

Abstract
Background
Although natriuretic peptides are considered cardioprotective, clinical heart failure trials with recombinant BNP (nesiritide) failed to prove it. Unsuspected proadrenergic effects might oppose the anticipated benefits of natriuretic peptides.
Methods and Results
We investigated whether natriuretic peptides induce catecholamine release in isolated hearts, sympathetic nerve endings (cardiac synaptosomes) and PC12 cells bearing a sympathetic neuron phenotype. Perfusion of isolated guinea pig hearts with BNP elicited a 3-fold increase in norepinephrine release which doubled in ischemia/reperfusion conditions. BNP and ANP also released norepinephrine from cardiac synaptosomes and dopamine from nerve-growth-factor-differentiated PC12 cells in a concentration-dependent manner. These catecholamine-releasing effects were associated with an increase in intracellular calcium, and abolished by blockade of calcium channels and calcium transients, demonstrating a calcium-dependent exocytotic process. Activation of the guanylyl cyclase-cGMP-protein-kinase-G system with nitroprusside or membrane-permeant cGMP analogs mimicked the proexocytotic effect of natriuretic peptides, an action associated with an increase in intracellular cAMP and protein-kinase-A activity. cAMP enhancement resulted from an inhibition of phosphodiesterase-type-3-induced cAMP hydrolysis. Collectively, these findings indicate that, by inhibiting phosphodiesterase-type-3, natriuretic peptides sequentially enhance intracellular cAMP levels, protein-kinase-A activity, intracellular calcium and catecholamine exocytosis.
Conclusions
Our results show that natriuretic peptides, at concentrations likely to be reached at cardiac sympathetic nerve endings in advanced congestive heart failure, promote norepinephrine release via a protein-kinase-G-induced inhibition of phosphodiesterase-type-3-mediated cAMP hydrolysis. We propose that this proadrenergic action may counteract the beneficial cardiac and hemodynamic effects of natriuretic peptides, and thus explain the ineffectiveness of nesiritide as a cardiac failure medication.
Keywords: catecholamines, heart failure, natriuretic peptides
Introduction
Plasma levels of Brain Natriuretic Peptide (BNP) and norepinephrine (NE) increase with the severity of congestive heart failure (CHF). In fact, BNP and NE are regarded as important biomarkers for the diagnosis, risk stratification and prediction of death in patients with CHF.1;2 Natriuretic peptides are generally viewed as cardioprotective since they were found to reduce blood pressure, plasma volume3;4 and myocardial infarct size.5 Yet, other investigators reported an enlargement of infarct size following Atrial Natriuretic Peptide (ANP) administration,6 and a reduction in infarct size when the natriuretic peptide type A receptor was abrogated or blocked,7 or when ANP was deleted.6 Moreover, although natriuretic peptides are thought to retard the progression of heart failure,8 the administration of recombinant BNP (nesiritide) was recently found to lack protective efficacy in acute heart failure patients.9 Thus, even though natriuretic peptides have been viewed as a compensatory neurohormonal system that is upregulated in the setting of heart failure, affording beneficial cardiac and hemodynamic effects,8 their role in alleviating cardiac ailments remains unsettled.
The beneficial actions of natriuretic peptides are attributed to the stimulation of particulate guanylyl cyclase and thus, to an increased formation of cGMP,10 which has smooth muscle relaxing and vasodilating effects,11 hypothetically related to decreased NE release from adrenergic nerves.12 A BNP-induced facilitation of cardiac vagal neurotransmission and bradycardia has also been reported.13 Nonetheless, injection of a membrane-permeable cGMP analog was found to increase heart rate before any changes in blood pressure,14 possibly due to enhanced NE release from sympathetic nerve endings. Indeed, the nitric oxide-guanylyl cyclase-cGMP system has been shown to promote NE outflow in the mesenteric vasculature.15 Therefore, whether cGMP has sympatholytic16–18 or pro-adrenergic14;19–21 effects remains controversial. Thus, it is not inconceivable that an increase in intracellular levels of cGMP mediated by natriuretic peptides could actually contribute to the heightened cardiac sympathetic drive and excessive NE release that characterize CHF2 and atrial fibrillation.22;23
Given that an enhanced NE release bears dysfunctional and arrhythmogenic consequences,24–26 we investigated the NE-releasing properties of natriuretic peptides and cGMP. We report that BNP increases NE release in the guinea-pig heart ex vivo, an effect which is further enhanced in ischemia/reperfusion. Furthermore, natriuretic peptides, sodium nitroprusside (SNP) and cell-permeable cGMP analogs all elicit catecholamine exocytosis in sympathetic nerves isolated from the guinea-pig heart (i.e., cardiac synaptosomes) and in nerve-growth factor(NGF)-differentiated PC12 cells, which bear a sympathetic nerve-ending phenotype. This pro-exocytotic effect likely results from an increase in intracellular calcium (Ca2+). The process involves a protein kinase G (PKG)-mediated inhibition of phosphodiesterase type-3 (PDE3), which increases cAMP and protein kinase A (PKA) activity.
Methods
Ex vivo hearts
Twenty one male Hartley guinea pigs (300–350 grams; Charles River Laboratories, Kingston, NY) were anesthetized with CO2 and euthanized by stunning with approval from the Institutional Animal Care and Use Committee. Isolated hearts were perfused at constant pressure (55 cm H2O) with oxygenated (5% CO2, 95% O2) Ringer at 37°C in a Langendorff apparatus (Radnoti Glass Technology, Monrovia, CA). After equilibration, a number of hearts were perfused with BNP for 6 minutes. Other hearts were subjected to 10 minutes of global ischemia followed by 10 minutes of reperfusion, in the absence or presence of BNP. Coronary flow was measured every 2 minutes; samples were assayed for NE. Surface ECG was obtained from the left ventricle and right atrium, recorded in digital format, and analyzed with Power Laboratory/8SP (AdInstrument, Colorado Springs, CO).
