

Abstract
Metastatic disease is the proximal cause of mortality for most cancers and remains a significant problem for the clinical management of neoplastic disease. Recent advances in global transcriptional analysis have enabled better prediction of individuals likely to progress to metastatic disease. However, minimal overlap between predictive signatures has precluded easy identification of key biological processes contributing to the prometastatic transcriptional state. To overcome this limitation, we have applied network analysis to two independent human breast cancer datasets and three different mouse populations developed for quantitative analysis of metastasis. Analysis of these datasets revealed that the gene membership of the networks is highly conserved within and between species, and that these networks predicted distant metastasis free survival. Furthermore these results suggest that susceptibility to metastatic disease is cell-autonomous in estrogen receptor-positive tumors and associated with the mitotic spindle checkpoint. In contrast, nontumor genetics and pathway activities-associated stromal biology are significant modifiers of the rate of metastatic spread of estrogen receptor-negative tumors. These results suggest that the application of network analysis across species may provide a robust method to identify key biological programs associated with human cancer progression.
Keywords: gene expression, mouse models
Recent advances in global transcriptome analysis has enabled better understanding of the different subtypes of breast cancer (1), as well as tumor prognosis and treatment (2). Gene signatures derived from these analyses have provided new opportunities for better tailoring of treatment options based on individual tumor biology. However, although these signatures are potentially important clinical tools, for the most part they do not provide novel insight regarding the underlying mechanisms. This lack of insight is in part because they were developed as prognostic classifiers, based on a minimum set of genes rather than to interrogate the mechanisms underlying tumor biology. The ability of these clinical classifiers to investigate molecular mechanism is further complicated by the minimal overlap between independent signatures derived from different studies. The lack of overlap is thought to be because of the fact that there are likely thousands of genes that correlate with tumor progression (3). Membership of the individual genes in each signature is therefore dictated by the transcriptional patterns derived from the specific patient populations. Subtle variations in those populations result in different gene sets meeting the statistical thresholds to be included in the final signature. Using conventional methods, it has been estimated that thousands of samples would be required to develop a robust, stable signature (4). Thus, although these signatures have important potential for clinical applications, comparisons of the signatures have not provided similar benefit for the elucidation of mechanisms of metastasis by identifying common molecular or cellular functions.
Recent advances in computational biology have provided new strategies to study biological processes as networks of coexpressed genes rather than collections of genes correlated to particular phenotypes (5). These methods have been successfully applied to animal models of neoplastic disease to identify both individual genes and cellular processes associated with cancer susceptibility (6). These results suggest that the rich public data available for human breast cancer might be used for similar analysis to identify critical biological processes and functions associated with breast cancer progression. Moreover, our laboratory has been generating similar datasets from modeling inherited metastatic susceptibility in murine systems. Identification of genes and cellular functions associated with metastatic disease in common between mouse and human networks would provide strong evidence of a causal role for these factors in tumor progression.
Here we describe the results of a cross-species network analysis of metastatic breast cancer. Using two publicly available human breast cancer gene-expression datasets that represent the natural progression of disease, as well as three experimental mouse populations used for identification of inherited metastasis susceptibility genes, we have identified two gene networks that are independent predictors of metastatic disease in a meta-analysis of 1,881 human tumors (7). Unlike previously described human prognostic signatures, the networks significantly overlap between the two human datasets. In addition, significant overlap was also observed for the networks generated from the mouse samples. Unexpectedly, these networks are specific for either estrogen receptor (ER+) or ER− breast cancer. Moreover, the results suggest that the network associated with metastatic progression in ER+ cancers is tumor-cell autonomous, but that of ER− represents a stromal component. Finally, by limiting the analysis to highly connected genes that are shared between overlapping mouse and human networks, the prognostic signatures were reduced to less than 10 genes each. These core signatures implicate the mitotic spindle checkpoint as a critical factor for metastatic progression in ER+ breast cancers, and suggests that inherent differences in immune response and stromal pathways modify the rate of metastatic disease progression in ER− patients.
Results
Network Analysis Identifies Multiple Coexpressed Networks Associated with Disease Progression.
