants,1a, 2e, 4–5 systematic studies of the thermotropic properties and the cytotoxicity of alkyl β-D-xylopyranosides have not been reported previously. As part of the present study, a series of simple hydrocarbon and fluorocarbon alkyl β-D-xylopyranosides was synthesized and their physicochemical properties and cytotoxicity were investigated to gain further insights into the usefulness of these promising next-generation surfactants for pharmaceutical and consumer product applications.
2. Results and discussion
2.1 Synthesis of alkyl β-D-xylopyranosides
The chemical synthesis of simple alkyl β-D-xylopyranosides and structurally related alkenyl and alkenoyl derivatives of D-xylose in poor-to-good yields has been reported previously.4, 9 In the present study, octyl β-D-xylopyranoside (9b) was synthesized in a four step synthesis from D-xylose (1) as outlined in Scheme 1. Briefly, D-xylose was converted into the corresponding peracetylated β-D-xylose 2 with acetic anhydride in pyridine. Subsequent bromination with HBr followed by glycosylation with alcohols in the presence of Ag2CO3 and CaSO4 in anhydrous dichloromethane yielded not only the desired octyl 2,3,4-tri-O-acetyl-β-D-xylopyranoside (4) but also significant amounts of 3,4-di-O-acetyl-1,2-O-(1-octyloxyethylidene)-α-D-xylopyranose (5) which could not be purified by column chromatography. The desired product, octyl β-D-xylopyranoside (9b), was obtained by deprotection of 4 with sodium methoxide in methanol and subsequent neutralization with Dowex 50W×8–100 ion exchange resin.
Scheme 1.

Synthesis of octyl β-D-xylopyranoside (9b) via 1,2,3,4-tetra-O-acetyl-β-D-xylopyranose (2).
Several reaction conditions were investigated to further optimize the glycosylation reaction (Table 1). Silver salts, including Ag2CO3, Ag2O and AgOTf, were employed because they are not only efficient promoters of glycosylation reactions but also neutralize the HBr released in the reaction. Iodine (I2), either alone or in combination with Ag2CO3 and 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ), is another effective, but less expensive activator of glycosylation reactions.10 I2 in combination with DDQ also avoids the need for a potentially toxic heavy metal salt as promoter. Finally, 1,1,3,3-tetramethyl urea (TMU) was investigated because it efficiently neutralizes the acid formed in the glycosylation reaction.11 In agreement with several earlier studies and our initial synthesis attempts,4, 9a, b the major drawback of all reaction conditions investigated was the unsatisfactory yield of the desired octyl tetra-O-acetyl-βD-xylopyranoside (4) (Table 1, entries 1 to 6). The highest yields, largest product to by-product ratios (i.e., ratios of 4:5) and the shortest reaction time were obtained with I2, DDQ, and 4 Å MS (entry 4). With Ag2CO3 as the base, the presence of I2 significantly decreases the side reaction but did not improve the yield of 4 (entries 1 vs. 2). Furthermore, CaSO4 as drying agent was more favorable compared to powdered 4 Å MS (entries 1 vs. 3). The use of AgOTf/TMU, either in the presence or absence of 4 Å MS, also did not improve the yield of 4 or reduce the formation of 5 (entries 5 and 6).
Table 1.
Synthesis of octyl 2,3,4-tri-O-acetyl-β-D-xylopyranoside (4) by reaction of 3 with 1-octanol.a
| Entry | Reaction conditions | Time [h] | Yield (%)b | Ratio of 4 to 5c | Reference |
|---|---|---|---|---|---|
| 1 | Ag2CO3, CaSO4 | 10 | 33 | 4.4 | 43 |
| 2 | Ag2CO3, CaSO4, I2 | 10 | 26 | 9.0 | 43 |
| 3 | Ag2CO3, 4 Å MS | 10 | 8 | 1.7 | 43 |
| 4 | I2, DDQ, 4 Å MS | 3 | 36 | 16 | 10 |
| 5 | AgOTf, TMU, 4 Å MS | 4 | 23 | 2.8 | 11 |
| 6 | AgOTf, TMU | 4 | 17 | 3.2 | 11 |
Reactions in anhydrous dichloromethane (1 mL/0.1 g) were performed at ambient temperature using the following amounts of the reagents: Ag2CO3 (1.05 mol equiv), CaSO4 (same weight as Ag2CO3), 4 Å MS (0.1 g/mL), AgOTf (2.2 mol equiv), and TMU (3 mol equiv). With exception of entry 4 all reactions were performed in the dark.
Isolated yield.
Determined by gas chromatography and based on relative peak area.
MS = molecular sieves; DDQ = 2,3-dichloro-5,6-dicyano-1,4-benzoquinone; TMU = 1,1,3,3-tetramethyl urea.
Subsequent efforts employed the benzoyl protecting group in order to optimize the reaction efficiency and to avoid the side-reaction of the acetyl group. As expected, improved total yields of octyl β-D-xylopyranoside (9b) were obtained when the benzoyl protecting group was used to minimize orthoester formation (25 % for acetyl versus 54 % for benzoyl protected 9b) (Scheme 2). This modified synthesis strategy was subsequently employed to prepare a series of alkyl β-D-xylopyranosides in total yields of 54–72 % from 1,2,3,4-tetra-O-benzoyl-α-D-xylopyranose (6). This approach can also be scaled-up to prepare gram quantities of alkyl β-D-xylopyranosides 9a–f.
Scheme 2.

Synthesis of alkyl β-D-xylopyranosides 9a–g from 1,2,3,4-tetra-O-benzoyl-α-D-xylopyranose (6). DDQ: 2,3-dichloro-5,6-dicyano-1,4-benzoquinone; MS: molecular sieves; DCM: dichloromethane.
In addition to the alkyl β-D-xylopyranosides 9a–f, the partially fluorinated alkyl β-D-xylopyranoside 9g was synthesized because fluorinated surfactants are highly surface active and, at the same time, biocompatible.2f–j The short perflourooctyl chain was selected as hydrophobic tail to obtain a moderately water-soluble surfactant for a preliminary biocompatibility assessment. The yield of the partially fluorinated alkyl β-D-xylopyranoside 9g synthesized from 1,2,3,4-tetra-O-benzoyl-α-D-xylopyranose (6) was lower than the yield of the corresponding hydrocarbon surfactants, with a total yield of only 27 %. Therefore, the alternate route reported by Petrovic et al. was explored to synthesize larger quantities of 9g.9c Briefly, boron trifluoride ethyl etherate was used to glycosylate 2 and 6, yielding 8g and 10, respectively (Scheme 3).2k, 12 The yield was higher for the reaction with the perbenzoate 6 (56 %) than with the peracetylated derivative 2 (33 %), as with the Koenigs-Knorr reaction described above. Both products were readily deprotected with sodium methoxide/Dowex 50W×8–100 ion exchange resin to provide 9g in 62 % yield. Comparable yields for both synthesis steps have been reported previously by Petrovic and co-workers for the synthesis of hydrocarbon alkyl β-D-xylopyranosides 9b–e.9c
Scheme 3.

Synthesis of 9g from 1,2,3,4-tetra-O-acetyl-β-D-xylopyranose (2) or 1,2,3,4-tetra-O-benzoyl-α-D-xylopyranose (6) using BF3-Et2O.
2.2 Crystal structure of decyl β-D-xylopyranoside (9c)
While crystal structures of simple alkyl α-D-pyranosides, including alkyl 2-deoxy-α-D-arabino-hexopyranosides13 and alkyl α-D-glucopyranosides,14 have been extensively studied, only relatively few crystal structures of alkyl β-D-pyranosides have been published. In the present study, crystals of the hemihydrate of 9c suitable for crystal structure determination were obtained by slow evaporation of the solvent from a saturated solution of 9c in acetone/hexane. Crystals of 9c were monoclinic (space group C2) with a = 13.2154(6), b = 4.3040(2), c = 29.9491(16) Å, and β = 102.665(3) ° (see Tables S1 and S2 for additional crystal data, structure refinement information as well as bond length and bond angles of 9c). Only two other crystal structures of alkyl β-D-xylopyranosides have been reported. Methyl β-D-xylopyranoside crystallized in the monoclinic space group (P21), with a = 7.893, b = 6.908, c = 7.709 Å, and β = 113.4 °.15 In contrast, heptyl β-D-xylopyranoside monohydrate crystallized in the orthorhombic space group P212121 (a = 53.215(8), b = 8.8301(9) and c = 6.5276(7) Å).16
The unit cell of 9c contained two independent molecules that form alternating bilayers of hydrophilic xyloside groups and hydrophobic decyl groups parallel to the a–b plane (Figure S30). The distance of the layers d was calculated as d = c × sinβ, where c is the length of the cell axis c and β the monoclinic angle. The distance of the layers calculated using this formula was 25.3 Å, which is comparable to the value of the corresponding glucoside of 26.4 Å.14 A relatively large d value of 26.6 Å was observed for the shorter chain heptyl β-D-xylopyranoside monohydrate.16
The formation of 9c bilayers in the solid state is not surprising because interactions between different parts of an organic molecule are usually less favorable than the interactions between similar parts of the molecule (i.e., the decyl chains and the carbohydrate groups of 9c).17 As with other carbohydrate surfactants,13–14, 16 the carbohydrate groups and decyl chains of 9c are packed in a manner that maximizes intermolecular interactions within the crystal. Specifically, the decyl chains have van der Waals contact by adopting a tilt angle of 41.7° relative to the a–b plane and the carbohydrate groups and the water molecule are connected by a network of hydrogen bonds. Similarly, the two independent molecules in the crystal structure of heptyl β-D-xylopyranoside monohydrate form an acute angle of 49.2° and 37.4° to the bilayer plane (i.e. the b–c plane).16 This separation into hydrophobic and hydrophilic regions has been reported for many carbohydrate surfactants displaying liquid-crystalline properties.18
2.3 Thermotropic liquid crystalline properties of alkyl β-D-xylopyranosides 9a–g
The liquid crystalline properties of biological lipids play an important role in biological structures and function, and in the case of simple surfactants, may correlate with their biological effects.18–19 Many pentose-based surfactants, such as alkyl arabinopyranosides20 and alkyl DL-xylitols,21 also display liquid-crystalline properties and, as first described by Noller and Rockwell for simple alkyl D-glucopyranosides,22 show double melting transitions Mp representing a change from a solid crystalline phase to a liquid crystalline phase, followed by the clearing point Cp (i.e., a transition from the liquid crystalline phase to a liquid melt phase). The present study uses DSC and TGA to gain initial insights into the chain length-dependent thermotropic behavior of hydrocarbon and fluorocarbon alkyl β-D-xylopyranosides 9a–g.