Preparation of cardiac synaptosomes
Thirty seven guinea pig hearts were isolated as described in the above section. Hearts were perfused at constant pressure in a Langendorff apparatus with oxygenated Ringer’s solution for 15 minutes to ensure that no blood traces remained in the coronary circulation.27 Hearts were subsequently freed from fat and connective tissue and minced in ice-cold 0.32 M sucrose containing 1 mM EGTA, pH 7.4. Synaptosomes (pinched-off sympathetic nerve endings) were isolated as previously described27 and incubated with natriuretic peptides, SNP (a nitric oxide donor) or 8-bromo-cGMP (a membrane-permeable cyclic GMP analog) for 10 minutes in a water bath at 37°C either in the absence or presence of the N-type Ca2+ channel blocker ω-conotoxin GVIA (ω-CTX; 100 nM), the L-type Ca2+ channel blocker nifedipine (5 μM), the intracellular Ca2+ chelator BAPTA-AM [1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis (acetoxymethyl ester)] (10 μM), the cell-permeable protein kinase inhibitor PKI14–22 amide myristoylated (20 nM), Rp-8-Br-cGMPS (a membrane-permeant cGMP analog which blocks cGMP-dependent protein kinase; 30 μM), the PDE3 activator insulin (30 nM) or the PDE3 inhibitor cilostamide [N-Cyclohexyl-N-methyl-4-(1,2-dihydro-2-oxo-6-quinolyloxy)butyramide] (10 μM). When these drugs were used, synaptosomes were pre-incubated with them for 10 minutes. Controls were incubated for an equivalent length of time without drugs. At the end of the incubation period each sample was centrifuged at 20,000 g for 20 min, 4°C. The supernatant was assayed for NE content by HPLC with electrochemical detection (EC); the pellet was analyzed for protein content, by a modified Lowry procedure.27
PC12 cells
The rat pheochromocytoma PC12 cell line was maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) plus 10% fetal bovine serum (FBS), 5% donor horse serum (DHS), 1% L-glutamine, and antibiotics at 37°C in 5% CO2.28 The differentiating protocol involved plating PC12 cells on tissue culture plates coated with collagen (rat tail type-VII, Sigma) combined with exposure to low serum medium containing 1% FBS, 0.5% DHS, 1% L-glutamine, and antibiotics supplemented with 7S-NGF (BD Biosciences, Bedford, MA). PC12 cells were grown to confluence in 100 mm plates or 6-well plates and differentiated with NGF (100 ng/mL) for 5–7 days. For each experiment, the culture medium was aspirated and cells were washed twice with Na Ringer, then incubated with natriuretic peptides, SNP, 8-pCPT-cGMP (a membrane-permeable cGMP analog), 8-bromo-cGMP or forskolin (an adenylyl cyclase activator) for 20 minutes in an incubator at 37°C either in the absence or presence of BAPTA (10 μM), PKI14–22 (20 nM), Rp-8-Br-cGMPS (0.5 μM), LY-83583 (a guanylyl cyclase inhibitor; 10 μM), insulin (100 nM) or cilostamide (10 μM). When these drugs were used, PC12 cells were pre-incubated with them for 10 minutes. Controls were incubated for an equivalent length of time without drugs. At the end of each experiment aliquots of the supernatant and cell lysates (after a 30-minute treatment with Triton X-100) were taken from each well and analyzed for dopamine (DA) content by HPLC-EC with a 6-minute retention time.28 Other cell lysates were analyzed for intracellular cAMP levels, PKA activity, PKG activity or PDE3 activity. We used two membrane-permeable cGMP analogs (8-bromo-cGMP and 8-pCPT-cGMP) because they are both resistant to hydrolysis by phosphodiesterases and activate PKG, but differ in that 8-pCPT-cGMP is more potent than 8-bromo-cGMP, and does not affect cGMP-regulated phosphodiesterase.29
Intracellular Ca2+ assay
Cells were washed twice with Na Ringer, and then treated with potassium (100 mM; 3 min) or BNP (100 nM; 10 min) in the presence or absence of Rp-8-Br-cGMPS (0.5 μM; 10 min), insulin (100 nM; 10 min) or cilostamide (10 μM; 10 min). Controls were incubated for an equivalent length of time without drugs. At the end of each experiment, cells were washed twice with Dulbecco’s phosphate buffered saline (PBS) containing 10 mM EGTA (to chelate external Ca2+) and then with normal PBS to remove the remaining EGTA. Cells were then lysed with addition of water and harvested with a scraper. Ca2+ content was determined using a Ca2+ assay kit (QuantiChromTM Ca2+ Assay Kit by BioAssay Systems, Hayward, CA). The Ca2+ content was adjusted by the protein content of the cells and expressed as mg of Ca2+/mg of protein.30
cAMP assay
PC12 cells were treated and lysed as described above. Intracellular cAMP levels were determined using a cAMP Biotrak EIA kit (GE Healthcare Bio-Sciences Corp., Piscataway, NJ) following the manufacturer’s protocol.28 This cAMP assay is highly specific and is based on competition between unlabeled cAMP and a fixed quantity of peroxidase-labeled cAMP for a limited number of binding sites on a cAMP specific antibody. The cross-reactivity for cGMP, AMP, ADP and ATP is below 0.01%, while cAMP is 100%.
PKA activity
PKA phosphorylation (i.e., an indication of PKA activation) was measured using a p-PKAα antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) in Western blot. Methods for Western blot analysis were as previously described.27
PKG activity
Phosphorylated vasodilator-stimulated phosphoprotein (VASP; a major substrate for PKG) at Ser239 is a sensitive biochemical marker for monitoring the activity of PKG.31 VASP phosphorylation (i.e., PKG activity) was measured using a p-VASP antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) in Western blot. Methods for Western blot analysis were as previously described.27
PDE3 activity
PDE3 activity was measured using a commercially available colorimetric PDE assay kit (Biomol International, Inc., Plymouth Meeting, PA) as previously described.32 PC12 cell lysates were prepared and then total protein concentration was measured as described above. Free phosphate contamination was removed according to manufacturer’s protocol. Samples were incubated for 10 minutes at 37°C and reactions were stopped with Biomol Green (Biomol). Samples were then put on a shaker for 20 minutes at room temperature. Results were measured using a Molecular Devices microplate reader (Sunnyvale, CA). PDE3-specific cAMP-hydrolytic activity was expressed as the difference between cAMP hydrolyzed (expressed as nmol/min/mg protein) in the presence and absence of the specific PDE3 inhibitor cilostamide.
Drugs and Chemicals
BNP and ANP were purchased from AnaSpec (Fremont, CA); SNP, ω-CTX, nifedipine, 8-bromo-cGMP, 8-pCPT-cGMP, forskolin, PKI14–22, Rp-8-Br-cGMPS, LY-83583, insulin and cilostamide were purchased from Sigma-Aldrich (St Louis, MO); BAPTA-AM was purchased from Invitrogen (Carlsbad, CA).
Statistics
Data are presented as mean ± SEM. Parametric tests were used throughout the study. For the type of experiment involving repeated measures over time, repeated measures ANOVA was performed for statistical analysis, followed by post-hoc pairwise comparisons of the time points. Dunnett’s multple comparison test was used to adjust the post-hoc pairwise comparisons of the time points (Figure 1A). For 2-group comparisons, the unpaired t test was used in all the other figures. In addition, to confirm the significance of the parametric findings, we have calculated P values using non-parametric tests (i.e., Mann-Whitney test and Kruskal-Wallis test followed by the post-hoc Dunn’s test). GraphPad Prism version 4.03 for Windows (GraphPad Software, San Diego, Calif) was used. Values of P<0.05 were considered statistically significant.
Figure 1.

Norepinephrine-releasing effects of BNP in ex vivo whole hearts. A. Continuous perfusion with BNP (100 nM) enhances NE overflow in Langendorff guinea pig hearts. Bars are means (± SEM; n=8). *, P<0.05 and **, P<0.01 from control by repeated measures ANOVA followed by post-hoc multiple comparisons. Overall P-value = 0.0067. B. guinea pig hearts were subjected to 10-minute global ischemia followed by 10-minute reperfusion (I/R), either in the absence or presence of BNP (100 nM). Bars are means (± SEM; n=6–13) of NE overflow in the first 2 minutes of reperfusion. ***, P<0.0001 from control by unpaired t-test. #, P=0.0420 from I/R by unpaired t-test.
Results
Myocardial ischemia/reperfusion enhances the norepinephrine-releasing effects of BNP
Continuous administration of BNP (100 nM) to Langendorff-perfused guinea-pig hearts ex vivo caused a ≥2-fold increase in NE overflow which reached a peak within 2–4 minutes and persisted until BNP administration was discontinued (Figure 1A). Notably, in hearts undergoing 10-minute global ischemia, NE release increased 3 fold in the first 2 minutes of reperfusion, and the addition of BNP (100 nM) caused a further ~2-fold increase in the same 2-minute period (Figure 1B). Hence, the NE-releasing effects of BNP are enhanced in the setting of ischemia/reperfusion.