Network analysis was performed on the GSE2034 (8) (n = 286) and GSE11121 (9) (n = 200) human breast cancer datasets. These datasets consist of lymph node-negative patients untreated with adjuvant therapy, representing the natural course of disease. In addition, three datasets from our mouse mammary tumor virus-polyoma middle T antigen (MMTV PyMT) transgenic mouse-based metastasis susceptibility screens were analyzed (10). The mouse datasets represent three different experimental cross-populations developed to map the inherited factors associated with metastatic mammary cancer (11). The tumors derived from these experiments are all induced by the expression of the PyMT antigen but have differing metastatic susceptibility because of the segregation of different genomic components from mice with low metastatic capacity. Network analysis was performed on microarray gene expression data of 56 samples from a PyMT × AKXD recombinant inbred cross (11, 12) and 68 samples from an NZB × PyMT backcross (11). In addition, transcriptome sequencing data from 30 samples of a MOLF × PyMT backcross were also included in the analysis. Normalized expression data were used to identify expression networks for each dataset individually using weighted gene coexpression network analysis based on topological overlap measure algorithms (13). Network structure was visualized using a minimum spanning tree and each network named based on the most highly connected gene (Fig. 1A). Fifteen to 20 networks of coexpressed genes were identified for each dataset (Table 1).
Fig. 1.

(A) Network diagram for the GSE2034 TPX2 network. Genes are indicated by circles. Lines connecting genes represent relationships between genes in the network based on correlations of gene expression. Red circles represent genes with more than one connection that were used in the GOBO Kaplan-Meier analysis to identify networks associated with DMFS. (B) GOBO Kaplan-Meier analysis result for the TPX2 network. The red curve represents patients with network expression higher than the median; gray is lower. The P value is Bonferroni-corrected for multiple testing.
Table 1.
Network Kaplan-Meier analysis summary
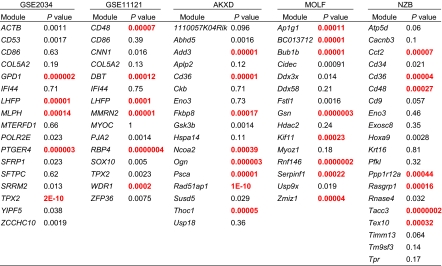 |
Modules were named after most connected gene in each network. Univariate P values based on GOBO analysis of genes with two or more connections in each module. P values less than 0.0006 were considered signficant after Bonferroni correction for multiple P values and are indicated in red.
To determine which of the networks were associated with disease progression, Kaplan-Meier analysis was performed using the gene expression-based outcome (GOBO) algorithm (7). Because the hub genes are expected to capture the majority of expression variation of each network, only genes with two or more connections were included in each network signature (Fig. 1A). Between five and eight networks per dataset were found to be significantly associated with distant metastasis-free survival (DMFS) after Bonferroni correction for multiple testing (Fig. 1B; Table 1, red text).
Transcriptional Networks Significantly Overlap Between Different Datasets.
We analyzed networks associated with DMFS that are replicated in multiple datasets, because these are more likely to represent core processes in the metastatic cascade. Because GSE2034 was the dataset with the largest number of samples, all subsequent overlap analysis was performed using this dataset as the reference. Two-hundred ninety significant overlaps were identified at a false-discovery rate < 0.05 after correction for multiple comparisons. To limit the analysis to the most significantly overlapping networks, these results were filtered for network pairs that shared at least 20 genes, and the shared genes constitute at least 25% of the members in one of the two networks. Ten of the 17 GSE2034 networks significantly overlap networks from other datasets (Table 2). Two of the GSED2034 networks were represented in all four of the other datasets (CD53 and TPX2) (Table 2). For each of these two networks, four of the five overlapping networks were significantly associated with DMFS (Table 1 and bold text in Table 2), consistent with the hypothesis that they represent critical processes in metastatic progression. All further analysis was therefore focused on the CD53 and TPX2 networks.
Table 2.