The DSC analysis of the hydrocarbon alkyl β-D-xylopyranosides showed two endothermic peaks in the case of 9b and 9f (Figure 2 and Table 2) and three endothermic peaks in the case of 9a, 9c, 9d and 9e. These phase transitions occurred in a relatively narrow temperature range. Decomposition of the hydrocarbon alkyl β-D-xylopyranosides 9a–f, but not the partially fluorinated analog 9g was observed at temperatures above 150 °C. In the case of octyl β-D-xylopyranoside (9b), the first phase transition (Mp), most likely a transition from a solid crystalline phase to a smectic A type phase,23 occurred at 72.5 °C. The clearing point of 9b was observed at 99.4 °C. Comparable temperatures for Mp (64.5 °C23; 67.5 °C24) and Cp (96.9 °C23; 103.3 °C24) have been reported previously for 9b. The putative Mp and Cp for the fluorinated β-D-xylopyranoside 9g occurred at much higher temperatures (105.1 °C and 147.0 °C, respectively) due to the perfluorinated hydrophobic tail. This increase in the phase transition temperatures of a fluorinated compound compared to the respective hydrocarbon analogue is consistent with other fluorinated compounds, for example perfluorinated carboxylic acids25 or partially fluorinated alkyl β-D-glucopyranosides,2k and is a result of the more dense packing of the rigid perfluorinated tail in the solid state.
Figure 2.
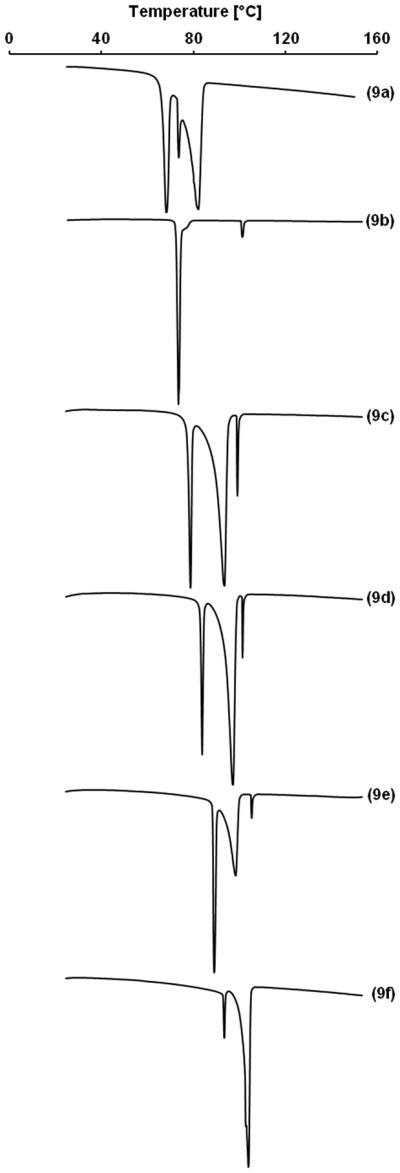
Representative thermograms of hydrocarbon alkyl β-D-xylopyranosides 9a–f.
Table 2.
Onset, maximum (Tm) and half-width of the phase transitions observed for the alkyl β-D-xylopyranosides 9a–g (see Figures 2 and 3 for representative thermograms).
| Compound | RF,H | Onset of phase transition [°C] | Tm [°C] | Half-width of phase transition [°] |
|---|---|---|---|---|
| 9a | n-C6H13 | 66.3±0.1 | 68.3±0.4 | 2.0±0.3 |
| 73.0±0.1 | 73.5±0.2 | 0.7±0.2 | ||
| 77.9±0.6 | 82.4±0.5 | 3.9±0.2 | ||
|
| ||||
| 9b | n-C8H17 | 71.5±0.1 | 72.5±0.3 | 1.3±0.3 |
| 98.8±0.9 | 99.4±0.9 | 0.9±0.1 | ||
|
| ||||
| 9c | n-C10H21 | 76.5±0.1 | 77.6±0.1 | 1.1±0.2 |
| 86.7±1.4 | 91.6±0.8 | 3.5±0.3 | ||
| 97.8±0.9 | 97.9±0.9 | 0.6±0.1 | ||
|
| ||||
| 9d | n-C12H25 | 81.8±0.1 | 82.5±0.0 | 0.9±0.1 |
| 91.3±1.3 | 94.9±1.0 | 2.9±0.1 | ||
| 100.2±1.1 | 100.4±1.2 | 0.5±0.2 | ||
|
| ||||
| 9e | n-C14H29 | 87.0±0.1 | 87.6±0.1 | 1.0±0.2 |
| 93.0±0.1 | 96.7±0.1 | 2.9±0.2 | ||
| 103.1±0.1 | 103.4±0.1 | 0.5±0.1 | ||
|
| ||||
| 9f | n-C16H33 | 91.4±0.1 | 91.9±0.1 | 0.7±0.1 |
| 100.4±0.1 | 101.7±0.8 | 2.0±0.4 | ||
|
| ||||
| 9g | (CH2)2C6F13 | 102.8±0.2 | 105.1±0.1 | 1.9±0.2 |
| 146.8±0.1 | 147.0±0.1 | 0.3±0.1 | ||
The first phase transition observed for the alkyl β-D-xylopyranosides 9a, 9c, 9d and 9e also corresponds to a change from a solid crystalline phase to a liquid crystalline phase. One likely explanation for the other two phase transitions is the loss of crystal water from the sealed DSC pan. As a result, the second, broad phase transition most likely represents the melting of the anhydrous alkyl β-D-xylopyranosides of 9a, 9c, 9d and 9e and the third phase transition is their Cp. A similar effect of the loss of crystal water has been reported previously for the phase transitions of alkyl α-D-glucopyranosides.14 Although the experimental parameters are quite different, the interpretation of the DSC results is supported by TGA experiments which suggest the loss of approximately half a water molecule from the alkyl β-D-xylopyranoside samples (Table 3). It is interesting to note that the decomposition temperature of the fluorinated alkyl βD-xylopyranoside 9g was lower compared to the hydrocarbon compounds in the TGA experiments (Table 3). In contrast to this partially fluorinated compound, perfluorocarbons and perfluorinated surfactants are thermally more stable than their hydrocarbon analogues.26
Table 3.
Weight loss and combustion temperature of alkyl β-D-xylopyranosides 9a–9g.
| Compound | RF,H | Temperature of solvent loss [°C] | Percent weight loss [%] | Number of water molecules | Decomposition Temperature [°C] |
|---|---|---|---|---|---|
| 9a | n-C6H13 | 81.74 ± 0.41 | 2.50±0.66 | 0.4 | 262.8 ± 4.34 |
| 9b | n-C8H17 | 85.32 ± 0.55 | 2.35±0.91 | 0.5 | 267.1 ± 9.15 |
| 9c | n-C10H21 | 96.63 ± 0.44 | 2.69±0.07 | 0.5 | 277.2 ± 11.5 |
| 9d | n-C12H25 | 86.35 ± 0.98 | 1.56±0.09 | 0.3 | 290.4 ± 10.5 |
| 9e | n-C14H29 | 100.1 ± 2.93 | 1.87±1.08 | 0.5 | 294.8 ± 7.15 |
| 9f | n-C16H33 | 103.4 ± 1.95 | 1.24±0.49 | 0.3 | 295.9 ± 2.86 |
| 9g | (CH2)2C6F13 | 50.57 ± 1.64 | 2.64±0.83 | 0.9 | 231.6 ± 4.57 |
Mp of the hydrocarbon alkyl β-D-xylopyranosides 9a–f displayed distinctive trends as a function of hydrophobic tail length (Figures 2 and 5). Mp linearly increased with chain length (y = 2.53x + 51.22; R2 = 0.999). A similar increase in the temperature of this phase transition has been observed for other carbohydrate surfactants, such as glucopyranosides2k, 14 and alkyl DL-xylitol surfactants;21a, 21c however, the increase was typically not as linear as observed for 9a–f.21c A clearing point was observed in all thermograms, with only 9f being an exception. The respective transition temperatures were near constant for alkyl β-D-xylopyranosides with a hydrophobic tail ≥ 8, and only occurred at a significantly lower temperature for hexyl β-D-xylopyranoside (9a) (Figure 5). The clearing temperatures of ether, thioether and ester-based xylitol surfactants show a comparable dependence on the length of the hydrophobic tail.21c
Figure 5.

Chain length-dependent changes in the transition from a solid crystalline phase to a liquid crystalline phase (Mp) and the transition from the liquid crystalline phase to a liquid melt phase (Cp) of hydrocarbon alkyl β-D-xylopyranosides 9a–f.
2.4 Cytotoxicity of alkyl β-D-xylopyranosides
Limited studies of the toxicity of carbohydrate surfactants2 suggest that the headgroup and the hydrophobic tail are both important structural determinants of surfactant-lipid interactions and, thus, their toxicity. However, systematic studies of the toxicity of alkyl β-D-xylopyranoside in mammalian cells in culture have not been reported. In the present study, the cytotoxicity of the alkyl β-D-xylopyranosides 9a–g was initially investigated in the spontaneously immortalized human keratinocyte cell line HaCaT using the MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) cell viability assay. This cell line was selected because it exhibits characteristics of normal skin keratinocytes27 and, therefore, provides insights into possible effects of alkyl β-D-xylopyranosides on skin cells. No effect on cell viability was observed for the shortest chain alkyl β-D-xylopyranoside 9a (CC50 > 1 mM) and the long chain alkyl β-D-xylopyranosides 9d–f (CC50 > 100 mM for 9d and 9e; CC50 > 25 mM for 9f). These four alkyl β-D-xylopyranosides were therefore not included in subsequent cell culture experiments with Jurkat and Hs27 cells. The octyl and decyl β-D-xylopyranosides 9b and 9c displayed moderate toxicity, with the longer chain xylopyranoside 9c being more toxic in the HaCaT cell line (Figure 6). The fluorinated octyl β-D-xylopyranosides 9g also displayed moderate toxicity in HaCaT cells and had a CC50 that was comparable to the CC50 of 9c. All three xylopyranosides were more toxic than octylthioglucoside (OTG), a hydrocarbon surfactant that has been used previously as positive control in cytotoxicity studies with simple, carbohydrate-based surfactants.2k, 12a
Figure 6. Dose-response curves and determination of CC50 values utilizing the MTS assay.