Guanylyl cyclase stimulation induces an increase in catecholamine release
We next evaluated this NE-releasing effect at the level of sympathetic nerve terminals (i.e., cardiac synaptosomes). In a concentration-dependent manner, BNP and ANP each elicited a ~12–35% increase in NE release from cardiac synaptosomes, (EC50’s ~20 nM; Figure 2A, panels a and b). Similarly, in a concentration-dependent manner, BNP and ANP each elicited a ~10–50% increase in DA release (EC50 ~5 nM; Figure 2B, panels a and b) from NGF-differentiated PC12 rat pheochromocytoma cells, a cellular model of sympathetic neuron.33
Figure 2.

Catecholamine-releasing effect of natriuretic peptides and sodium nitroprusside (SNP). A. Concentration-response curves for the NE-releasing effects elicited by BNP, ANP and SNP (3–100 nM) in sympathetic nerve terminals (cardiac synaptosomes) isolated from guinea pig hearts. B: Concentration-response curves for the DA-releasing effects elicited by BNP, ANP and SNP (0.1–100 nM) in NGF-differentiated PC12 cells. In A, points are mean increases in NE release above basal level (± SEM; n=10–16, 12 and 8–12 for a, b and c, respectively). **, P<0.01 and ***, P<0.001 from lowest concentrations of BNP, ANP or SNP by unpaired t-test. In B, points are mean increases in DA release above basal level (± SEM; n=7–10, 6–7 and 6–10 for a, b and c, respectively). *, P<0.05, **, P<0.01 and ***, P<0.001 from lowest concentrations of BNP, ANP or SNP by unpaired t-test. Basal NE and DA levels were 1.44 ± 0.04 (± SEM; n=46) and 6.82 ± 0.75 pmol/mg protein (± SEM; n=7), respectively.
Since BNP and ANP are both activators of particulate guanylyl cyclase,4 we next assessed whether activation of soluble guanylyl cyclase would also increase catecholamine release. We found that incubation with SNP (3–100 nM; a soluble guanylyl cyclase activator) induced a ~6–28% increase in NE from cardiac synaptosomes (EC50 ~20 nM; Figure 2A, panel c) and a ~5–25% increase in DA release from PC12 cells (EC50 ~5 nM; Figure 2B, panel c). Collectively, these results suggested that stimulation of guanylyl cyclase induces an increase in catecholamine release.
The catecholamine-releasing effect of cGMP is Ca2+-dependent
Since guanylyl cyclase activators elicited catecholamine release from sympathetic nerve endings (See Figures 1 and 2), we next assessed whether cGMP would also promote NE and DA release. We found that, as a function of its concentration, the membrane-permeable analog of cGMP, 8-bromo-cGMP (0.1–100 μM), induced an increase in the release of NE and DA from cardiac synaptosomes (10–35%; ED50 ~10 μM) and PC12 cells (25–80%; ED50 ~3 μM), respectively (Figure 3A–D). In the presence of the N-type Ca2+ channel inhibitor ω-CTX GVIa (100 nM)34 or the L-type Ca2+ channel inhibitor nifedipine (5 μM),35 the concentration-response curve for the NE-releasing effects of 8-bromo-cGMP was markedly shifted downward, indicating that Ca2+ entry via N- and L-type channels is important for the cGMP-induced NE release (Figure 3A and B).
Figure 3.

Calcium dependence of the catecholamine-releasing effect of cGMP. A–C. release of endogenous NE from guinea pig heart synaptosomes by 8-Br-cGMP (1 to 100 μM) in the absence (control) and presence of the N- and L-type Ca2+ channel blockers ω-CTX (100 nM) and nifedipine (5 μM), and of the intracellular Ca2+ chelator BAPTA (10 μM). D. DA release from NGF-differentiated PC12 cells in the absence (control) and presence of BAPTA (10 μM). Points are mean increases in NE and DA release above basal level (± SEM; n=12 for A and B; n=8 for C; n=4–15 for D). Basal NE and DA levels were 1.42 ± 0.05 and 6.94 ± 0.92 pmol/mg protein, n=32 and 20, respectively. *, P<0.05, **, P<0.01 and ***, P<0.001 from corresponding control point by unpaired t-test. E. Intracellular Ca2+ content of NGF-differentiated PC12 cells treated with Na-Ringer, K+ (100 mM) or BNP (100 nM) in the absence or presence of PKG inhibitor (Rp-8-Br-cGMPS; 0.5 μM) or the PDE3 activator insulin (100 nM), with or without the PDE3 inhibitor cilostamide (10 μM; Cilo). Bars represent mean quantitative values (±SEM; n=8–12 for E). **, P<0.01 and ***, P<0.0001 from control, ##, P<0.01 from BNP and †††, P<0.0001 from the combination of BNP, insulin and cilostamide by unpaired t-test.
Moreover, we found that in the presence of the intracellular Ca2+ chelator BAPTA-AM36 the concentration-response curves for the 8-bromo-cGMP-induced release of NE and DA from cardiac synaptosomes and PC12 cells, respectively, were markedly shifted downward, indicating that Ca2+ transients also play a role in the exocytotic effect of cGMP (Figure 3C and D).
Indeed, incubation with BNP (100 nM, for 10 minutes) elicited a 2.5-fold increase in intracellular Ca2+ in NGF-differentiated PC12 cells. This Ca2+ increase was prevented by the PKG inhibitor (0.5 μM, for 10 minutes) and by the PDE3 activator insulin (100 nM, for 10 minutes) (Figure 3E). Inhibition of PDE3 (cilostamide 10 μM, 10 minutes) reversed the effect of insulin. Notably, depolarization with K+ (100 mM, 3 minutes) caused a 3-fold increase in intracellular Ca2+ which was not significantly modified by PKG inhibition or PDE3 stimulation (Figure 3E). This strengthened the findings with Ca2+ channel inhibitors and chelator indicating that an increase in intracellular Ca2+ mediates the promotion of catecholamine release by natriuretic peptides and cGMP analogs.
The catecholamine-releasing effect of BNP and cGMP relies on the activation of the cAMP-PKA pathway
The administration of BNP (100 nM), and the two membrane-permeable cGMP analogs 8-bromo-cGMP (1 μM) and 8-pCPT-cGMP (3 μM) to PC12 cells induced in each case a ~2-fold increase in intracellular cAMP level, while the adenylyl cyclase activator forskolin (10 μM) elicited a ~30-fold increase in intracellular cAMP (Figure 4A). Comparably, the administration of BNP (100 nM), 8-bromo-cGMP (1 μM), 8-pCPT-cGMP (3 μM) and forskolin (10 μM) to PC12 cells induced in each case a ~90% increase in PKA activity (Figure 4B). Notably, the 8-bromo-cGMP-induced increase in neurotransmitter release in cardiac synaptosomes and PC12 cells was markedly attenuated by PKA inhibition with PKI14–22 (20 nM)37 (Figure 4C). Collectively, these data indicated that BNP- and cGMP-mediated enhancements of catecholamine exocytosis rely on an increase in PKA activity due to an increase in intracellular cAMP.
Figure 4.