Module overlap analysis
|
GSE2034 |
Overlapping networks |
|||||
| Network | No. of genes | Data set | Network | No. of genes | No. of shared genes | Overlap P value |
| ACTB | 147 | GSE11121 | WDR1 | 144 | 55 | 8.39E-35 |
| ACTB | 147 | NZB | Cct2 | 1586 | 75 | 1.71E-03 |
| CD53 | 151 | GSE11121 | CD48 | 138 | 115 | 9.70E-168 |
| CD53 | 151 | MOLF | BC013712 | 92 | 50 | 2.14E-51 |
| CD53 | 151 | NZB | Cd48 | 69 | 29 | 1.01E-25 |
| CD53 | 151 | AKXD | Psca | 840 | 42 | 6.04E-06 |
| CD86 | 61 | AKXD | Ogn | 144 | 23 | 4.5E-45 |
| CD86 | 61 | GSE11121 | CD86 | 99 | 55 | 1.6E-44 |
| COL5A2 | 220 | GSE11121 | COL5A2 | 188 | 156 | 1.1E-193 |
| COL5A2 | 220 | MOLF | Fstl1 | 254 | 63 | 1.1E-35 |
| COL5A2 | 220 | AKXD | Ogn | 144 | 44 | 1.0E-21 |
| COL5A2 | 220 | MOLF | Serpinf1 | 82 | 29 | 4.3E-17 |
| COL5A2 | 220 | NZB | Cd34 | 77 | 29 | 3.7E-10 |
| LHFP | 91 | GSE11121 | LHFP | 113 | 40 | 4.1E-40 |
| LHFP | 91 | GSE11121 | MMRN2 | 74 | 34 | 1.4E-38 |
| LHFP | 91 | AKXD | Ogn | 144 | 24 | 1.8E-14 |
| MLPH | 169 | GSE11121 | SOX10 | 75 | 21 | 9.4E-12 |
| POLR2E | 124 | AKXD | Fkbp8 | 269 | 54 | 1.6E-31 |
| SFRP1 | 74 | GSE11121 | SOX10 | 75 | 45 | 9.8E-66 |
| SFRP1 | 74 | NZB | Timm13 | 913 | 24 | 0.020 |
| SFRP1 | 74 | NZB | Cct2 | 1586 | 23 | 7.1E-28 |
| TPX2 | 299 | GSE11121 | TPX2 | 575 | 274 | 1.8E-226 |
| TPX2 | 299 | MOLF | Bub1b | 244 | 106 | 8.0E-60 |
| TPX2 | 299 | NZB | Tacc3 | 85 | 60 | 7.3E-50 |
| TPX2 | 299 | AKXD | Rad51ap1 | 134 | 48 | 1.0E-22 |
| YIPF5 | 63 | GSE11121 | WDR1 | 144 | 28 | 4.5E-17 |
Networks in bold were significant in the GOBO Kaplan-Meier analysis after Bonferroni correction.
TPX2 Network Predict DMFS Specifically in ER+ Tumors.
To better understand the nature of the human-mouse network comparisons, the structure of the overlaps was evaluated. Pair-wise comparisons between the GSE2034 TPX2 and overlapping partner networks revealed nine common hub genes (BUB1, UBE2C, CDC20, CCNB2, KIF2C, BUB1B, CEP55, CENPA, TPX2) (Fig. 2A). The conservation of these genes across the five datasets and their positions within the network suggested that they may play a critical role in the metastatic process. The TPX2 gene signature was therefore reduced to these nine genes and the Kaplan-Meier analysis repeated. As seen in Fig. 2B, the nine-gene signature was also capable of discriminating patient outcome. Unexpectedly, stratification of the dataset by ER status revealed that the nine-gene TPX2 signature was significantly associated with DMFS in ER+ (Fig. 2C) but not ER− patients (Fig. 2D). To confirm that these results were not specific to the GOBO analysis, the minimal TPX2 signature was analyzed from the glass slide-based Netherlands Cancer Institute (NKI) dataset (14) using BRB ArrayTools (http://linus.nci.nih.gov/BRB-ArrayTools.html). Consistent with the GOBO analysis, the TPX2 minimal signature was prognostic in ER+ tumors (Fig. 2E) but not ER− (Dataset S1).
Fig. 2.

Analysis of the core TPX2 network. (A) The TPX2 network showing the positions of the nine node genes (red text) conserved among all five datasets. (B–D) Kaplan-Meier analysis of the TPX2 nine-gene signature on all tumors, (B) ER+ only, (C) ER− only, or (D) of the GOBO meta-analysis. (E) Analysis of the nine-gene TPX2 signature on the ER+ patients of the independent NKI dataset. Red curves in all plots represent patients with higher expression of the nine genes. (F) Heat map showing differential expression of the Tpx2 core genes in Mvt1 cells transfected with either the long or short isoform of Brd4.
TPX2 Signature Is Tumor-Cell Autonomous.