Dose-response effect and CC50 were determined in HaCaT, Hs27 and Jurkat cells for alkyl β-D-xylopyranosides 9b (A), 9c (B), 9g (C) and octylthioglucoside (OTG) (D). Cells were exposed for 20 h to increasing concentrations of 9b, 9c, 9g or OTG. The cellular viability is shown in the y axis while the concentration of compounds (μM) is shown in the x axis. Data are represented as the mean percentage of viable cells at each dose ± SD of four replicates. CC50 values of the experimental compounds for each cell line are summarized in (E).
The observation that the short (9a) and long chain alkyl β-D-xylopyranosides (9d–f) had no effect on HaCaT cell viability in the concentration range investigated is in agreement with previous structure-activity studies with other carbohydrate surfactants in B16F10 cells2k, 12a and various anesthetic and antimicrobial compounds in non-mammalian systems.28 The low toxicity of carbohydrate surfactants with a short hydrophobic tail, such as 9a, is likely due to the limited partitioning of these surfactants into the cell membrane.12a The low toxicity of the long chain alkyl β-D-xylopyranosides 9d–f appears to be counterintuitive because the cellular uptake of surfactants and, therefore, their toxicity should increase with increasing length of the hydrophobic tail. However, a number of factors (e.g. a decrease in aqueous and lipid solubility, an increase in binding to protein in the culture medium, a decrease in the critical micelle concentration and a decrease in the diffusion through the cell membrane with increasing length of the hydrophobic tail) can reduce the cellular uptake and, thus, the toxicity of the long chain alkyl β-D-xylopyranosides 9d–f.12a, 28
Subsequent cell culture experiments investigated the effect of the carbohydrate surfactants 9b, 9c, 9g and OTG on the cell viability in Jurkat and Hs27 using the MTS assay. The human Jurkat T-cell lymphoblastic leukemia is a transformed cell line that grows in suspension and was selected because it is highly sensitive to a variety of chemical treatments.29 Finally, HS27 normal human fibroblasts of newborn foreskin were studied because they are commonly used as “normal” cell controls when compared to transformed immortal cancer lines.29b, 30 Experiments with these two cell lines also revealed moderate toxicity for all three alkyl β-D-xylopyranosides and OTG (Figure 6). In the Jurkat cell line, the CC50 values decreased in the order 9b > 9g ~ 9c. This trend is comparable to the one observed for the HaCaT cell line. A slightly different rank order was observed for the Hs27 cell line, with 9b > 9g > 9c. The three cell lines displayed different sensitivity towards alkyl β-D-xylopyranoside-mediated toxicity. For the three hydrocarbon surfactants 9b, 9c and OTG, the HaCaT cells were less sensitive compared to the other two cell lines and displayed the highest CC50 values. In the case of the hydrocarbon alkyl β-D-xylopyranosides 9b and 9c, the Hs27 cell line seemed to be more sensitive than the other two cell lines. A different rank order was observed for the partially fluorinated alkyl β-D-xylopyranoside 9g, with the Hs27 cell line being less sensitive toward 9g compared to the HaCaT and Jurkat cell lines.
Several studies have reported that fluorinated surfactants are less toxic than the analogous hydrocarbon surfactant due to a protective effect of the perfluorinated tail.2f–j In contrast to these earlier findings, surfactant 9g was more cytotoxic compared to the analogous hydrocarbon compound 9b. One likely explanation for the comparatively high toxicity of 9g is that the degree of fluorination is not sufficient to convey a protective effect in the cell lines under investigation. In contrast, the CC50 values of 9g and 9c were comparable in the HaCaT and Jurkat cell lines and only slightly different in the Hs27 cell lines (Figure 6E), despite the difference in the length of the hydrophobic tail (C8 versus C10). This finding can be explained with the “1.5 rule” which predicts that fluorinated surfactants behave like analogous hydrocarbon surfactants with a longer hydrophobic tail, with one CF2 group approximating 1.5 CH2 groups.31 According to this rule, the fluorinated alkyl β-D-xylopyranosides 9g should behave like undecyl β-D-xylopyranoside (i.e., 2 × CH2 + 1.5 × 6 × CF2 = 11) and thus display properties comparable to decyl β-D-xylopyranoside (9c). Indeed, in the CC50 values of 9g and 9c are similar, which suggests that physicochemical parameters, such as the partitioning of alkyl β-D-xylopyranoside surfactants into the cell membrane, may play an important role in their cytotoxicity.
The reduction in cell viability observed with 9b, 9c, 9g and OTG can be due to necrosis or apoptosis, the two major forms of cell death. There is evidence that the effect of carbohydrate surfactants on cell viability in mammalian cell lines is not due to a massive disruption of phospholipid membranes but caused by more specific mechanisms. Previous studies with carbohydrate surfactants demonstrate that the CC50 values for cell viability in B16G10 cells are significantly smaller than the corresponding values for haemolytic activity in rabbit red blood cells.2k, 12a Furthermore, carbohydrate surfactants with only minor differences in the structure of the head group have different CC50 values12a and, in the case of several glucopyranosides, induce apoptosis in B16G10 cells.2k In the present study the number of apoptotic Jurkat cells after exposure to the alkyl β-D-xylopyranosides 9b, 9c and 9g as well as OTG was quantified using Annexin V-FITC, a molecule that binds to the phosphatidylserine present on the extracellular side of the cell membrane of cells in the early stage of apoptosis. As shown in Figure 7, all four surfactants decrease cell viability in Jurkat cells by apoptosis and not necrosis. This observation provides further evidence that carbohydrate surfactants cause toxicity by specific mechanisms, such as a selective interaction with lipids rafts in the cell membrane, which ultimately results in a receptor-independent activation of intracellular signaling pathways, such as Fas-mediated apoptosis cascades.32
Figure 7. Induction of apoptosis by alkyl β-D-xylopyranosides 9b, 9c, 9g and OTG.

Jurkat cells were evaluated for apoptosis via Annexin-FITC and propidium iodide staining after exposure for 20 h to alkyl β-D-xylopyranosides 9b, 9c, 9g or OTG at their respective CC50. Positive controls for apoptosis and necrosis included 2 μM Stauroporine and 0.5% Tween 20, respectively. Untreated cells and DMSO treated cells were used as negative and solvent controls, respectively. Data are represented as the mean ± SD of three replicates.
2.5 Conclusions
A series of simple alkyl xyloside surfactants was synthesized from xylose and readily-available alcohols, using an optimized Koenigs-Knorr reaction as the key glycosylation step. These surfactants were shown to be non-toxic in several established cell lines. Furthermore, all xylosides exhibited liquid-crystalline behavior based on DSC experiments. In agreement with the thermotropic behavior observed in the DSC studies, the hemihydrate of alkyl xyloside 9b formed bilayers in the solid state. These compounds have potential as environmentally friendly surfactants due to their low toxicity and renewable precursors, but further studies are needed to fully characterize their phase behavior, investigate their surface properties and assess their toxicity.
3. Experimental
3.1 General methods
The 1H and 13C NMR spectra were recorded on a multinuclear Bruker Avance 300 or a Bruker DRX 400 Digital NMR spectrometer at ambient temperature. 19F spectra were recorded using a Bruker Avance 300 NMR spectrometer. Mass spectra, including accurate mass measurements, were recorded by the High Resolution Mass Spectrometry Facility at the University of California, Riverside. Elemental analyses were obtained from Atlantic Micro Lab Microanalysis Service (Atlanta, Georgia, USA). X-ray diffraction data were collected at 90.0(2) K using MoKα x-rays on a Nonius KappaCCD as described previously.33 Melting points were determined using a MelTemp apparatus, and are uncorrected. All reactions were monitored by thin layer chromatography, followed by visualization with UV and anisaldehyde-H2SO4. 1,2,3,4- Tetra-O-acetyl-β-D-xylopyranose (2),34 2,3,4-tri-O-acetyl-α-D-xylopyranosyl bromide (3),35 1,2,3,4-tetra-O-benzoyl-α-D-xylopyranose (6),36 and 2,3,4-tri-O-benzoyl-α-D-xylopyranosyl bromide (7)36 were prepared according to known literature procedures. Their 1H NMR, 13C NMR, and melting points matched literature values.
3.2 General method for the glycosylation of 335
1-Octanol (2 mol equiv) in dichloromethane (1 mL per 0.1 g of 3) was stirred with the respective reagents (see Table 1) for 30 minutes. 2,3,4-Tri-O-acetyl-α-D-xylopyranosyl bromide (3) (2 - 10 mmol) was then added and the reaction mixture was stirred at room temperature. The reaction mixture was filtered through Celite and washed with saturated NaHCO3 (3 × 50 mL) and NaCl solution (3 × 50 mL). The only exception was the reaction with I2/DDQ/MS (entry 4), which was extracted first with 2 % (w/v) Na2S2O4 solution to remove excess I2. The crude product was purified by column chromatography on silica gel using hexane:ethyl acetate (6:1, v/v, then 3:1, v/v) as eluent to yield 4 as a white solid: mp 43–45 °C, lit.9a 52–53 °C (ethanol-water); 1H NMR (CDCl3, 300 MHz): δ 0.85 (t, 3H, J = 6.7 Hz, H-8′), 1.24 (br. s, 10H, 5 × CH2), 1.51–1.59 (m, 2H, H-2′), 2.01–2.03 (3 × s, 9H, 3 × CH3CO), 3.33 (dd, 1H, J5a,5e = 11.7 Hz, J4,5a = 7.5 Hz, H-5a), 3.42 (dt, 1H, J1′a,1′b = 9.6 Hz, J1′a,2′ = 6.6 Hz, H-1′a), 3.78 (dt, 1H, J1′a,1′b = 9.5 Hz, J1′a,2′ = 6.4 Hz, H-1′b), 4.09 (dd, 1H, J5a,5e = 11.7 Hz, J5e,4 = 5.1 Hz, H-5e), 4.43 (d, 1H, J1,2 = 6.9 Hz, H-1), 4.88 (dd, 1H, J1,2 = 6.8 Hz, J2,3=8.7 Hz, H-2), 4.90–4.96 (m, 1H, H-4), 5.13 (pseudo t, 1H, J2,3 = J3,4 = 8.6 Hz, H-3); 13C NMR (CDCl3, 75 MHz): δ 14.3, 21.0, 22.9, 26.1 (C-3′), 29.5–29.7, 32.0, 62.2, 69.2, 70.0, 70.1, 71.7, 100.9, 169.7, 170.1, 170.4; EIMS m/z (relative abundance %): 170 (58), 157 (42), 139 (12), 128 (100), 115 (32), 97 (24), 86 (20), 69 (44), 57 (13).
