

Abstract
Vulnerable plaque remains clinically undetectable, and there is no accepted in vitro model. We characterize the calcific nodules produced by calcifying vascular cells (CVC) in ApoE-null mice, demonstrating increased destabilization of cultured nodules in the presence of oxidized low-density lipoprotein (oxLDL) and monocytes under pulsatile shear stress. CVC implanted in the subcutaneous space of hyperlipidemic mice produced nodules revealing features of calcific atherosclerotic plaque including a fibrous cap, cholesterol clefts, thin shoulder, lipids, and calcium mineral deposits. CVC nodules seeded in the pulsatile flow channel (τavg = 23 dyn/cm2, ∂τ/∂t = 71 dyn·cm−2·s−1) underwent deformation and destabilization. Computational fluid dynamics revealed distinct shear force profiles on the nodules. Presence of oxLDL or monocytic THP-1 cells significantly increased the numbers of nodules destabilized from the substrate. Both oxLDL and THP-1 increased matrix metalloproteinase (MMP) activity in CVC. The MMP inhibitor GM6001 significantly reversed oxLDL- and THP-1-induced nodule destabilization, whereas overexpression of MMP-9 increased destabilization. These findings demonstrate that CVC-derived nodules resembled calcific atherosclerotic plaque and were destabilized in the presence of active lipids and monocytes via induction of MMPs.
Keywords: atherosclerosis, plaque rupture, calcifying vascular cells, calcification
vascular calcification develops in arterial regions prone to disturbed flow and oxidative stress. As an actively regulated process, vascular cells differentiate to osteoblast-like phenotype (34). Oxidative stress induces Bmp-Msx2-Wnt signaling pathway to regulate the pathogenesis of vascular calcification (6, 29, 36). In human aortic valve stenosis, dysregulation of antioxidant genes contributes to increased oxidative stress and valvular calcification (28). In the intima, calcification has been identified as a distinct, but relevant process to atherosclerosis (1, 37, 38, 42), beginning as early as the second decade of life, soon after fatty streak formation, and increases with age and lesion progression (39).
The vascular cells that appear to be responsible for producing calcium mineral undergo chondrogenic, leiomyogenic (smooth muscle), and stromogenic (marrow stromal) lineage acquisition in culture (5). In embryogenesis, immature mesenchymal cells aggregate into patterns and differentiate to form mature tissues. Later in life, this process may be recapitulated: raised cellular aggregates form atherosclerotic lesions in a patchy distribution throughout the vascular system and the cardiac valve leaflets (41). In many such lesions, architecturally complete, ectopic bone tissue arises, often even including bone marrow (46). While intimal calcification is clearly associated with atherosclerosis, its role on mechanical plaque stability remains controversial.
Atherosclerotic plaque rupture typically occurs at the shoulder regions (24). This phenomenon may be due to concentration of solid tensile stress at a thin edge or due to dissection by fluid shear stress under the edges of lesions. Calcification may contribute by introducing compliance mismatch (9). We previously showed that calcifying vascular cells (CVC, with characteristics of vascular mesenchymal stem cells) form raised nodules when grown on glass or plastic substrates and expressed collagenous extracellular matrix and mineral deposits within 6–10 days (5). These calcifying nodules detached from their substrate in proportion to the magnitude of shear over a range of 4.9 to 400 dyn/cm2 (27).
These nodules have also been likened to atherosclerotic plaque. Gimbrone and Cotran (14) first reported that advanced smooth muscle cell cultures exhibit focal proliferation and secrete excess extracellular matrix, giving rise to “nodular protrusions resembling atherosclerotic lesions” in vitro. In this context, detachment of calcified nodules in response to shear forces provides an entry point to characterize mechanical destabilization of calcific atherosclerotic plaque in the presence of oxidative stress and inflammatory responses.