PKA dependence of the catecholamine-releasing effect of BNP and cGMP. A. intracellular cAMP levels in NGF-differentiated PC12 cells treated with BNP (100 nM), 8-Br-cGMP (1 μM), 8-pCPT-cGMP (3 μM) or forskolin (10 μM). Bars are means of absolute values (± SEM; n=7–11). ***, P<0.001 from control by unpaired t-test. B. PKA activity in NGF-differentiated PC12 cells treated with BNP (100 nM), 8-Br-cGMP (1 μM), 8-pCPT-cGMP (3 μM) or forskolin (10 μM). Upper strips, representative immunoblot of PC12 cell lysate probed with anti-phosphorylated PKA antibody. Lower strips, same immunoblot probed with β-actin antibody. Bars represent mean quantitative values (± SEM; n=6–8). **, P<0.01 from control by unpaired t-test. C. Concentration-response curves for the NE- and DA-releasing effect of the cell-permeable analog of cGMP, 8-Br-cGMP (0.1–100 μM) in synaptosomes isolated from guinea pig hearts (panel a) and NGF-differentiated PC12 cells (panel b) in the absence (control) or presence of the PKA inhibitor PKI14–22 (20 nM). Points are means (± SEM; a, n=12; b, n=4–15). *, ** and ***, P<0.05, P<0.01 and P<0.0001 respectively, from corresponding control point by unpaired t-test. Basal NE and DA levels were 1.4 ± 0.08 and 6.94 ± 0.92 pmol/mg protein, n=12 and 20, respectively.
The catecholamine-releasing effect of BNP and cGMP relies on a PKG-induced activation of the cAMP-PKA pathway
Our results suggested that an activation of the cAMP-PKA pathway plays a role in the catecholamine-releasing effect of BNP and cGMP (see Figure 4). Since natriuretic peptides are known to signal via PKG,3;4 we next investigated whether PKG activation might in turn activate the cAMP-PKA pathway and thus, promote NE and DA exocytosis. We found that in the presence of the PKG inhibitor Rp-8-Br-cGMPS (cardiac synaptosomes 30 μM; PC12 cells, 0.5 μM)38 the concentration-response curves for the BNP-induced release of NE and DA from cardiac synaptosomes and PC12 cells, respectively, were shifted downward, indicating a PKG involvement in the exocytotic effects of BNP (Figure 5A).
Figure 5.

PKG dependence of the catecholamine-releasing effect of BNP. A. Concentration-response curves for the NE- and DA-releasing effect of BNP (0.1–100 nM) in synaptosomes isolated from guinea pig hearts (panel a) and NGF-differentiated PC12 cells (panel b) in the absence (control) or presence of the PKG inhibitor Rp-8-Br-cGMPS (30 and 0.5 μM in panel a and b, respectively). Points are means (± SEM; a, n=10–16; b, n=7–11). *, ** and ***, P<0.05, P<0.01 and P<0.001 respectively, from corresponding control point by unpaired t-test. Basal NE and DA levels were 1.35 ± 0.02 and 6.98 ± 0.81 pmol/mg protein, n=22 and 10, respectively. B. PKG activity in PC12 cells in response to BNP (100 nM) in the absence (control) or presence of the guanylyl cyclase inhibitor (LY83583; 10 μM) or the PKG inhibitor (Rp-8-Br-cGMPS; 0.5 μM), and in response to 8-Br-cGMP (1μM). Upper strip, representative immunoblot of PC12 cell lysate probed with anti-phosphorylated VASP (a major substrate for PKG) antibody. Lower strip, same immunoblot probed with anti-β-actin antibody. Columns (means ± SEM; n = 4–9) represent quantitative data. ** and ***, P=0.0012 and P=0.0004 respectively, from control by unpaired t-test. #, P<0.05 from BNP by unpaired t-test. C. intracellular levels of cAMP in PC12 cells in response to BNP (100 nM) and cGMP analogs (8-pCPT-cGMP; 3 μM, 8-Br-cGMP; 1 μM) in the absence or presence of PKG inhibitor (Rp-8-Br-cGMPS; 0.5 μM); positive control: forskolin (10 μM). Columns are means ± SEM (n = 4–11). ***, P<0.001 from control by unpaired t-test. #, ## and ###, P=0.0302, P=0.0016 and P<0.0001 respectively, from respective controls by unpaired t-test. D. PKA activity in PC12 cells in response to BNP (100 nM), cGMP analogs (8-pCPT-cGMP; 3 μM, 8-Br-cGMP; 1 μM) or forskolin (10 μM) in the absence or presence of PKG inhibitor (Rp-8-Br-cGMPS; 0.5 μM). Upper strips, representative immunoblots of PC12 cell lysate probed with anti-phosphorylated PKA antibody. Lower strips, same immunoblots probed with anti-β-actin antibody. Columns (means ± SEM, n = 5–9) represent quantitative data. * and ***, P=0.0107 and P<0.01 from control by unpaired t-test. # and ##, P=0.0481, and P<0.01 respectively, from own controls by unpaired t-test.
In fact, BNP (100 nM) elicited a ~60% increase in PKG activity of PC12 cell lysate, which was prevented by the PKG inhibitor (0.5 μM) and by an inhibitor of guanylyl cyclase, LY83583 (10 μM).39 As a positive control, 8-bromo-cGMP (1 μM) increased PKG activity by ~50% (Figure 5B). Furthermore, BNP (100 nM) caused a ~2.5-fold increase in cAMP level in PC12 cells, which was mimicked by both 8-bromo-cGMP (1 μM) and 8-pCPT-cGMP (3 μM), but prevented by the PKG inhibitor (0.5 μM). In contrast, the forskolin-induced ~30-fold increase in cAMP in PC12 cells (10 μM) was unaffected by PKG inhibition (Figure 5C). Similarly, BNP (100 nM), 8-bromo-cGMP (1 μM) and 8-pCPT-cGMP (3 μM) each caused a ~90% increase in PKA activity in PC12 cells which was prevented by the PKG inhibitor (0.5 μM). In contrast, the ~90% increase in PKA activity induced by forskolin (10 μM) was unmodified by PKG inhibition (Figure 5D). Collectively, these data suggested that the BNP-induced promotion of catecholamine exocytosis involves an increase in PKG activity followed by a rise in intracellular cAMP and augmented PKA activity.
The catecholamine-releasing effect of BNP depends on an increase in intracellular cAMP resulting from an inhibition of PDE3-induced cAMP hydrolysis
Since cGMP is known to inhibit PDE3,4 we next investigated whether the cAMP-dependent pro-exocytotic effects of BNP may result from PDE3 inhibition. We first used insulin and cilostamide, known to stimulate and inhibit PDE3, respectively.40;41 We found that insulin (30 nM and 100 nM) markedly inhibited the increase in NE and DA release induced by BNP (100 nM) in cardiac synaptosomes and PC12 cells, respectively, and that the effect of insulin was reversed by cilostamide (10 μM) (Figure 6A).
Figure 6.