In addition, crude 3,4-di-O-acetyl-1,2-O-(1-octyloxyethylidene)-α-D-xylopyranose (5) was obtained as a colorless oil: 1H NMR (CDCl3, 400 MHz): δ 0.85 (t, 3H, J = 7.1, Hz, H-8′), 1.26–1.28 (m, 13H, 5 × CH2 and CH3), 1.57–1.62 (m, 2H, H-2′), 2.00 (s, 3H, CH3CO), 2.07 (s, 3H, CH3CO), 3.39–3.57 (m, 5H, H-1′, H-2, H-5), 4.80 (d, 1H, J1,2 = 4 Hz, H-1), 4.88 (ddd, 1H, J4,5a = 10.6, J3,4 = 9.7, J4,5e = 5.8, H-4), 5.19 (pseudo t, 1H J2,3 = J3,4 = 9.7 Hz, H-3); 13C NMR (CDCl3, 75 MHz): δ 14.3, 20.9–21.2 (2 × CH3CO and CH3), 22.8, 26.3, 29.4–29.6, 32.0, 58.8, 68.9, 69.1, 71.2, 73.2, 98.4, 103.2, 170.2, 171.3; mass spectrum m/z (relative abundance %): 217 (4), 157 (9), 128 (68), 115 (24), 97 (39), 86 (32), 69 (100), 57 (36).
3.3 General procedure for the synthesis of alkyl xylosides 8
A solution of the respective alkyl alcohol (12 mmol, 2.0 equiv.) in dichloromethane (30 mL, 1 mL per 0.1 g of 3) was stirred with I2 (1.45 g, 6 mmol), DDQ (0.65 g, 3.0 mmol) and powdered 4 Å molecular sieve for 30 minutes at room temperature. 2,3,4-Tri-O-benzoyl-α-D-xylopyranosyl bromide (7) (3.0 g, 6.0 mmol) was added and the reaction mixture was stirred at room temperature for 3 hours. The reaction mixture was filtered through Celite, washed with an aqueous solution of Na2S2O4 (2 %, w/v), saturated NaHCO3 (3 × 30 mL) and saturated NaCl solution (2 × 30 mL). The crude product was purified by column chromatography on silica gel to yield 8.
3.3.1 Hexyl 2,3,4-tri-O-benzoyl-β-D-xylopyranoside (8a)
Prepared from 1-hexanol (1.16 g, 11.4 mmol, 2.0 equiv.) and 7 (3.0 g, 5.7 mmol). Purified by flash column chromatography as described above (hexane-ethyl acetate, 6:1, v/v, then 3:1, v/v) to afford 8a (2.75 g, 88%): Colorless oil; Rf 0.61 (hexane-ethyl acetate, 3:1, v/v); 1H NMR (CDCl3, 400 MHz) δ 0.78 (t, 3H, J = 7.0 Hz, CH3), 1.12–1.24 (m, 4H, 2 × CH2), 1.24–1.32 (m, 2H), 1.52–1.63 (m, 2H), 3.53 (dt, 1H, J1′a,1′b = 9.6 Hz, J1′a,2′ = 6.8 Hz, H-1′a), 3.69 (dd, 1H, J5a,5e = 12.0 Hz, J4,5a = 7.0 Hz, H-5a), 3.88 (dt, 1H, J1′a,1′b = 9.6 Hz, J1′b,2′ = 6.8 Hz, H-1′b), 4.42 (dd, 1H, J5a,5e = 12.0 Hz, J4,5e = 4.4 Hz, H-5e), 4.81 (d, 1H, J1,2 = 5.4 Hz, H-1), 5.29 (pseudo td, 1H, J3,4 = J4,5a = 7.0 Hz, J4,5e = 4.4 Hz, H-4), 5.37 (dd, 1H, J2,3 = 7.0 Hz, J1,2 = 5.4 Hz, H-2), 5.76 (pseudo t, 1H, J2,3 = J3,4 = 7.0 Hz, H-3), 7.30–7.40 (m, 6H), 7.44–7.56 (m, 3H), 7.96–8.00 (m, 6H); 13C NMR (CDCl3, 100 MHz): δ 14.2, 22.7, 25.9, 29.7, 30.1, 31.7, 61.4, 69.4, 69.8, 70.5, 100.3, 128.51, 128.54, 128.6, 129.4, 129.5, 129.6, 130.0, 130.07, 130.1, 133.4, 133.5, 133.54, 165.4, 165.6, 165.8; HRESIMS m/z: calcd for C32H34O8Na [M+Na]+: 569.2146; found: 569.2134.
3.3.2 Octyl 2,3,4-tri-O-benzoyl-β-D-xylopyranoside (8b)
Prepared from 1-octanol (0.50 g, 3.8 mmol, 2.0 equiv.) and 7 (1.0 g, 1.9 mmol). Purified by flash column chromatography (hexane-ethyl acetate, 6:1, v/v, then 3:1, v/v) to yield 8b as a colorless oil (0.82 g, 75 %) that was used directly in the next reaction. Recrystallization from ethanol gave a white solid (28 %); Rf 0.61 (hexane-ethyl acetate, 4:1, v/v); mp 47–48°C; 1H NMR (CDCl3, 300 MHz) δ 0.84 (t, 3H, J = 6.9 Hz, CH3), 1.14–1.27 (m, 10H, 5 × CH2), 1.51–1.62 (m, 2H), 3.52 (dt, 1H, J1′a,1′b = 9.6 Hz, J1′a,2′ = 6.5 Hz, H-1′a), 3.72 (dd, 1H, J5a,5e = 12.1 Hz, J4,5a = 7.0 Hz, H-5a), 3.88 (dt, 1H, J1′a,1′b = 9.6 Hz, J1′b,2′ = 6.5 Hz, H-1′b), 4.44 (dd, 1H, J5a,5e = 12.1 Hz, J4,5e = 4.3 Hz, H-5e), 4.84 (d, 1H, J1,2 = 5.4 Hz, H-1), 5.32 (pseudo td, 1H, J3,4 = J4,5a = 7.0 Hz, J4,5e = 4.3 Hz, H-4), 5.40 (dd, 1H, J2,3 = 7.0 Hz, J1,2 = 5.4 Hz, H-2), 5.79 (pseudo t, 1H, J2,3 = J3,4 = 7.0 Hz, H-3), 7.28–7.35 (m, 6H), 7.43–7.51 (m, 3H), 7.98–8.01 (m, 6H); 13C NMR (CDCl3, 75 MHz): δ 14.1, 22.7, 26.1, 29.2, 29.4, 29.7, 31.8, 61.3, 69.4, 69.6, 70.5, 100.2, 128.38, 128.41, 128.5, 129.36, 129.4, 129.6, 129.91, 129.94, 129.96, 133.2, 133.3, 133.4, 165.2, 165.5, 165.6; HRESIMS m/z: calcd for C34H38O8Na [M+Na]+: 597.2459; found: 597.2450.
3.3.3 Decyl 2,3,4-tri-O-benzoyl-β-D-xylopyranoside (8c)
Prepared from 1-decanol (1.90 g, 12.0 mmol, 2.0 equiv.) and 7 (3.0 g, 5.7 mmol). Purified by flash column chromatography as described above (hexane-ethyl acetate, 9:1, v/v, then 6:1, v/v) to afford 8c (3.43 g, 99 %): White solid; Rf 0.62 (hexane-ethyl acetate, 3:1, v/v); mp 59–60°C; 1H NMR (CDCl3, 400 MHz) δ 0.88 (t, 3H, J = 7.0 Hz, CH3), 1.10–1.36 (m, 14H, 7 × CH2), 1.52–1.59 (m, 2H), 3.51 (dt, 1H, J1′a,1′b = 9.5 Hz, J1′a,2′ = 6.5 Hz, H-1′a), 3.71 (dd, 1H, J5a,5e = 12.1 Hz, J4,5a = 7.0 Hz, H-5a), 3.88 (dt, 1H, J1′a,1′b = 9.5 Hz, J1′b,2′ = 6.4 Hz, H-1′b), 4.42 (dd, 1H, J5a,5e = 12.1 Hz, J4,5e = 4.4 Hz, H-5e), 4.82 (d, 1H, J1,2 = 5.6 Hz, H-1), 5.29 (pseudo td, 1H, J3,4 = J4,5a = 7.0 Hz, J4,5e = 4.4 Hz, H-4), 5.37 (dd, 1H, J2,3 = 7.0 Hz, J1,2 = 5.6 Hz, H-2), 5.76 (pseudo t, 1H, J2,3 = J3,4 = 7.0 Hz, H-3), 7.31–7.39 (m, 6H), 7.46–7.54 (m, 3H), 7.96–8.00 (m, 6H); 13C NMR (CDCl3, 100 MHz): δ 14.3, 22.9, 26.2, 29.5, 29.6, 29.7, 29.72, 29.75, 31.1, 32.1, 61.4, 69.4, 69.8, 70.5, 100.3, 128.51, 128.54, 128.6, 129.4, 129.5, 129.6, 130.0, 130.08, 130.09, 133.4, 133.48, 133.54, 165.4, 165.6, 165.8; HRESIMS m/z: calcd for C36H42O8Na [M+Na]+: 625.2772; found: 625.2756.