This nine-gene signature consisted primarily of genes that are associated with microtubule and mitotic spindle biology. Four of the encoded proteins physically interact (BUB1, BUB1B, UBE2C, CDC20) and are important factors in chromosomal segregation (15, 16). This finding suggests that the effect of the TPX2 network would be tumor-cell autonomous. To investigate this result, the TPX2 signature was examined in tumors resulting from implanting the highly metastatic Mvt1 mouse mammary tumor cell line (17) into mice with high inherited susceptibility ([AKR/J × FVB/NJ]F1) or low susceptibility ([DBA/2J × FVB/NJ]F1) (18) to metastatic disease. Only 3 of the 299 genes in the GSE2034 TPX2 network were significantly different in these samples, consistent with this network being tumor autonomous.
If the TPX2 network is tumor-cell autonomous and associated with metastatic progression, it would be expected to correlate with the metastatic propensity of the tumor cells. To test this hypothesis, transcriptional analysis of the metastasis susceptibility gene Bromodomain 4 (Brd4) (19) was performed. Brd4 is transcribed as two isoforms, the longer of which is antimetastatic (19) and induces a gene signature associated with longer DMFS, but the short isoform is prometastatic and induces a gene signature indicative of poorer DMFS (20). The expression of the TPX2 network was therefore examined in the highly metastatic Mvt1 mouse mammary tumor cell line (17) stably expressing either the long or short Brd4 isoforms. Transcriptional profiling demonstrated that the two isoforms had opposing effects on the TPX2 network (Fig. 2F). Furthermore, the prometastatic short isoform up-regulated the majority of the prometastatic TPX2 network, but the antimetastatic long isoform down-regulated the majority of the network. These results are consistent with the known role of the Brd4 isoforms and demonstrate that the relative activity of the TPX2 network correlates with metastatic capacity, at least within this model system. The precise role of the TPX2 network and its relationship between Brd4 and other metastasis-modifying factors, however, is unclear at present and currently under investigation.
CD53 Network Predicts DMFS Specifically in ER− Tumors.
A similar analysis was carried out for the CD53 network. A three-gene signature was identified for the CD53 network that was common between the overlapping mouse and human networks (CD48, CD53, IL10RA) (Fig. 3A). Like the TPX2 minimal signature, the three-gene CD53 signature was significantly associated with DMFS in the full GOBO dataset (Fig. 3B). In contrast, however, the CD53 minimal signature was significant only in ER− patients (Fig. 3D), not ER+ patients (Fig. 3C). The association of the CD53 signature with ER− patients replicated in the NKI dataset (Fig. 3E), but not in the ER+ patients (Dataset S2).
Fig. 3.

Analysis of the core CD53 network. (A) The CD53 network showing the positions of the three node genes (red text) conserved among all five datasets. (B–D) Kaplan-Meier analysis of the TPX2 nine gene signature on all tumors, (B) ER+ only, (C) ER− only, or (D) of the GOBO meta-analysis. (E) Analysis of the three gene CD53 signature on the ER− patients of the independent NKI dataset. (F) Heat map showing differential expression of the CD53 network genes in Mvt1 tumors transplanted into AKR or DBA F1 animals.
Gene set enrichment analysis of the full CD53 signature revealed a significant association with immune response (9.0 × 10−23). The ability of patients with ER− breast cancers to mount an effective DMFS-associated immune response might be because of either differential production of tumor-derived factors or an inherent difference in the ability of the stromal cells to recognize and respond to the tumor. To investigate this possibility, the Mvt1 tumor transplants into AKR or DBA F1 mice was reanalyzed for the CD53 network. Thirty-one of 151 of the CD53 network genes were found to be differentially expressed at a significance threshold of P < 0.01 between the tumors implanted into AKR or DBA F1 animals. All but one of these genes was overexpressed in the low-metastatic DBA genotype compared with the high-metastatic genotype (Fig. 3F).
Discussion
The network analysis of independent human/mouse gene-expression datasets described here has resulted in reproducible sets of genes associated with metastatic disease. Previous efforts to identify prognostic genes have resulted in some overlaps between independent datasets and have identified general cellular functions, such as proliferation (8, 21, 22), associated with disseminated disease. However, these previous studies have not provided an understanding of the underlying transcriptional program that would inform investigators of particular molecular complexes and network nodes that are necessary for control of complex networks (23). Here we present an initial view of the underlying transcriptional network structures associated with metastatic susceptibility using two human breast cancer datasets that represent the natural course of disease. Further refinement of these structures by addition of similar human datasets will undoubtedly further enhance our understanding of the metastasis-associated network structures and reveal additional opportunities for investigation and intervention.