The catecholamine-releasing effect of BNP depends on PDE3 inhibition. A. PDE3 activation with insulin (30 nM and 100 nM in synaptosomes and PC12 cells, respectively) attenuates BNP (100 nM)-induced increase in NE and DA in synaptosomes isolated from guinea pig hearts (panel a) and NGF-differentiated PC12 cells (panel b). Pretreatment with the PDE3 inhibitor cilostamide (10 μM; Cilo) prevents the insulin-mediated attenuation of the NE-releasing effect of BNP. Columns are means (± SEM; a, n=12–16; b, n=10–17). ***, P<0.001 from BNP, ## and ###, P=0.0011 and P<0.0001 respectively, from the combination of BNP, insulin and cilostamide, by unpaired t-test. Basal NE and DA levels were 1.35 ± 0.02 and 6.44 ± 0.43 pmol/mg protein, n=22 and 10, respectively. B. intracellular levels of cAMP in PC12 cells in response to BNP (100 nM) in the absence or presence of insulin (100 nM). Pretreatment with cilostamide (10 μM; Cilo) prevents the insulin-mediated attenuation of the cAMP-enhancing effect of BNP. Columns are means ± SEM (n = 9–26). ***, P<0.0001 from control; ##, P=0.0059 from BNP; ††, P=0.0027 from the combination of BNP, insulin and cilostamide, by unpaired t-test. C. BNP (100 nM) decreases PDE3 activity (expressed as the rate of cAMP hydrolyzed in terms of nmol/minute/mg protein) in PC12 cells, PKG inhibitor (Rp-8-Br-cGMPS; 0.5 μM) blocks the PDE3-inhibiting effect of BNP. Insulin (100 nM) and cilostamide (10 μM) alone are controls. Combination of BNP (100 nM) and insulin (100 nM) blocks the PDE3-inhibiting effect of BNP, pretreatment with cilostamide (10 μM) restores the PDE3-inhibiting effect of BNP. Columns are means (± SEM; n=7–10). ***, P<0.0001 from control; ###, P<0.0001 from BNP; †††, P=0.0001 from the combination of BNP and insulin, by unpaired t-test. D. PKA activity in PC12 cells in response to BNP (100 nM) in the absence or presence of insulin (100 nM). Pretreatment with cilostamide (10 μM) prevents the insulin-mediated attenuation of the PKA-enhancing effect of BNP. Forskolin (10 μM) is the positive control. Upper strip, representative immunoblot of PC12 cell lysate probed with anti-phosphorylated PKA antibody. Lower strip, same immunoblot probed with anti-β-actin antibody. Columns (means ± SEM, n = 5–9) represent quantitative data. ***, P<0.0001 from control; ###, P<0.0001 from BNP; †††, P<0.0001 from the combination of BNP, insulin and cilostamide, by unpaired t-test.
As these findings indirectly suggested a BNP-mediated inhibition of PDE3, we next measured PDE3 activity in PC12 cell lysate and its response to BNP. As expected,40;41 insulin (100 nM) stimulated PDE3 activity by ~60 %, whereas cilostamide (10 μM) inhibited it by ~30 % (Figure 6C). Similar to cilostamide, BNP (100 nM) also inhibited PDE3 activity by ~30 %, an effect that was prevented by the PKG inhibitor Rp-8-Br-cGMPS (0.5 μM) (Figure 6C). Stimulation of PDE3 with insulin (100 nM) prevented the inhibitory effect of BNP, an effect that was abolished by cilostamide (10 μM) (Figure 6C). These findings suggested that BNP inhibits PDE3 in a PKG-dependent manner.
Having demonstrated that BNP inhibits PDE3 activity, we next assessed the effect of this action on cellular cAMP level. We found that BNP (100 nM) enhanced cAMP level in PC12 cells by ~120 % (Figure 6B). PDE3 stimulation with insulin (100 nM) prevented the BNP-induced increase in cAMP, an effect that was reversed by inhibition of PDE3 with cilostamide (10 μM) (Figure 6B). Similarly, BNP (100 nM) enhanced PKA activation by ≥3 fold and this effect was prevented by insulin (100 nM) but restored by cilostamide (10 μM) (Figure 6D). Notably, the BNP-induced increase in PKA activity was similar to that elicited by forskolin (10 μM), used here as a positive control (Figure 6D). Collectively, these findings indicated that, by inhibiting PDE3, BNP sequentially enhances intracellular cAMP levels, PKA activity, Ca2+ content and catecholamine exocytosis.
Discussion
The purpose of this study was to characterize a novel and unsuspected property of natriuretic peptides; i.e., the promotion of NE exocytosis. We report that not only natriuretic peptides, but also SNP and cell-permeable cGMP analogs all elicit catecholamine exocytosis. This effect is mediated by an increase in intracellular Ca2+, and associated with an increase in intracellular cAMP and PKA activity resulting from a PKG-mediated inhibition of PDE3 (Figure 7).
Figure 7.
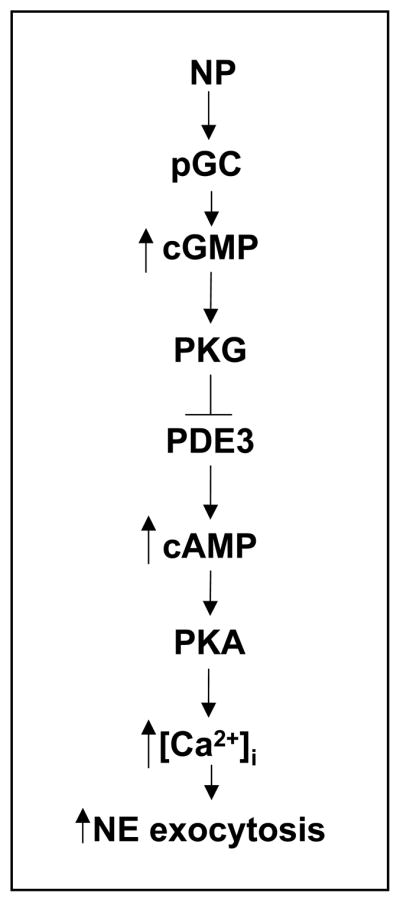
Natriuretic peptides elicit Ca2+-dependent norepinephrine exocytosis from cardiac sympathetic nerves via a cGMP-PKG-mediated inhibition of PDE3-induced cAMP hydrolysis. NP, natriuretic peptides; pGC, particulate guanylyl cyclase; PKG, protein kinase G; PDE3, phosphodiesterase type-3; PKA, protein kinase A; [Ca2+]i, intracellular calcium; NE, norepinephrine.
What prompted our investigation were the previous experimental evidence that ANP administration increases infarct size and mortality in mice with coronary ligation,6 and the recent clinical report that the administration of nesiritide, a recombinant form of BNP, failed to affect rehospitalization and risk of death in patients with acute decompensated heart failure.9 These findings conflicted with the generally held view that natriuretic peptides afford beneficial cardiac and hemodynamic effects.3;8 We questioned whether the lack of clinical efficacy of natriuretic peptides may derive from enhanced sympathetic activation and NE release, given that natriuretic peptides increase intracellular cGMP,4 and that injection of a membrane-permeable cGMP analog increases heart rate before any changes in blood pressure.14
BNP and ANP significantly enhanced the basal release of NE and DA in cardiac synaptosomes and PC12 cells with EC50’s ≅20 and 5 nM, respectively. Notably, patients with NYHA class III heart failure have mean BNP plasma levels of 2–3 nM.42 Considering that BNP is produced by ventricular myocytes,4;8;10 it is very likely that in advanced cardiac failure high concentrations of BNP will be present at the interface between ventricular myocytes and sympathetic nerve endings and reach levels capable of promoting NE release, particularly if ischemia is present, thus exacerbating heart failure symptoms. Hence, our findings in the ex vivo heart subjected to ischemia/reperfusion, isolated sympathetic nerve endings and PC12 cells are compatible with what may occur in human pathophysiology.