3.3.4 Dodecyl 2,3,4-tri-O-benzoyl-β-D-xylopyranoside (8d)
Prepared from 1-dodecanol (2.23 g, 12.0 mmol, 2.0 equiv.) and 7 (3.0 g, 5.7 mmol). Purified by flash column chromatography as described above (hexane-ethyl acetate, 9:1, v/v, then 6:1, v/v) to afford 8d (3.14 g, 87 %): White solid; Rf 0.63 (hexane-ethyl acetate, 3:1, v/v); mp 64–66°C; 1H NMR (CDCl3, 400 MHz) 0.86 (t, 3H, J = 6.4 Hz, CH3), 1.23–1.28 (m, 18H, 9 × CH2), 1.54–1.59 (m, 2H), 3.50 (dt, 1H, J1′a,1′b = 9.5 Hz, J1′a,2′ = 6.5 Hz, H-1′a), 3.69 (dd, 1H, J5a,5e = 12.1 Hz, J4,5a = 7.0 Hz, H-5a), 3.86 (dt, 1H, J1′a,1′b = 9.5 Hz, J1′b,2′ = 6.3 Hz, H-1′b), 4.41 (dd, 1H, J5a,5e = 12.1 Hz, J4,5e = 4.4 Hz, H-5e), 4.80 (d, 1H, J1,2 = 5.5 Hz, H-1), 5.28 (pseudo td, 1H, J3,4 = J4,5a = 7.0 Hz, J4,5e = 4.4 Hz, H-4), 5.35 (dd, 1H, J2,3 = 7.0 Hz, J1,2 = 5.5 Hz, H-2), 5.74 (pseudo t, 1H, J2,3 = J3,4 = 7.0 Hz, H-3), 7.31–7.38 (m, 6H), 7.46–7.63 (m, 3H), 7.95–7.99 (m, 6H); 13C NMR (CDCl3, 100 MHz): δ 14.3, 22.9, 26.2, 29.5, 29.6, 29.7, 29.75, 29.8, 29.81, 29.84, 31.1, 32.1, 61.4, 69.4, 69.8, 70.5, 100.3, 128.51, 128.54, 128.6, 129.4, 129.5, 129.6, 130.0, 130.1, 133.4, 133.48, 133.53, 165.4, 165.6, 165.8; HRESIMS m/z: calcd for C38H46O8Na [M+Na]+: 653.3085; found: 653.3066.
3.3.5 Tetradecyl 2,3,4-tri-O-benzoyl-β-D-xylopyranoside (8e)
Prepared from 1-tetradecanol (2.57 g, 12.0 mmol, 2.0 equiv.) and 7 (3.0 g, 5.7 mmol). Purified by flash column chromatography as described above (hexane-ethyl acetate, 9:1, v/v, then 6:1, v/v) to afford 8e (3.57 g, 95 %): White solid; Rf 0.66 (hexane-ethyl acetate, 3:1, v/v); mp 67–68°C; 1H NMR (CDCl3, 400 MHz) 0.86 (t, 3H, J = 6.9 Hz, CH3), 1.12–1.24 (m, 22H, 11 × CH2), 1.52–1.58 (m, 2H), 3.50 (dt, 1H, J1′a,1′b = 9.5 Hz, J1′a,2′ = 6.5 Hz, H-1′a), 3.68 (dd, 1H, J5a,5e = 12.1 Hz, J4,5a = 7.0 Hz, H-5a), 3.86 (dt, 1H, J1′a,1′b = 9.5 Hz, J1′b,2′ = 6.4 Hz, H-1′b), 4.41 (dd, 1H, J5a,5e = 12.1 Hz, J4,5e = 4.3 Hz, H-5e), 4.79 (d, 1H, J1,2 = 5.4 Hz, H-1), 5.28 (pseudo td, 1H, J 3,4 = J4,5a = 7.0 Hz, J4,5e = 4.3 Hz, H-4), 5.35 (dd, 1H, J2,3 = 7.0 Hz, J1,2 = 5.4 Hz, H-2), 5.74 (pseudo t, 1H, J2,3 = J3,4 = 7.0 Hz, H-3), 7.31–7.37 (m, 6H), 7.46–7.64 (m, 3H), 7.95–8.00 (m, 6H); 13C NMR (CDCl3, 100 MHz): δ 14.3, 22.9, 26.2, 29.5–29.9, 32.1, 61.4, 69.4, 69.8, 70.6, 100.4, 128.53, 128.56, 128.63, 129.4, 129.5, 129.7, 130.07, 130.10, 130.11, 133.4, 133.5, 133.6, 165.4, 165.6, 165.8; HRESIMS m/z: calcd for C40H50O8Na [M+Na]+: 681.3398; found: 681.3380.
3.3.6 Hexadecyl 2,3,4-tri-O-benzoyl-β-D-xylopyranoside (8f)
Prepared from 1-hexadecanol (2.77 g, 12.0 mmol, 2.0 equiv.) and 7 (3.0 g, 5.7 mmol). Purified by flash column chromatography as described above (hexane-ethyl acetate, 9:1, v/v, then 6:1, v/v) to afford 8f (3.54 g, 90 %): White solid; Rf 0.63 (hexane-ethyl acetate, 5:1, v/v); mp 67–69°C; 1H NMR (CDCl3, 400 MHz) 0.86 (t, 3H, J = 6.8 Hz, CH3), 1.12–1.24 (m, 26H, 13 × CH2), 1.52–1.57 (m, 2H), 3.50 (dt, 1H, J1′a,1′b = 9.6 Hz, J1 a,2 = 6.5 Hz, H-1′a), 3.68 (dd, 1H, J5a,5e = 12.1 Hz, J4,5a = 7.0 Hz, H-5a), 3.86 (dt, 1H, J1′a,1′b = 9.6 Hz, J1′b,2′ = 6.4 Hz, H-1′b), 4.41 (dd, 1H, J5a,5e = 12.1 Hz, J4,5e = 4.3 Hz, H-5e), 4.79 (d, 1H, J1,2 = 5.4 Hz, H-1), 5.28 (pseudo td, 1H, J3,4 = J4,5a = 7.0 Hz, J4,5e = 4.3 Hz, H-4), 5.35 (dd, 1H, J2,3 = 7.0 Hz, J1,2 = 5.4 Hz, H-2), 5.73 (pseudo t, 1H, J2,3 = J3,4 = 7.0 Hz, H-3), 7.31–7.37 (m, 6H), 7.48–7.54 (m, 3H), 7.95–7.99 (m, 6H); 13C NMR (CDCl3, 100 MHz): δ 14.6, 23.1, 26.5, 29.5–30.0, 32.2, 61.4, 69.4, 69.8, 70.6, 100.4, 128.54, 128.57, 128.6, 129.4, 129.5, 129.7, 130.1, 130.11, 130.12, 133.4, 133.5, 133.6, 165.4, 165.6, 165.8; HRESIMS m/z: calcd for C42H54O8Na [M+Na]+: 709.3711; found: 709.3692.
3.3.7 3,3,4,4,5,5,6,6,7,7,8,8,8-Tridecafluorooctyl 2,3,4-tri-O-benzoyl-β-D-xylopyranoside (8g)
Prepared from 1H,1H,2H,2H-perfluorooctanol (2.1 g, 5.7 mmol, 1.5 equiv.) and 7 (2.0 g, 3.8 mmol) to afford 8g (0.37 g, 9 %) after flash column chromatography followed by recrystalization from ethanol: White solid; Rf 0.61 (hexane-ethyl acetate, 3:1, v/v); mp 119–120°C; 1H NMR (CDCl3, 400 MHz) δ 2.42 (tt, 2H, J2′, 3′ = 18.8 Hz, J1′,2′ = 6.5 Hz, H-2′), 3.74 (dd, 1H, J5a,5e = 12.2 Hz, J4,5a = 6.7 Hz, H-5a), 3.83 (dt, 1H, J1′a,1′b = 10.3 Hz, J1′a,2′ = 6.7 Hz, H-1′a), 4.16 (dt, 1H, J1′a,1′b = 10.3, J1′b,2′ = 6.5 Hz, H-1′b), 4.43 (dd, 1H, J5a,5e = 12.2 Hz, J4,5e = 4.2 Hz, H-5e), 4.85 (d, 1H, J1,2 = 5.21 Hz, H-1), 5.28 (pseudo td, 1H, J3,4 = J4,5a = 7.0, J4,5e = 4.2 Hz, H-4), 5.35 (dd, 1H, J2,3 = 7.0 Hz, J1,2 = 5.2 Hz, H-2), 5.73 (pseudo t, 1H, J2,3 = J3,4 = 7.0 Hz, H-3), 7.30–7.39 (m, 6H), 7.47–7.55 (m, 3H), 7.94–8.01 (m, 6H); 13C NMR (CDCl3, 100 MHz): δ 31.5, 61.1, 68.9, 69.87, 69.92, 100.1, 128.30, 128.34, 128.4, 129.1, 129.17, 129.21, 129.83, 129.88, 129.9, 133.3, 133.38, 133.41, 165.2, 165.3, 165.5; 19F NMR (282 MHz, CDCl3): δ −81.3 (CF3), −113.9 (CF2), −122.5 (CF2), −123.4 (CF2), −124.2 (CF2), −126.7 (CF2); HRESIMS m/z: calcd for C34H25O8F13Na [M+Na]+: 831.1234; found: 831.1241.
3.4 General procedure for the synthesis of 9a–g2k, 37
A solution of sodium methoxide (0.32 g, 6.0 mmol, 3.0 equiv.) in methanol (20 mL) was added drop wise to a solution of 4 or 8a–g (2 mmol) in methanol or dichloromethane (10 mL). The mixture was stirred at ambient temperature for 30 min followed by neutralization with Dowex 50W×8–100 ion exchange resin. The ion exchange resin was filtered off and the solvent was removed under reduced pressure. The crude product was dissolved in acetone. The residue was filtered and the crude product was purified by column chromatography and/or recrystallized to give 9a–g.
3.4.1 Hexyl β-D-xylopyranoside (9a)
Prepared from 8a (2.3 g, 4.2 mmol) to afford 9a (0.75 g, 76 %). The crude product was purified by recrystallization from acetone to yield white, flaky crystals (0.45 g, 46 %; 72% from 6 without purification of intermediates): Rf 0.53 (CH2Cl2-MeOH, 8:1, v/v); mp 84–85°C, lit.37 90–91 °C (acetone); 1H NMR (d4-MeOH, 400 MHz): δ 0.86 (t, 3H, J = 6.9, CH3), 1.24–1.37 (m, 6H, 3 × CH2), 1.52–1.59 (m, 2H, H-2′), 3.08–3.16 (m, 2H, H-2 and H-5a), 3.22–3.27 (m, 1H, H-3), 3.39–3.50 (m, 2H, H-1′a and H-4), 3.72–3.81 (m, 2H, H-1′b and H-5e), 4.13 (d, 1H, J1,2 = 7.5, H-1); 13C NMR (d4-MeOH, 100 MHz) δ 14.5, 23.8, 26.9, 30.9, 32.9, 67.0, 71.0, 71.4, 75.0, 78.0, 105.2; HRESIMS m/z: calcd for C11H26NO5 [M+NH4]+: 252.1806; found: 252.1801.