The inclusion of the mouse datasets provides an additional filter to focus on critical genes and cellular functions. By identifying genes shared between overlapping mouse and human networks that were independent predictors of DMFS, highly significant prognostic signatures were developed based on relatively few genes. In addition, the differences of network structure between the mouse and human networks permitted the refinement of signatures down to the highly connected nodes in the human networks. In the TPX2 network, these shared genes implicate the mitotic spindle checkpoint as a critical factor in metastatic spread in ER+ disease and specifically implicate the UBE2C/BUB1/BUB1B/CDC20 complex. Furthermore, because many of the gene-expression differences in the mouse datasets are likely the result of inherited polymorphisms, as all of the tumors are induced by the same oncogenic transgene, differences in transcription of these key node genes in humans may also be partially a result of inherited factors. Consistent with this possibility, examination of the gene list for the TPX2 network reveals the presence of two genes previously identified in our mouse mammary tumor genetic screens [CDC25A (24), RRP1B (25)]. Additionally, four TPX2 network genes [RFC3, RFC4 (26), JMJD6 (27), CNOT1 (28)] are known to physically interact with the metastasis susceptibility protein BRD4 (19). Further investigation into the role of these critical node genes may reveal interesting and important insights into the etiology of metastatic breast cancer.
In contrast to the tumor cell-autonomous genes associated with the ER+-associated TPX2 network, the ER−-associated CD53 network is comprised primarily of stromal immune cell components. Many of the most highly connected nodes within the GSE2034 network are associated with T-cell receptors (CD3D, CD2) or are related to the T-cell receptor (CD48) (29). Also included in this network are a number of immune-related cytokines and cytokine receptors, suggesting a direct protective role for these cytokines in ER− disease. Combined with the data obtained from the transplantation of tumors into the high- and low-metastatic mouse genotypes, these results suggest an important role for inherited differences in immune response to the primary tumor in the ultimate development of metastatic disease in patients with ER− breast cancer. Consistent with this hypothesis, recent evidence suggests that polymorphisms in CD53 are associated with regulation of TNF-α levels (30), which may affect the ability of the immune system to respond to the primary tumor. Although the identity of the infiltrating tumor cells is not defined by the CD53 network, the results of this study may provide a valuable starting point for further investigations into the role of the immune system in ER− breast cancer progression.
The role of the TPX2 and CD53 networks in ER+ and ER− breast cancer have additional important implications in understanding inherited predisposition to metastatic disease. Previous studies have implicated differences in gene expression of stromal components in the progression of breast cancer (31, 32). However, the factors driving these differences were not identified. The inherited differences in immune components in the mouse that predict DMFS in ER− patients is, to the best of our knowledge, unique evidence that polymorphisms in stromal tissue play an important role in predisposing women to disseminated disease. Our previous efforts to identify metastasis susceptibility genes have focused on tumor-autonomous factors (25, 33–35) rather than nonautonomous factors, such as immune response. Interestingly, epidemiology studies have demonstrated that polymorphisms in the human orthologs of these cell-autonomous metastasis susceptibility genes are prognostic only in ER+ patients (36), consistent with the finding that the cell-autonomous TPX2 mitotic spindle checkpoint network is significant only in ER+ patients. Thus, the critical inherited factors for metastatic susceptibility targets for ER+ patients appear to function primarily within tumor cells, but is the most relevant for ER− disease polymorphisms within the stromal tissues.
Finally, we believe that these results provide a compelling argument for the continued use of mouse models to investigate the inherited components of metastatic breast cancer. Despite the difference in network structure, the three mouse datasets generated by array platform and next-generation sequencing reproducibly identified the critical nodes within the human network networks. As indicated above, the differences in network structure likely is because of both decreased somatic and inherited genetic heterogeneity within the animal samples. Utilization of additional tumor-initiating systems and more complex population models like the Collaborative Cross (37), designed to more closely resemble human genetic diversity, may result in network structure more closely resembling those from human datasets. Importantly, the cross-species analysis permits the identification the critical hub genes within human networks. Furthermore, the mouse model enables the generation of large sample sets that represent the natural course of disease without any confounding factors associated with adjuvant therapy or differences in treatment regimens. Thus, we believe the combined use of the mouse- and human-expression datasets in network analysis provides a powerful tool for investigating the molecular basis of metastasis, and further analysis will yield additional important insights into the etiology of disseminated disease.