In as much as most actions of natriuretic peptides are mediated by the guanylyl cyclase-cGMP-PKG pathway,4;43 we investigated whether their catecholamine–releasing effects are also mediated by this signaling pathway. We found that PKG inhibition not only antagonized the catecholamine-releasing effects of BNP in cardiac synaptosomes and PC12 cells, but also attenuated the BNP-induced increase in PKG activity, and reduced the increase in intracellular cAMP, PKA activation and Ca2+ content. These findings suggest that PKG activation is a pivotal step in the BNP-induced stimulation of the cAMP-PKA pathway culminating in the Ca2+-mediated exocytotic process. Indeed, PKA inhibition markedly diminished the catecholamine-releasing effect of 8-bromo-cGMP and so did the N- and L-type Ca2+ channel inhibitors, ω-CTX34 and nifedipine35, respectively. Yet, since the intracellular Ca2+ chelator BAPTA-AM44 also greatly reduced the catecholamine-releasing effect of 8-bromo-cGMP, the relative contribution of Ca2+ entry via voltage-gated channels and of Ca2+ release from the endoplasmic reticulum remains unclear. Activation of the phospholipase C-phosphatidylinositol-IP3 pathway could also contribute to the increase in intracellular Ca2+ which promotes the NE-releasing effect of natriuretic peptides. In fact, an interaction of ANP with its C-receptor was found to activate phospholipase Cβ3 via Gβγ i. 45
cAMP is an important intracellular messenger in the regulation of neurotransmitter release in the mesenteric vasculature.46 Among the various phosphodiesterases which hydrolyze cAMP, PDE3 is known to be inhibited by cGMP.47;48 Since we found that the pro-exocytotic effects of BNP are associated with a PKG-dependent increase in intracellular cAMP, PKA activity and Ca2+ content, we questioned whether this increase might be due to a cGMP/PKG-induced inhibition of PDE3. Other investigators had reported that the guanylyl cyclase-cGMP pathway regulates cAMP levels in atrial myocytes via PDE3 inhibition.49 Indeed, we found that incubation of PC12 cells with BNP decreased PDE3 activity, and this effect was prevented by PKG inhibition. Moreover, stimulation of PDE3 markedly reduced the catecholamine–releasing effect of BNP, and the associated increase in PKA activity and intracellular Ca2+, effects that were all reversed by inhibition of PDE3. Therefore, it is likely that the pro-exocytotic effect of BNP involves an increase in intraneuronal cAMP resulting from a cGMP-PKG-mediated inhibition of PDE3. It is conceivable that cGMP might also directly enhance PKA activity as previously demonstrated in PKG-deficient mice,50 although the mechanisms remain uncertain.47
Interestingly, PDE3 activity is significantly reduced in failing human hearts and murine hearts with chronic pressure overload.51 Thus, the findings that nesiritide failed to alleviate decompensated heart failure9 could be explained by a further BNP-induced PDE3 inhibition. This would ultimately result in additional redundant NE release, thus preventing natriuretic peptides from correcting the symptoms of CHF.
In conclusion, our findings demonstrate that natriuretic peptides, in a concentration range likely to be reached at cardiac sympathetic nerve endings in advanced CHF, promote Ca2+-dependent NE release via a PKG-induced inhibition of PDE3-mediated cAMP hydrolysis. Thus, despite the traditional view that natriuretic peptides are cardioprotective,3;8 our findings may help explaining the conclusions of the recent large clinical trial that treatment with recombinant BNP fails to protect heart failure patients from rehospitalization and death.9 Indeed, long-term inhibition of PDE3 was previously shown to be associated with a 40% increase in mortality, primarily as a result of arrhythmias and sudden death,52;53 which our findings may now help ascribe to excessive sympathetic activation. The corollary of these and our investigations is that agents that preserve PDE3 function, rather than inhibiting it, may offer an alternative approach to treat cardiac dysfunctions associated with excessive sympathetic activity.
Acknowledgments
Funding sources: This work was supported by NIH grants HL034215, HL47073, and by a Pharmaceutical Research and Manufacturers of America Foundation Pre-doctoral Fellowship (Noel Yan-Ki Chan). We thank Paul J. Christos, DrPH for statistical help, partially supported by the Clinical Translational Science Center Grant UL1-RR024996.
Abbreviations
- ANP
atrial natriuretic peptide
- BNP
brain natriuretic peptide
- Ca2+
calcium
- CHF
congestive heart failure
- DA
dopamine
- NE
norepinephrine
- NGF
nerve-growth factor
- PDE3
phosphodiesterase type 3
- PKA
protein kinase A
- PKG
Protein kinase G
- SNP
sodium nitroprusside
Footnotes
Conflict of Interest Disclosures: None
References
- 1.Anand IS, Fisher LD, Chiang YT, Latini R, Masson S, Maggioni AP, Glazer RD, Tognoni G, Cohn JN. Changes in brain natriuretic peptide and norepinephrine over time and mortality and morbidity in the Valsartan Heart Failure Trial (Val-HeFT) Circulation. 2003;107:1278–1283. doi: 10.1161/01.cir.0000054164.99881.00. [DOI] [PubMed] [Google Scholar]
- 2.Braunwald E. Biomarkers in heart failure. N Engl J Med. 2008;358:2148–2159. doi: 10.1056/NEJMra0800239. [DOI] [PubMed] [Google Scholar]
- 3.Molkentin JD. A friend within the heart: natriuretic peptide receptor signaling. J Clin Invest. 2003;111:1275–1277. doi: 10.1172/JCI18389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Potter LR, Abbey-Hosch S, Dickey DM. Natriuretic peptides, their receptors, and cyclic guanosine monophosphate-dependent signaling functions. Endocr Rev. 2006;27:47–72. doi: 10.1210/er.2005-0014. [DOI] [PubMed] [Google Scholar]
- 5.Burley DS, Baxter GF. B-type natriuretic peptide at early reperfusion limits infarct size in the rat isolated heart. Basic Res Cardiol. 2007;102:529–541. doi: 10.1007/s00395-007-0672-1. [DOI] [PubMed] [Google Scholar]