3.4.2 Octyl β-D-xylopyranoside (9b)2k
Synthesized from 8b (1.16 g, 2.02 mmol) and purified by column chromatography on silica gel (eluent: dichloromethane-methanol, 8/1, v/v) yielded 9b (0.44 g, 56 %; 54% from 6 without purification of intermediates). Recrystallization from acetone and acetone-hexane gave a white solid (0.24 g, 31 %): Rf 0.71 (6:1 CH2Cl2-MeOH); mp 90–91°C, lit.9a 91–92 °C; 1H NMR (d4-MeOH, 300 MHz): δ 0.87 (t, 3H, J = 6.9, CH3), 1.27 (br s, 10H, 5 × CH2), 1.53–1.60 (m, 2H, H-2′), 3.09–3.18 (m, 2H, H-2 and H-5a), 3.26–3.28 (m, 1H, H-3), 3.40–3.53 (m, 2H, H-1′a and H-4), 3.73–3.84 (m, 2H, H-1′b and H-5e), 4.19 (d, 1H, J1,2 = 7.5, H-1); 13C NMR (d4-MeOH, 75 MHz): δ 14.6, 23.9, 27.3, 30.4–31.0, 33.2, 67.1, 71.1, 71.4, 75.1, 78.0, 105.3; HRESIMS m/z: calcd for C13H30NO5 [M+NH4]+: 280.2119; found: 280.2122. Anal. Calcd for C13H26O5: C, 59.52; H, 9.99. Found: C, 59.72; H, 10.00.
3.4.3 Decyl β-D-xylopyranoside (9c)
Prepared from 8c (3.0 g, 5.0 mmol) to afford 9c (1.12 g, 78 %). The crude product was purified by recrystallization from acetone to yield white, flaky crystals (0.83 g, 58 %; 64% from 6 without purification of intermediates): Rf 0.50 (CH2Cl2-MeOH, 8:1, v/v); mp 94–95°C, lit.37 98.5–99.5 °C (acetone/ethanol); 1H NMR (d4-MeOH, 400 MHz): δ 0.84 (t, 3H, J = 6.6, CH3), 1.24–1.31 (m, 14 H, 7 × CH2), 1.51–1.58 (m, 2H, H-2′), 3.07–3.14 (m, 2H, H-2 and H-5a), 3.21–3.27 (m, 1H, H-3), 3.39–3.49 (m, 2H, H-1′a and H-4), 3.71–3.81 (m, 2H, H-1′b and H-5e), 4.12 (d, 1H, J1,2 = 7.5, H-1); 13C NMR (d4-MeOH, 100 MHz) δ 14.6, 23.9, 27.2, 30.6, 30.7, 30.83, 30.86, 30.9, 33.2, 67.0, 71.0, 71.3, 75.0, 78.0, 105.2; HRESIMS m/z: calcd for C15H34NO5 [M+NH4]+: 308.2432; found: 308.2431.
3.4.4 Dodecyl β-D-xylopyranoside (9d)
Prepared from 8d (2.64 g, 4.19 mmol) to afford 9d (1.11 g, 84 %). The crude product was purified by recrystallization from acetone to yield a white flaky solid (0.61 g, 47 %; 61% from 6 without purification of intermediates): Rf 0.47 (CH2Cl2-MeOH, 6:1, v/v); mp 96–97°C, lit.37 101.5–102 °C (methanol); 1H NMR (d4-MeOH, 400 MHz): δ 0.84 (t, 3H, J = 7.0, CH3), 1.23–1.32 (m, 18 H, 9 × CH2), 1.47–1.63 (m, 2H, H-2′), 3.07–3.16 (m, 2H, H-2 and H-5a), 3.19–3.30 (m, 1H, H-3), 3.37–3.49 (m, 2H, H-1′a and H-4), 3.71–3.81 (m, 2H, H-1′b and H-5e), 4.12 (d, 1H, J1,2 = 7.2, H-1); 13C NMR (d4-MeOH, 100 MHz) δ 14.6, 23. 9, 27.2, 30.6, 30.7, 30.8–31.0, 33.2, 67.1, 71.0, 71.4, 75.1, 78.0, 105.2; HRESIMS m/z: calcd for C17H38NO5 [M+NH4]+: 336.2745; found: 336.2743.
3.4.5 Tetradecyl β-D-xylopyranoside (9e)
Prepared from 8e (3.30 g, 5.01 mmol) to afford 9e (1.58 g, 91 %). The crude product was purified by recrystallization from ethanol to yield a flaky, white solid (0.77 g, 45 %; 72% from 6 without purification of intermediates): Rf 0.50 (CH2Cl2-MeOH, 9:1, v/v); mp 100–101°C, lit.37 103–104 °C (ethanol); 1H NMR (d4-MeOH, 400 MHz): δ 0.85 (t, 3H, J = 6.7, CH3), 1.23–1.31 (m, 22 H, 11 × CH2), 1.51–1.59 (m, 2H, H-2′), 3.07–3.15 (m, 2H, H-2 and H-5a), 3.21–3.26 (m, 1H, H-3), 3.39–3.49 (m, 2H, H-1′a and H-4), 3.72–3.81 (m, 2H, H-1′b and H-5e), 4.12 (d, 1H, J1,2 = 7.2, H-1); 13C NMR (d4-MeOH, 100 MHz) δ 14.6, 23.9, 27.2, 30.6, 30.7, 30.8–31.0, 33.2, 67.1, 71.0, 71.4, 75.1, 78.0, 105.3; HRESIMS m/z: calcd for C19H38O5Na [M+Na]+: 369.2611; found: 369.2606.
3.4.6 Hexadecyl β-D-xylopyranoside (9f)
Prepared from 8f (3.28 g, 4.76 mmol) to afford 9f after recrystallization from acetone (1.36 g, 76 %). The product was further purified by recrystallization from methanol to yield a white, flaky solid (0.55 g, 31 %; 70% from 6 without purification of intermediates): Rf 0.39 (9:1 CH2Cl2-MeOH); mp 102–103°C, lit.37 105.5–107; 1H NMR (d4-MeOH, 400 MHz): δ 0.84 (t, 3H, J = 6.7, CH3), 1.23–1.31 (m, 26 H, 13 × CH2), 1.52–1.57 (m, 2H, H-2′), 3.08–3.15 (m, 2H, H-2 and H-5a), 3.22–3.26 (m, 1H, H-3), 3.39–3.49 (m, 2H, H-1′a and H-4), 3.72–3.81 (m, 2H, H-1′b and H-5e), 4.13 (d, 1H, J1,2 = 7.2, H-1); 13C NMR (d4-MeOH, 100 MHz) δ 14.6, 23.9, 27.2, 30.6, 30.7, 30.8, 30.8–31.0, 33.2, 67.1, 71.1, 71.4, 75.1, 78.0, 105.3; HRESIMS m/z: calcd for C21H46NO5 [M+NH4]+: 392.3371; found: 392.3369.
3.4.7 3,3,4,4,5,5,6,6,7,7,8,8,8-Tridecafluorooctyl β-D-xylopyranoside (9g)
Prepared as described in the general procedure from crude 8g (3.35 g, 4.14 mmol) to afford 9g after crystallization from acetone/hexane as a white solid (0.54 g, 27 % from 6 without purification of intermediates): Rf 0.49 (CH2Cl2-MeOH, 8:1, v/v); mp 103–104°C; 1H NMR (d4-MeOH,400 MHz): δ 2.44–2.56 (m, 2H, H-2′), 3.10–3.19 (m, 2H, H-2 and H-5a), 3.23–3.28 (m, 1H, H-3), 3.43 (ddd, 1H, J4,5a = 10.2 Hz, J3,4 = 8.7 Hz, J4,5e = 5.3 Hz, H-4), 3.78–3.84 (m, 2H, H-1′a and H-5e), 4.05 (dt, 1H, J1′a,1′b = 10.4 Hz, J1′b,2′ = 6.9 Hz, H-1′b), 4.19 (d, 1H, J1,2 = 7.5 Hz, H-1); 13C NMR (d4-MeOH, 100 MHz) δ 32.7, 62.7, 67.1, 71.3, 74.9, 77.9, 105.5; 19F NMR (282 MHz, CDCl3): δ −80.8 (CF3), −112.8 (CF2), −121.2 (CF2), −122.2 (CF2), −123.0 (CF2), −125.6 (CF2); HRESIMS m/z: calcd for C13H17NO5F13 [M+NH4]+: 514.0894; found: 514.0890.
3.4.8 3,3,4,4,5,5,6,6,7,7,8,8,8-Tridecafluorooctyl 2,3,4-tri-O-acetyl-β-D-xylopyranoside (10)2k, 9c, 12
1,2,3,4-Tetra-O-acetyl-β-D-xylopyranose (2) (662 mg, 2.08 mmol) and 3,3,4,4,5,5,6,6,7,7,8,8,8-tridecafluorooctanol (544 μL, 909 mg, 2.49 mmol, 1.2 eq.) were dissolved in CH2Cl2 (8 mL) at room temperature. The stirring solution was cooled in an ice-water bath. BF3·Et2O (385 μL, 443 mg, 3.12 mmol, 1.5 eq.) was added drop wise. The ice-bath was removed and the solution was allowed to warm to room temperature overnight. The reaction was quenched with saturated NaHCO3 (10 mL). After separation of the layers the aqueous phase was extracted twice with CH2Cl2 (15 mL). The combined extracts were dried over Na2SO4, filtered and concentrated. The crude residue was purified by silica gel flash column chromatography using hexanes-EtOAc as eluent (5:1, v/v) to yield 10 (434 mg, 34 %) as a white solid. Rf 0.75 (4:1 hexanes:EtOAc, v:v); mp 94–95 °C; 1 H NMR (CDCl3, 400 MHz): δ 2.04 (s, 3H, CH3CO), 2.04 (s, 3H, CH3CO), 2.06 (s, 3H, CH3CO), 2.42 (tt, 2H, J2′,3′ = 18.8 Hz, J1′,2′ = 6.5 Hz, H-2′), 3.40 (dd, 1H, J5a,5b = 11.8, J4,5a = 8.6 Hz, 5a), 3.79 (dt, 1H, J1′a,1′b = 10.4 Hz, J1′a,2′ = 6.7 Hz, H-1′a), 4.07–4.16 (m, 2H, H-1′b and H-5e), 4.52 (d, 1H, J1,2 = 6.6 Hz, H-1), 4.90–4.97 (m, 2H, H2 and H4), 5.16 (pseudo t, 1H, J2,3 = J3,4 = 8.3 Hz, H-3); 13C NMR (CDCl3, 100 MHz,): δ 20.5, 20.6, 20.7, 31.4, 61.3, 62.0, 68.7, 70.3, 71.0, 100.6, 169.4, 169.8, 170.0; 19F NMR (282 MHz, CDCl3): δ −81.3 (CF3), −113.9 (CF2), −122.4 (CF2), −123.3 (CF2), −124.1 (CF2), −126.6 (CF2); HRESIMS m/z: calcd for C19H19O8F13Na [M+Na]+: 645.0765; found: 645.0781; Anal. Calc for C19H19O8F13: C 37.67; H 3.08; found: C 36.75; H 2.96.