Materials and Methods
Mouse Samples.
The [AKXD RI × PyMT]F1 (AKXD) and (FVB/NJ × [NZB/B1NJ × PyMT])N2 (NZB) backcrosses have been previously described (11). The thirty MOLF samples represented the phenotypic extremes of a 194 animal (FVB/NJ × [MOLF/Ei × PyMT])N2 backcross developed to map modifiers of mammary tumor latency, tumor growth and pulmonary metastasis. All animal experiments were performed under approved Animal Care and Use protocols at either the Fox Chase Cancer Center (AKXD and NZB) or the National Cancer Institute (NCI; MOLF).
Gene-Expression Analysis.
Affymetrix MOE430 gene-expression analysis was performed as previously described (38). mRNA-Seq of the MOLF samples was performed on the Illumina Genome Analyzer II or High Seq platform at the NCI Center for Cancer Research Sequencing Facility. All sequence reads were mapped to mouse reference genome (NCBI37/mm9), AceView (39), and a custom Refseq-based alternative splicing database. The best mapping for each read was chosen to form a consensus set of mappings. Reads with questionable alignments were remapped using sim4 (40). The gene-expression level for each transcript annotated in Refseq was estimated by the reads per kilo exonic bases per million reads (41) value, a within-sample normalized metric correcting for differences in gene length and total mapped reads in the transcriptome. Simple normalization by total reads was performed before network analysis. The data are available through the Gene Expression Omnibus, accession no. GSE30866.
Brd4 Cell Line Transcriptional Profiling.
The Brd4-expressing cell line has been previously described (19, 20). Briefly, the highly metastatic Mvt1 cell line (17) was transfected with long isoform Brd4 expression vectors (19) or transduced with short isoform-expressing lentiviruses (20). Individual clones were generated, total RNA isolated, and arrayed on Affymetrix MOE430 v2 chips by the K.W.H. laboratory, as previously described (19) or by the Microarray Core in the NCI Laboratory of Molecular Technology (20). Expression data were normalized using the Partek Genomics Suite then loaded into BRB ArrayTools version 4.2.0-Beta_2 (42). The relative expression of the TPX2 network was determined using the Class Comparison tool of BRB ArrayTools.
AKR and DBA F1 Transplant Tumor-Expression Profiling.
AKR and DBA F1 animals were generated by breeding FVB/NJ male animals to either DBA/2J or AKR/J females. The highly metastatic Mvt1 cell line (17) was orthotopically transplanted into the fourth inguinal mammary gland into 8-wk-old animals and the animals aged for 28 d before harvesting tumors (18). Total RNA from three independent AKR or DBA F1 tumors were arrayed on Affymetrix MOE430 v2 chips and the relative expression of the TPX2 or CD53 networks determined using the Class Comparison tool of BRB ArrayTools version 4.2.0-Beta_2.
Network Analysis.
Network analysis was performed on the top 4,000 most variable genes present in the microarray data. The R package “weighted correlation network analysis” (WGCNA) (43) was used to find clusters (network networks) of high-correlation genes. The network for each network was generated with the minimum-spanning tree with dissimilarity matrix from WGCNA. This study used the high-performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health, Bethesda, MD (http://biowulf.nih.gov). Figures for publication were generated using the software package Cytoscape (44).
Supplementary Material
Acknowledgments
We thank Drs. Lalage Wakefield, Glenn Merlino, and Thomas Geiger for critical reading of this manuscript. Analyses of the Netherlands Cancer Institute dataset, Brd4 cell lines, and AKR and DBA F1 tumors were performed using BRB-ArrayTools developed by Dr. Richard Simon and BRB-ArrayTools Development Team. This research was supported in part by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE30866).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1117872109/-/DCSupplemental.