- 6.Houng AK, McNamee RA, Kerner A, Sharma P, Mohamad A, Tronolone J, Reed GL. Atrial natriuretic peptide increases inflammation, infarct size and mortality after experimental coronary occlusion. Am J Physiol Heart Circ Physiol. 2009;296:H655–H661. doi: 10.1152/ajpheart.00684.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Izumi T, Saito Y, Kishimoto I, Harada M, Kuwahara K, Hamanaka I, Takahashi N, Kawakami R, Li Y, Takemura G, Fujiwara H, Garbers DL, Mochizuki S, Nakao K. Blockade of the natriuretic peptide receptor guanylyl cyclase-A inhibits NF-kappaB activation and alleviates myocardial ischemia/reperfusion injury. J Clin Invest. 2001;108:203–213. doi: 10.1172/JCI12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Munagala VK, Burnett JC, Jr, Redfield MM. The natriuretic peptides in cardiovascular medicine. Curr Probl Cardiol. 2004;29:707–769. doi: 10.1016/j.cpcardiol.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 9.O’Connor CM, Starling RC, Hernandez AF, Armstrong PW, Dickstein K, Hasselblad V, Heizer GM, Komajda M, Massie BM, McMurray JJ, Nieminen MS, Reist CJ, Rouleau JL, Swedberg K, Adams KF, Jr, Anker SD, Atar D, Battler A, Botero R, Bohidar NR, Butler J, Clausell N, Corbalan R, Costanzo MR, Dahlstrom U, Deckelbaum LI, Diaz R, Dunlap ME, Ezekowitz JA, Feldman D, Felker GM, Fonarow GC, Gennevois D, Gottlieb SS, Hill JA, Hollander JE, Howlett JG, Hudson MP, Kociol RD, Krum H, Laucevicius A, Levy WC, Mendez GF, Metra M, Mittal S, Oh BH, Pereira NL, Ponikowski P, Wilson WH, Tanomsup S, Teerlink JR, Triposkiadis F, Troughton RW, Voors AA, Whellan DJ, Zannad F, Califf RM. Effect of nesiritide in patients with acute decompensated heart failure. N Engl J Med. 2011;365:32–43. doi: 10.1056/NEJMoa1100171. [DOI] [PubMed] [Google Scholar]
- 10.Daniels LB, Maisel AS. Natriuretic peptides. J Am Coll Cardiol. 2007;50:2357–2368. doi: 10.1016/j.jacc.2007.09.021. [DOI] [PubMed] [Google Scholar]
- 11.Murad F. Shattuck Lecture. Nitric oxide and cyclic GMP in cell signaling and drug development. N Engl J Med. 2006;355:2003–2011. doi: 10.1056/NEJMsa063904. [DOI] [PubMed] [Google Scholar]
- 12.Greenberg SS, Cantor E, Diecke FP, Peevy K, Tanaka TP. Cyclic GMP modulates release of norepinephrine from adrenergic nerves innervating canine arteries. Am J Hypertens. 1991;4:173–176. doi: 10.1093/ajh/4.2.173. [DOI] [PubMed] [Google Scholar]
- 13.Herring N, Zaman JA, Paterson DJ. Natriuretic peptides like NO facilitate cardiac vagal neurotransmission and bradycardia via a cGMP pathway. Am J Physiol Heart Circ Physiol. 2001;281:H2318–H2327. doi: 10.1152/ajpheart.2001.281.6.H2318. [DOI] [PubMed] [Google Scholar]
- 14.Whalen EJ, Saurer TB, Johnson AK, Lewis SJ. Intracellular cGMP may promote Ca2+-dependent and Ca2+-independent release of catecholamines from sympathetic nerve terminals. Vascul Pharmacol. 2006;45:102–111. doi: 10.1016/j.vph.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 15.Yamamoto R, Wada A, Asada Y, Yuhi T, Yanagita T, Niina H, Sumiyoshi A. Functional relation between nitric oxide and noradrenaline for the modulation of vascular tone in rat mesenteric vasculature. Naunyn Schmiedebergs Arch Pharmacol. 1994;349:362–366. doi: 10.1007/BF00170881. [DOI] [PubMed] [Google Scholar]
- 16.Brunner-La Rocca HP, Kaye DM, Woods RL, Hastings J, Esler MD. Effects of intravenous brain natriuretic peptide on regional sympathetic activity in patients with chronic heart failure as compared with healthy control subjects. J Am Coll Cardiol. 2001;37:1221–1227. doi: 10.1016/s0735-1097(01)01172-x. [DOI] [PubMed] [Google Scholar]
- 17.O’Tierney PF, Tse MY, Pang SC. Elevated renal norepinephrine in proANP gene-disrupted mice is associated with increased tyrosine hydroxylase expression in sympathetic ganglia. Regul Pept. 2007;143:90–96. doi: 10.1016/j.regpep.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 18.Wang L, Henrich M, Buckler KJ, McMenamin M, Mee CJ, Sattelle DB, Paterson DJ. Neuronal nitric oxide synthase gene transfer decreases [Ca2+]i in cardiac sympathetic neurons. J Mol Cell Cardiol. 2007;43:717–725. doi: 10.1016/j.yjmcc.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 19.Cubeddu L, Barnes E, Weiner N. Release of norepinephrine and dopamine-beta-hydroxylase by nerve stimulation. IV. An evaluation of a role for cyclic adenosine monophosphate. J Pharmacol Exp Ther. 1975;193:105–127. [PubMed] [Google Scholar]
- 20.Yanagihara N, Okazaki M, Terao T, Uezono Y, Wada A, Izumi F. Stimulatory effects of brain natriuretic peptide on cyclic GMP accumulation and tyrosine hydroxylase activity in cultured bovine adrenal medullary cells. Naunyn Schmiedebergs Arch Pharmacol. 1991;343:289–295. doi: 10.1007/BF00251128. [DOI] [PubMed] [Google Scholar]
- 21.Martire M, Altobelli D, Cannizzaro C, Preziosi P. Effects of nitric oxide donors on basal and K+-evoked release of [3H]noradrenaline from rat cerebral cortex synaptosomes. Eur J Pharmacol. 1998;350:345–351. doi: 10.1016/s0014-2999(98)00269-6. [DOI] [PubMed] [Google Scholar]
- 22.Kalman JM, Munawar M, Howes LG, Louis WJ, Buxton BF, Gutteridge G, Tonkin AM. Atrial fibrillation after coronary artery bypass grafting is associated with sympathetic activation. Ann Thorac Surg. 1995;60:1709–1715. doi: 10.1016/0003-4975(95)00718-0. [DOI] [PubMed] [Google Scholar]
- 23.Chang CM, Wu TJ, Zhou SM, Doshi RN, Lee MH, Ohara T, Fishbein MC, Karagueuzian KS, Chen PS, Chen LS. Nerve sprouting and sympathetic hyperinnervation in a canine model of atrial fibrillation produced by prolonged right atrial pacing. Circulation. 2001;103:22–25. doi: 10.1161/01.cir.103.1.22. [DOI] [PubMed] [Google Scholar]
- 24.Schomig A. Catecholamines in myocardial ischemia. Systemic and cardiac release. Circulation. 1990;82:II13–II22. [PubMed] [Google Scholar]
- 25.Meredith IT, Broughton A, Jennings GL, Esler MD. Evidence of a selective increase in cardiac sympathetic activity in patients with sustained ventricular arrhythmias. N Engl J Med. 1991;325:618–624. doi: 10.1056/NEJM199108293250905. [DOI] [PubMed] [Google Scholar]
- 26.Levi R, Smith NCE. Histamine H3-receptors: A new frontier in myocardial ischemia. J Pharmacol Exp Ther. 2000;292:825–830. [PubMed] [Google Scholar]
- 27.Seyedi N, Mackins CJ, Machida T, Reid AC, Silver RB, Levi R. Histamine H3-receptor-induced attenuation of norepinephrine exocytosis: a decreased PKA activity mediates a reduction in intracellular calcium. J Pharmacol Exp Ther. 2005;312:272–280. doi: 10.1124/jpet.104.072504. [DOI] [PubMed] [Google Scholar]
- 28.Morrey C, Estephan R, Abbott GW, Levi R. Cardioprotective Effect of Histamine H3-Receptor Activation: Pivotal Role of Gbg-Dependent Inhibition of Voltage-Operated Ca2+ Channels. J Pharmacol Exp Ther. 2008;326:871–878. doi: 10.1124/jpet.108.137919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Geiger J, Nolte C, Butt E, Sage SO, Walter U. Role of cGMP and cGMP-dependent protein kinase in nitrovasodilator inhibition of agonist-evoked calcium elevation in human platelets. Proc Natl Acad Sci U S A. 1992;89:1031–1035. doi: 10.1073/pnas.89.3.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hernandez L, Park KH, Cai SQ, Qin L, Partridge N, Sesti F. The antiproliferative role of ERG K+ channels in rat osteoblastic cells. Cell Biochem Biophys. 2007;47:199–208. doi: 10.1007/s12013-007-0006-9. [DOI] [PubMed] [Google Scholar]