3.5 Differential Scanning Calorimetry
Alkyl β-D-xylopyranosides 9a–g were stored under vacuum to avoid moisture adsorption. Before starting the DSC experiments the TA Instruments Q200 instrument (TA Instruments, New Castle, DE) was calibrated with indium for temperature and enthalpy accuracy. Samples were prepared by weighing 1.0 to 1.5 mg of the dry surfactant into pre-weighed Tzero aluminum pans. Tzero hermetic lids were then pressed onto the pan. Empty Tzero pans with Tzero hermetic lid were used as the reference sample. The thermal analysis was performed between 20°C and 250°C with a ramp rate of 2.5°C/min. The phase transitions were analyzed using Universal Analysis software (TA Instruments).
3.6 Thermogravimetric Analysis
A Mettler Toledo TGA/SDTA851e instrument (Mettler-Toledo Inc., Columbus, OH) was used to perform thermogravimetric analysis (TGA) on the alkyl β-D-xylopyranosides. The instrument was calibrated with indium and aluminum before the experiments were started. Between 2.5 mg to 4.5 mg of each alkyl β-D-xylopyranosides 9a–f were added to the alumina crucible. TGA analysis was performed by heating the sample from 25 °C to 200 °C at a ramp rate of 10 °C/min under flowing nitrogen at 2 mL/min. Data were analyzed by STARe software (Mettler-Toledo Inc.).
3.7 Cell Culture Experiments
3.7.1 Stock solutions
One-hundred millimolar stock solutions were prepared from all compounds and the positive control, octylthioglucoside (OTG; Thermo Scientific, Rockford, IL), which were readily soluble in dimethyl sulfoxide (DMSO; Sigma-Aldrich, St. Louis, MO) at that concentration. To verify the solubility of the compounds at experimental conditions, 2 μL of each stock were added to 200 μL of cell culture medium, vortexed for 5 sec and centrifuged at 1,400 rpm for 1 min for visual inspection of eventual precipitation. While 9a, 9b and OTG remained soluble, 9c–g precipitated out of solution and was further diluted. Therefore, the following stock solutions were used for cytotoxicity analyses: 9a (100 mM), 9b (100 mM), 9c (50 mM), 9d (10 mM), 9e (10 mM), 9f (2.5 mM), 9g (50 mM), and OTG (100 mM).
3.7.2 Cell lines & cell culture
Three cell lines were used for the cytotoxicity assays: spontaneously immortalized human keratinocytes (HaCaT);38 normal non-transformed human foreskin fibroblasts (Hs27); and human acute leukemia T lymphocytes (Jurkat).39 The culture medium for HaCaT and Hs27 cells was Dulbecco’s Modified Eagle Medium (DMEM; HyClone, Logan, UT) with 10 % heat-inactivated newborn calf serum (NCS; HyClone), while that for Jurkat cells was Roswell Park Memorial Institute medium (RPMI; HyClone) with 10 % heat inactivated fetal bovine serum (FBS; HyClone). Both media were supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin, and 0.25 μg/mL amphotericin B (Lonza, Walkersville, MD). The incubation conditions of the cells were at 37 °C in humidified 5 % carbon dioxide (CO2) atmosphere, in a regular water jacketed incubator.
3.7.3 MTS assay
HaCaT and Hs27 cells were seeded in flat bottom MICROTEST™ tissue culture 96-well plates (Becton Dickinson Labware, Franklin Lakes, NJ) at a density of 5,000, while Jurkat cells were seeded at a density of 25,000 cells per well. Compounds were tested at a concentration range from 1 μM to 1000 μM. After 18 h of incubation, 20 μL of the MTS reagent (CellTiter 96 AQueousOne Solution Cell Proliferation Assay; Promega, Madison, WI) were added to each well of HaCaT and Hs27 cells, which were subsequently incubated for an additional 2 h. For Jurkat cells, the MTS reagent was added after 16 h of incubation with the test compounds and was then incubated for an additional 4 h in the presence of the MTS reagent. Jurkat cells were seeded at higher density and were incubated longer in the presence of the MTS reagent since lymphocytes appear to produce less formazan than other cell types40 and these adjustments were implemented to achieve significant absorbance changes. For all three cell lines the total period of incubation was 20 h. The colored formazan product was measured by absorbance at 490 nm with a reference wavelength of 650 nm using a microplate reader (VERSAmax tunable microplate reader, MDS, Inc., Toronto, Canada). Control wells containing the same volumes of culture medium and MTS reagent were used to subtract background absorbance. In addition, 300 μM hydrogen peroxide (H2O2; Acros Organics, NJ) was used as positive control. DMSO treated cells as solvent control and untreated cells were included in each plate. Data were expressed as percentage of cell viability relative to DMSO treated cells. A series of quadruplicates were processed for experimental concentrations that were used to obtain the mean and standard deviation (SD) values.
3.7.4 Generation of dose-response curves and determination of CC50
Data derived from the MTS assay was used to create dose-response curves and determine the 50 % cytotoxic concentration (CC50). CC50 was defined as a concentration of compound causing loss of membrane integrity of 50 % of the cell population as compared to solvent treated cells after 20 h of incubation. The CC50 values were obtained as previously described.41 Briefly, data was normalized by subtracting from each experimental value the average percentage of dead cells from six wells treated with 1 % DMSO (solvent control). The two cytotoxicity values closest to the 50 % value were plotted with its associated chemical compound concentration and the equation of the regression line was utilized to determine the CC50.
3.7.5 Apoptosis assay
A day prior to the assay, the viability of Jurkat cells was increased by using Ficoll-Paque™ PLUS density gradient centrifugation.42 Briefly, after centrifugation at 400 x g for 30 min at room temperature, live cells at the interface were collected and washed with cell culture medium and were then expanded by starting a new culture. On the next day, Jurkat cells were seeded in a flat bottom 96-well MICROTEST™ plates at a cell density of 25,000 cells per well in 200 μL RPMI medium supplemented with10 % FBS and antibiotics as described above. Alkyl β-D-xylopyranosides 9b, 9c, 9g and OTG were added to the cells at their respective CC50; as well as 2 μM Staurosporine (Sigma-Aldrich) and 0.5 %Tween 20 (Acros Organics) as positive controls for apoptosis and necrosis, respectively. In addition, DMSO and untreated controls were included. All treatments including controls were run in series of triplicates. After 20 h exposure to compounds, cells were processed as previously described with minor modifications.41 Briefly, cells were collected in an ice-cold tube, centrifuged at 1,400 rpm for 5 min at 4°C and stained by resuspension in 100 μL binding buffer (0.1 M HEPES, pH 7.4, 140 mM NaCl, and 2.55 mM CaCl2) containing 1 μl of 25 μg/mL Annexin V-FITC and 5 μL of 250 μg/mL propidium iodide (PI; Beckman Coulter, Miami, FL). After incubation for 15 min on ice in the dark, cell suspensions were added with 200 μl of ice-cold binding buffer, gently homogenized and immediately analyzed using a Cytomics FC 500 flow cytometer (Beckman Coulter, Miami, FL). For each sample, 3,000 individual events were collected and analyzed using CXP software (Beckman Coulter, Miami, FL). Prior to data acquisition, the flow cytometer was set up and calibrated utilizing unstained, single- (PI or Annexin V-FITC) and double- (PI and Annexin V-FITC) stained cells.
Supplementary Material
Figure 1.

Molecular structure of the hemihydrate of decyl β-D-xylopyranoside (9c). Compound 9c was crystallized from acetone-hexane. Displacement ellipsoids are drawn at the 50 % probability level.
Figure 3.

Comparison of the thermograms of the hydrocarbon octyl xyloside 9b and corresponding fluorocarbon octyl xyloside 9g.
Figure 4.

TGA thermogram of (a) the hydrocarbon octyl xyloside 9b and (b) the corresponding fluorocarbon octyl xyloside 9g.
Acknowledgments
This work was supported by the National Science Foundation (CBET-0967381/0967390), the U.S. Department of Agriculture Biomass Research and Development Initiative (Grant Agreement 68-3A75-7-608) and the Department of Energy Development and Independence, Energy and Environment Cabinet of The Commonwealth of Kentucky. The cell culture work was supported by grant 5G12RR008124 to the Border Biomedical Research Center (BBRC), granted to the University of Texas at El Paso from the National Center for Research Resources (NCRR) of the NIH.