References
- 1.Perou CM, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 2.Driouch K, Landemaine T, Sin S, Wang S, Lidereau R. Gene arrays for diagnosis, prognosis and treatment of breast cancer metastasis. Clin Exp Metastasis. 2007;24:575–585. doi: 10.1007/s10585-007-9110-x. [DOI] [PubMed] [Google Scholar]
- 3.Ein-Dor L, Kela I, Getz G, Givol D, Domany E. Outcome signature genes in breast cancer: Is there a unique set? Bioinformatics. 2005;21:171–178. doi: 10.1093/bioinformatics/bth469. [DOI] [PubMed] [Google Scholar]
- 4.Ein-Dor L, Zuk O, Domany E. Thousands of samples are needed to generate a robust gene list for predicting outcome in cancer. Proc Natl Acad Sci USA. 2006;103:5923–5928. doi: 10.1073/pnas.0601231103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schadt EE, et al. An integrative genomics approach to infer causal associations between gene expression and disease. Nat Genet. 2005;37:710–717. doi: 10.1038/ng1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quigley DA, et al. Genetic architecture of mouse skin inflammation and tumour susceptibility. Nature. 2009;458:505–508. doi: 10.1038/nature07683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ringnér M, Fredlund E, Häkkinen J, Borg A, Staaf J. GOBO: Gene expression-based outcome for breast cancer online. PLoS ONE. 2011;6:e17911. doi: 10.1371/journal.pone.0017911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y, et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365:671–679. doi: 10.1016/S0140-6736(05)17947-1. [DOI] [PubMed] [Google Scholar]
- 9.Schmidt M, et al. The humoral immune system has a key prognostic impact in node-negative breast cancer. Cancer Res. 2008;68:5405–5413. doi: 10.1158/0008-5472.CAN-07-5206. [DOI] [PubMed] [Google Scholar]
- 10.Guy CT, Cardiff RD, Muller WJ. Induction of mammary tumors by expression of polyomavirus middle T oncogene: A transgenic mouse model for metastatic disease. Mol Cell Biol. 1992;12:954–961. doi: 10.1128/mcb.12.3.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hunter KW, et al. Predisposition to efficient mammary tumor metastatic progression is linked to the breast cancer metastasis suppressor gene Brms1. Cancer Res. 2001;61:8866–8872. [PubMed] [Google Scholar]
- 12.Yang H, et al. Caffeine suppresses metastasis in a transgenic mouse model: A prototype molecule for prophylaxis of metastasis. Clin Exp Metastasis. 2004;21:719–735. doi: 10.1007/s10585-004-8251-4. [DOI] [PubMed] [Google Scholar]
- 13.Li A, Horvath S. Network module detection: Affinity search technique with the multi-node topological overlap measure. BMC Res Notes. 2009;2:142. doi: 10.1186/1756-0500-2-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van de Vijver MJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- 15.Tang Z, Shu H, Oncel D, Chen S, Yu H. Phosphorylation of Cdc20 by Bub1 provides a catalytic mechanism for APC/C inhibition by the spindle checkpoint. Mol Cell. 2004;16:387–397. doi: 10.1016/j.molcel.2004.09.031. [DOI] [PubMed] [Google Scholar]
- 16.Reddy SK, Rape M, Margansky WA, Kirschner MW. Ubiquitination by the anaphase-promoting complex drives spindle checkpoint inactivation. Nature. 2007;446:921–925. doi: 10.1038/nature05734. [DOI] [PubMed] [Google Scholar]
- 17.Pei XF, et al. Explant-cell culture of primary mammary tumors from MMTV-c-Myc transgenic mice. In Vitro Cell Dev Biol Anim. 2004;40:14–21. doi: 10.1290/1543-706X(2004)40<14:ECOPMT>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 18.Lukes L, Crawford NP, Walker R, Hunter KW. The origins of breast cancer prognostic gene expression profiles. Cancer Res. 2009;69:310–318. doi: 10.1158/0008-5472.CAN-08-3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crawford NP, et al. Bromodomain 4 activation predicts breast cancer survival. Proc Natl Acad Sci USA. 2008;105:6380–6385. doi: 10.1073/pnas.0710331105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alsarraj J, et al. Deletion of the proline-rich region of the murine metastasis susceptibility gene Brd4 promotes epithelial-to-mesenchymal transition- and stem cell-like conversion. Cancer Res. 2011;71:3121–3131. doi: 10.1158/0008-5472.CAN-10-4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van 't Veer LJ, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 22.Sotiriou C, et al. Gene expression profiling in breast cancer: Understanding the molecular basis of histologic grade to improve prognosis. J Natl Cancer Inst. 2006;98:262–272. doi: 10.1093/jnci/djj052. [DOI] [PubMed] [Google Scholar]