- 31.Gill RM, Braz JC, Jin N, Etgen GJ, Shen W. Restoration of impaired endothelium-dependent coronary vasodilation in failing heart: role of eNOS phosphorylation and CGMP/cGK-I signaling. Am J Physiol Heart Circ Physiol. 2007;292:H2782–H2790. doi: 10.1152/ajpheart.00831.2006. [DOI] [PubMed] [Google Scholar]
- 32.Farrow KN, Groh BS, Schumacker PT, Lakshminrusimha S, Czech L, Gugino SF, Russell JA, Steinhorn RH. Hyperoxia increases phosphodiesterase 5 expression and activity in ovine fetal pulmonary artery smooth muscle cells. Circ Res. 2008;102:226–233. doi: 10.1161/CIRCRESAHA.107.161463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci U S A. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sher E, Biancardi E, Passafaro M, Clementi F. Physiopathology of neuronal voltage-operated calcium channels. FASEB J. 1991;5:2677–2683. doi: 10.1096/fasebj.5.12.1655547. [DOI] [PubMed] [Google Scholar]
- 35.Vater W, Kroneberg G, Hoffmeister F, Saller H, Meng K, Oberdorf A, Puls W, Schlossmann K, Stoepel K. Pharmacology of 4-(2′-nitrophenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylic acid dimethyl ester (Nifedipine, BAY a 1040) Arzneimittelforschung. 1972;22:1–14. [PubMed] [Google Scholar]
- 36.Harrison SM, Bers DM. The effect of temperature and ionic strength on the apparent Ca-affinity of EGTA and the analogous Ca-chelators BAPTA and dibromo-BAPTA. Biochim Biophys Acta. 1987;925:133–143. doi: 10.1016/0304-4165(87)90102-4. [DOI] [PubMed] [Google Scholar]
- 37.Glass DB, Cheng HC, Mende-Mueller L, Reed J, Walsh DA. Primary structural determinants essential for potent inhibition of cAMP-dependent protein kinase by inhibitory peptides corresponding to the active portion of the heat-stable inhibitor protein. J Biol Chem. 1989;264:8802–8810. [PubMed] [Google Scholar]
- 38.Moretto M, Lopez FJ, Negro-Vilar A. Nitric oxide regulates luteinizing hormone-releasing hormone secretion. Endocrinology. 1993;133:2399–2402. doi: 10.1210/endo.133.5.8104781. [DOI] [PubMed] [Google Scholar]
- 39.Vives D, Farage S, Motta R, Lopes AG, Caruso-Neves C. Atrial natriuretic peptides and urodilatin modulate proximal tubule Na(+)-ATPase activity through activation of the NPR-A/cGMP/PKG pathway. Peptides. 2010;31:903–908. doi: 10.1016/j.peptides.2010.02.018. [DOI] [PubMed] [Google Scholar]
- 40.Watanabe T, Satoo H, Kohara K, Takami R, Motoyashiki T, Morita T, Ueki H. Orthovanadate stimulates cAMP phosphodiesterase 3 activity in isolated rat hepatocytes through mitogen-activated protein kinase activation dependent on cAMP-dependent protein kinase. Biol Pharm Bull. 2004;27:789–796. doi: 10.1248/bpb.27.789. [DOI] [PubMed] [Google Scholar]
- 41.Hidaka H, Hayashi H, Kohri H, Kimura Y, Hosokawa T, Igawa T, Saitoh Y. Selective inhibitor of platelet cyclic adenosine monophosphate phosphodiesterase, cilostamide, inhibits platelet aggregation. J Pharmacol Exp Ther. 1979;211:26–30. [PubMed] [Google Scholar]
- 42.Pfisterer M, Buser P, Rickli H, Gutmann M, Erne P, Rickenbacher P, Vuillomenet A, Jeker U, Dubach P, Beer H, Yoon SI, Suter T, Osterhues HH, Schieber MM, Hilti P, Schindler R, Brunner-La Rocca HP. BNP-guided vs symptom-guided heart failure therapy: the Trial of Intensified vs Standard Medical Therapy in Elderly Patients With Congestive Heart Failure (TIME-CHF) randomized trial. JAMA. 2009;301:383–392. doi: 10.1001/jama.2009.2. [DOI] [PubMed] [Google Scholar]
- 43.Hofmann F, Bernhard D, Lukowski R, Weinmeister P. cGMP Regulated Protein Kinases (cGK) Handb Exp Pharmacol. 2009:137–162. doi: 10.1007/978-3-540-68964-5_8. [DOI] [PubMed] [Google Scholar]
- 44.Tsien RY. A non-disruptive technique for loading calcium buffers and indicators into cells. Nature. 1981;290:527–528. doi: 10.1038/290527a0. [DOI] [PubMed] [Google Scholar]
- 45.Murthy KS, Makhlouf GM. Identification of the G protein-activating domain of the natriuretic peptide clearance receptor (NPR-C) J Biol Chem. 1999;274:17587–17592. doi: 10.1074/jbc.274.25.17587. [DOI] [PubMed] [Google Scholar]
- 46.Mutafova-Yambolieva VN, Smyth L, Bobalova J. Involvement of cyclic AMP-mediated pathway in neural release of noradrenaline in canine isolated mesenteric artery and vein. Cardiovasc Res. 2003;57:217–224. doi: 10.1016/s0008-6363(02)00648-x. [DOI] [PubMed] [Google Scholar]
- 47.Rybalkin SD, Yan C, Bornfeldt KE, Beavo JA. Cyclic GMP phosphodiesterases and regulation of smooth muscle function. Circ Res. 2003;93:280–291. doi: 10.1161/01.RES.0000087541.15600.2B. [DOI] [PubMed] [Google Scholar]
- 48.Zaccolo M, Movsesian MA. cAMP and cGMP signaling cross-talk: role of phosphodiesterases and implications for cardiac pathophysiology. Circ Res. 2007;100:1569–1578. doi: 10.1161/CIRCRESAHA.106.144501. [DOI] [PubMed] [Google Scholar]
- 49.Wen JF, Cui X, Jin JY, Kim SM, Kim SZ, Kim SH, Lee HS, Cho KW. High and low gain switches for regulation of cAMP efflux concentration: distinct roles for particulate GC- and soluble GC-cGMP-PDE3 signaling in rabbit atria. Circ Res. 2004;94:936–943. doi: 10.1161/01.RES.0000123826.70125.4D. [DOI] [PubMed] [Google Scholar]
- 50.Sausbier M, Schubert R, Voigt V, Hirneiss C, Pfeifer A, Korth M, Kleppisch T, Ruth P, Hofmann F. Mechanisms of NO/cGMP-dependent vasorelaxation. Circ Res. 2000;87:825–830. doi: 10.1161/01.res.87.9.825. [DOI] [PubMed] [Google Scholar]
- 51.Ding B, Abe Ji, Wei H, Huang Q, Walsh RA, Molina CA, Zhao A, Sadoshima J, Blaxall BC, Berk BC, Yan C. Functional Role of Phosphodiesterase 3 in Cardiomyocyte Apoptosis: Implication in Heart Failure. Circulation. 2005;111:2469–2476. doi: 10.1161/01.CIR.0000165128.39715.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Packer M, Carver JR, Rodeheffer RJ, Ivanhoe RJ, DiBianco R, Zeldis SM, Hendrix GH, Bommer WJ, Elkayam U, Kukin ML. Effect of oral milrinone on mortality in severe chronic heart failure. The PROMISE Study Research Group. N Engl J Med. 1991;325:1468–1475. doi: 10.1056/NEJM199111213252103. [DOI] [PubMed] [Google Scholar]
- 53.Nony P, Boissel JP, Lievre M, Leizorovicz A, Haugh MC, Fareh S, de Breyne B. Evaluation of the effect of phosphodiesterase inhibitors on mortality in chronic heart failure patients. A meta-analysis. Eur J Clin Pharmacol. 1994;46:191–196. doi: 10.1007/BF00192547. [DOI] [PubMed] [Google Scholar]