Footnotes
Complete crystallographic data for the structural analysis of compound 9c have been deposited with the Cambridge Crystallographic Data Centre, CCDC no. 854184. Copies of this information may be obtained free of charge from the director, Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge, CB2 1EZ, UK. (fax: +44-1223-336033, deposit@ccdc.cam.ac.uk or via: www.ccdc.cam.ac.uk).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.(a) Hill K, LeHen-Ferrenbach C. Sugar-based surfactants for consumer products and technical applications. In: Ruiz CC, editor. Surfactant Sci Ser. Vol. 143. CRC Press; Boca Raton: 2009. pp. 1–20. [Google Scholar]; (b) Savic S, Tamburic S, Savic MM. Expert Opin Drug Delivery. 2010;7:353–369. doi: 10.1517/17425240903535833. [DOI] [PubMed] [Google Scholar]; (c) Rojas OJ, Stubenrauch C, Lucia LA, Habibi Y. Interfacial properties of sugar-based surfactants. AOCS Press; 2009. pp. 449–472. [Google Scholar]
- 2.(a) Söderlind E, Wollbratt M, von Corswant C. Int J Pharm. 2003;252:61–71. doi: 10.1016/s0378-5173(02)00599-9. [DOI] [PubMed] [Google Scholar]; (b) Söderlind E, Karlsson L. Eur J Pharm Biopharm. 2006;62:254–259. doi: 10.1016/j.ejpb.2005.08.012. [DOI] [PubMed] [Google Scholar]; (c) Isomaa B, Haegerstrand H, Paatero G, Engblom AC. Biochim Biophys Acta. 1986;860:510–524. doi: 10.1016/0005-2736(86)90548-1. [DOI] [PubMed] [Google Scholar]; (d) Isomaa B, Haegerstrand H, Paatero G. Biochim Biophys Acta. 1987;899:93–103. doi: 10.1016/0005-2736(87)90243-4. [DOI] [PubMed] [Google Scholar]; (e) Shinoyama H, Gama Y, Nakahara H, Ishigami Y, Yasui T. Bull Chem Soc Jpn. 1991;64:291–292. [Google Scholar]; (f) Milius A, Greiner J, Riess JG. New J Chem. 1991;15:337–344. [Google Scholar]; (g) Zarif L, Greiner J, Riess JG. J Fluorine Chem. 1989;44:73–85. [Google Scholar]; (h) Zarif L, Greiner J, Pace S, Riess JG. J Med Chem. 1990;33:1262–1269. doi: 10.1021/jm00166a028. [DOI] [PubMed] [Google Scholar]; (i) Kasuya MCZ, Cusi R, Ishihara O, Miyagawa A, Hashimoto K, Sato T, Hatanaka K. Biochem Biophys Res Commun. 2004;316:599–604. doi: 10.1016/j.bbrc.2004.02.094. [DOI] [PubMed] [Google Scholar]; (j) Kasuya MCZ, Ito A, Cusi R, Sato T, Hatanaka K. Chem Lett. 2005;34:856–857. [Google Scholar]; (k) Li X, Turánek J, Knötigová P, Kudlácková H, Mašek J, Pennington DB, Rankin SE, Knutson BL, Lehmler HJ. New J Chem. 2008;32:2169–2179. [Google Scholar]
- 3.Sun R, Lawther JM, Banks WB. Industrial Crops and Products. 1995;4:127–145. [Google Scholar]
- 4.Damez C, Bouquillon S, Harakat D, Henin F, Muzart J, Pezron I, Komunjer L. Carbohydr Res. 2007;342:154–162. doi: 10.1016/j.carres.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 5.Bouxin F, Marinkovic S, Le BJ, Estrine B. Carbohydr Res. 2010;345:2469–2473. doi: 10.1016/j.carres.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 6.Matsumura S, Sakiyama K, Toshima K. Biotechnol Lett. 1999;21:17–22. [Google Scholar]
- 7.Shinoyama H, Kamiyama Y, Yasui T. Agric Biol Chem. 1988;52:2197–2202. [Google Scholar]
- 8.Nakamura T, Toshima K, Matsumura S. Biotechnol Lett. 2000;22:1183–1189. [Google Scholar]
- 9.(a) de Bruyne CK, Loontiens FG. Nature. 1966;209:396–397. [Google Scholar]; (b) Satge C, Le Bras J, Henin F, Muzart J. Tetrahedron. 2005;61:8405–8409. [Google Scholar]; (c) Petrovic ZD, Andjelkovic D, Spasojevic A. Indian J Chem. 2006;45B:272–275. [Google Scholar]
- 10.Kartha KPR, Aloui M, Field RA. Tetrahedron Lett. 1996;37:8807–8810. [Google Scholar]
- 11.Hanessian S, Banoub J. Carbohydr Res. 1977;53:C13–C16. [Google Scholar]
- 12.(a) Li X, Turanek J, Knoetigova P, Kudlackova H, Masek J, Parkin S, Rankin SE, Knutson BL, Lehmler HJ. Colloids Surf, B. 2009;73:65–74. doi: 10.1016/j.colsurfb.2009.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rowan AS, Nicely NI, Cochrane N, Wlassoff WA, Claiborne A, Hamilton CJ. Org Biomol Chem. 2009;7:4029–4036. doi: 10.1039/b909729e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Singh MK, Swain D, Guru RTN, Jayaraman N. Carbohydr Res. 2009;344:1993–1998. doi: 10.1016/j.carres.2009.06.024. [DOI] [PubMed] [Google Scholar]
- 14.Hoffmann B, Milius W, Voss G, Wunschel M, van Smaalen S, Diele S, Platz G. Carbohydr Res. 2000;323:192–201. doi: 10.1016/s0008-6215(99)00264-5. [DOI] [PubMed] [Google Scholar]
- 15.Brown CJ, Cox G, Llewellyn FJ. J Chem Soc A. 1966:922–927. [Google Scholar]
- 16.Ishigami Y, Goto M, Takizawa Y, Shinoyama H. J Oleo Sci. 2001;50:555–560. [Google Scholar]
- 17.Lehmler HJ, Parkin S, Brock CP. Acta Cryst B. 2004;60:325–332. doi: 10.1107/S0108768104005609. [DOI] [PubMed] [Google Scholar]
- 18.Goodby JW, Goertz V, Cowling SJ, Mackenzie G, Martin P, Plusquellec D, Benvegnu T, Boullanger P, Lafont D, Queneau Y, Chambert S, Fitremann J. Chem Soc Rev. 2007;36:1971–2032. doi: 10.1039/b708458g. [DOI] [PubMed] [Google Scholar]
- 19.Singh MK, Jayaraman N. J Indian Inst Sci. 2009;89:113–135. [Google Scholar]
- 20.van Doren HA, van der Heijden AM, de Goede ATJW, van Rantwijk F, van Bekkum H. Liq Cryst. 2000;27:63–68. [Google Scholar]
- 21.(a) Goodby JW, Haley JA, Watson MJ, Mackenzie G, Kelly SM, Letellier P, Gode P, Goethals G, Ronco G, Harmouch B, Martin P, Villa P. Liq Cryst. 1997;22:497–508. [Google Scholar]; (b) Harmouch B, Gode P, Goethals G, Goodby J, Haley J, Kelly S, Letellier P, Mackenzie G, Martin P, Ronco G, Watson M, Villa P. J Carbohydr Chem. 1997;16:479–497. [Google Scholar]; (c) Goodby JW, Haley JA, Watson MJ, Mackenzie G, Kelly SM, Letellier P, Douillet O, Gode P, Goethals G, Ronco G, Villa P. Liq Cryst. 1997;22:367–378. [Google Scholar]
- 22.Noller CR, Rockwell WC. J Am Chem Soc. 1938;60:2076–2077. [Google Scholar]
- 23.Martin P, Gode P, Villa P, Goethals G. J Therm Anal Calorim. 2001;63:339–344. [Google Scholar]
- 24.Harmouch B, Gode P, Goethals G, Martin P, Ronco G, Villa P. Comun Jorn Com Esp Deterg. 1997;27:379–390. [Google Scholar]
- 25.Lehmler HJ, Oyewumi MO, Jay M, Bummer PM. J Fluorine Chem. 2001;107:141–146. [Google Scholar]
- 26.Kissa E. Fluorinated surfactants and repellents (Surfactant Science Series, Vol. 97) Vol. 97. Marcel Dekker; New York: 2001. p. 615. [Google Scholar]
- 27.Deyrieux AF, Wilson VG. Cytotechnology. 2007;54:77–83. doi: 10.1007/s10616-007-9076-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Balgavy P, Devinsky F. Adv Colloid Interf Sci. 1996;66:23–63. doi: 10.1016/0001-8686(96)00295-3. [DOI] [PubMed] [Google Scholar]
- 29.(a) Shaik N, Martinez A, Augustin I, Giovinazzo H, Varela-Ramirez A, Sanau M, Aguilera RJ, Contel M. Inorg Chem. 2009;48:1577–1587. doi: 10.1021/ic801925k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Martinez A, Rajapakse CSK, Sanchez-Delgado RA, Varela-Ramirez A, Lema C, Aguilera RJ. J Inorg Biochem. 2010;104:967–977. doi: 10.1016/j.jinorgbio.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pavillard V, Drbal AAA, Swaine DJ, Phillips RM, Double JA, Nicolaou A. Biochem Pharmacol. 2004;67:1587–1599. doi: 10.1016/j.bcp.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 31.(a) Mukerjee P. J Am Oil Chem Soc. 1982;59:573–578. [Google Scholar]; (b) Shinoda K, Hato M, Hayashi T. J Phys Chem. 1972;76:909–914. [Google Scholar]
- 32.Mollinedo F, Gajate C. Drug Resistance Updates. 2006;9:51–73. doi: 10.1016/j.drup.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 33.Lehmler HJ, Parkin S, Robertson LW. Chemosphere. 2002;46:485–488. doi: 10.1016/s0045-6535(01)00177-1. [DOI] [PubMed] [Google Scholar]
- 34.(a) Son JB, Hwang MH, Lee W, Lee DH. Org Lett. 2007;9:3897–3900. doi: 10.1021/ol7015115. [DOI] [PubMed] [Google Scholar]; (b) Camponovo J, Hadad C, Ruiz J, Cloutet E, Gatard S, Muzart J, Bouquillon S, Astruc D. J Org Chem. 2009;74:5071–5074. doi: 10.1021/jo900554b. [DOI] [PubMed] [Google Scholar]
- 35.Mitchell SA, Pratt MR, Hruby VJ, Polt R. J Org Chem. 2001;66:2327–2342. doi: 10.1021/jo005712m. [DOI] [PubMed] [Google Scholar]
- 36.Fletcher HG, Jr, Hudson CS. J Am Chem Soc. 1947;69:921–4. doi: 10.1021/ja01196a054. [DOI] [PubMed] [Google Scholar]
- 37.Hori R. Yakugaku Zasshi. 1958;78:523–526. [Google Scholar]
- 38.Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. J Cell Biol. 1988;106:761–771. doi: 10.1083/jcb.106.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schneider U, Schwenk HU, Bornkamm G. Int J Cancer. 1977;19:621–626. doi: 10.1002/ijc.2910190505. [DOI] [PubMed] [Google Scholar]
- 40.Chen CH, Campbell PA, Newman LS. Int Arch Allergy Appl Immunol. 1990;93:249–255. doi: 10.1159/000235309. [DOI] [PubMed] [Google Scholar]
- 41.Varela-Ramirez A, Costanzo M, Carrasco YP, Pannell KH, Aguilera RJ. Cell Biol Toxicol. 2011;27:159–168. doi: 10.1007/s10565-010-9178-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boyum A. Scand J Clin Lab Invest, Suppl. 1968;21:77–89. [PubMed] [Google Scholar]
- 43.Kasuya MC, Ikeda M, Hashimoto K, Sato T, Hatanaka K. J Carbohydr Chem. 2005;24:705–715. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.