- 23.Liu YY, Slotine JJ, Barabási AL. Controllability of complex networks. Nature. 2011;473:167–173. doi: 10.1038/nature10011. [DOI] [PubMed] [Google Scholar]
- 24.Cozma D, et al. A bioinformatics-based strategy identifies c-Myc and Cdc25A as candidates for the Apmt mammary tumor latency modifiers. Genome Res. 2002;12:969–975. doi: 10.1101/gr.210502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crawford NP, et al. Rrp1b, a new candidate susceptibility gene for breast cancer progression and metastasis. PLoS Genet. 2007;3:e214. doi: 10.1371/journal.pgen.0030214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maruyama T, et al. A mammalian bromodomain protein, brd4, interacts with replication factor C and inhibits progression to S phase. Mol Cell Biol. 2002;22:6509–6520. doi: 10.1128/MCB.22.18.6509-6520.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rahman S, et al. The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol Cell Biol. 2011;31:2641–2652. doi: 10.1128/MCB.01341-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lau NC, et al. Human Ccr4-Not complexes contain variable deadenylase subunits. Biochem J. 2009;422:443–453. doi: 10.1042/BJ20090500. [DOI] [PubMed] [Google Scholar]
- 29.Elishmereni M, Levi-Schaffer F. CD48: A co-stimulatory receptor of immunity. Int J Biochem Cell Biol. 2011;43:25–28. doi: 10.1016/j.biocel.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 30.Bos SD, et al. A genome-wide linkage scan reveals CD53 as an important regulator of innate TNF-alpha levels. Eur J Hum Genet. 2010;18:953–959. doi: 10.1038/ejhg.2010.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Finak G, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med. 2008;14:518–527. doi: 10.1038/nm1764. [DOI] [PubMed] [Google Scholar]
- 32.Teschendorff AE, Miremadi A, Pinder SE, Ellis IO, Caldas C. An immune response gene expression module identifies a good prognosis subtype in estrogen receptor negative breast cancer. Genome Biol. 2007;8:R157. doi: 10.1186/gb-2007-8-8-r157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alsarraj J, et al. Deletion of the proline-rich region of the murine metastasis susceptibility gene Brd4 promotes epithelial-to-mesenchymal transition- and stem cell-like conversion. Cancer Res. 2011;71:3121–3131. doi: 10.1158/0008-5472.CAN-10-4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crawford NP, et al. The diasporin pathway: A tumor progression-related transcriptional network that predicts breast cancer survival. Clin Exp Metastasis. 2008;25:357–369. doi: 10.1007/s10585-008-9146-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park YG, et al. Sipa1 is a candidate for underlying the metastasis efficiency modifier locus Mtes1. Nat Genet. 2005;37:1055–1062. doi: 10.1038/ng1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hsieh SM, Look MP, Sieuwerts AM, Foekens JA, Hunter KW. Distinct inherited metastasis susceptibility exists for different breast cancer subtypes: A prognosis study. Breast Cancer Res. 2009;11:R75. doi: 10.1186/bcr2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Churchill GA, et al. Complex Trait Consortium The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat Genet. 2004;36:1133–1137. doi: 10.1038/ng1104-1133. [DOI] [PubMed] [Google Scholar]
- 38.Yang H, et al. Metastasis predictive signature profiles pre-exist in normal tissues. Clin Exp Metastasis. 2005;22:593–603. doi: 10.1007/s10585-005-6244-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thierry-Mieg D, Thierry-Mieg J. AceView: A comprehensive cDNA-supported gene and transcripts annotation. Genome Biol. 2006;7(Suppl 1):S12.1–S12.14. doi: 10.1186/gb-2006-7-s1-s12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Florea L, Hartzell G, Zhang Z, Rubin GM, Miller W. A computer program for aligning a cDNA sequence with a genomic DNA sequence. Genome Res. 1998;8:967–974. doi: 10.1101/gr.8.9.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 42.Wright GW, Simon RM. A random variance model for detection of differential gene expression in small microarray experiments. Bioinformatics. 2003;19:2448–2455. doi: 10.1093/bioinformatics/btg345. [DOI] [PubMed] [Google Scholar]
- 43.Langfelder P, Horvath S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shannon P, et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.