In the current study, we hypothesized that CVC nodules resemble calcific atherosclerotic plaque and are destabilized in the presence of monocytes and oxidized low-density lipoprotein (oxLDL) via increasing matrix metalloproteinase (MMP) activity under mechanical shear force. We characterized calcific nodules implanted in ApoE-null mice and quantified in vitro nodule destabilization from the substrate or extracellular matrix in response to metabolically active lipids and monocytes under a well-defined hemodynamic shear stress simulating that found in the common carotid arteries (2). Implanted CVC nodules resembled calcified atherosclerotic plaque, harboring thin fibrous cap, collagen type II, smooth muscle cells, and lipids. Calcific nodules were destabilized in the presence of oxLDL or coculture with monocytic THP-1 cells. Both oxLDL and monocytes activated MMP, rendering the calcific nodules prone to destabilization.
MATERIALS AND METHODS
Materials and reagents.
THP-1 cells were obtained from American Type Culture Collection. DMEM and RPMI-1640 were purchased from Invitrogen. Fetal bovine serum (FBS) was purchased from Phenix Research. GM6001 was obtained from Santa Cruz Biotechnology. Real-time PCR reagents were obtained from Applied Biological Materials. Human LDL was kindly provided by Dr. Judith Berliner (Atherosclerosis Research Unit, Univ. of California, Los Angeles). Preparation of oxLDL was performed as previously described (40). Control and recombinant human MMP-9 adenoviruses were kindly provided by Dr. Jack Gauldie (McMaster Univ., Hamilton, ON, Canada).
Cell culture.
CVC, derived by dilutional cloning from bovine aortic smooth muscle cell cultures (5), were maintained in DMEM with 15% FBS. CVC were seeded onto glass slides at 100,000 cells/cm2 and cultured for 3–6 days to allow nodule formation in preparation for exposure to dynamic flow conditions. Monocytic THP-1 cells were cultured in RPMI with 10% FBS supplemented with 50 μM β-mercaptoethanol.
CVC implantation in hyperlipidemic ApoE-null mice.
CVC (1 × 106 cells) were injected into individual 0.2 μm pore-size diffusion chambers, which selectively allowed for interstitial fluid exchange without cellular ingress or egress. Chambers were sealed with methylmethacrylate and implanted into the subcutaneous space of four control (C57BL6 mice) and four hyperlipidemic ApoE-null recipient mice using sterile technique. After 8 wk of regular chow diet, the chambers were removed and specimens were embedded in OCT. Frozen sections were examined by standard histochemical and immunohistochemical staining. All of the protocols were approved by the Institutional Animal Care and Use Committee and done in accordance with the Guiding Principles in the Care and Use of Animals of the American Physiological Society.
CVC nodules in the pulsatile shear stress system.
A LabView-driven stepper motor was programmed to expose CVC to fluid shear in well-defined flow patterns simulating the shear stress profile found in human common carotid arteries (2, 20). The system provides precise, reproducible flow profiles across the width of the chamber with physiological temporal variations in shear stress (∂τ/∂t), frequency, and amplitude (35). The pulsatile flow system is composed of a NEMA 34 stepper motor (model N32HRLG-LEK-M2–00, Pacific Scientific), stepper drive (model P70530, National Instruments), universal motion interface (model UMI-7772, National Instruments), and peristaltic pumps (model HV-77200–60, Cole-Parmer). Time-averaged flow rate was monitored downstream by a coriolis flow meter (model MASS 2100 DI 1.5, Siemens).
CVC grown on glass slides (2.5 cm × 1 cm) for 3 days to allow for nodule formation were then preincubated in the presence or absence of 5 × 105/ml of monocytic THP-1 cells or 50 μg/ml of oxLDL for 3 days. The CVC-derived nodules were exposed to pulsatile shear stress (PSS) at a time-averaged shear stress (τave) of 23 dyn/cm2, a temporal gradient (∂τ/∂t) of 71 dyn·cm−2·s−1, and a pulse pressure (ΔP) of 10 psi for 2.5 h. The flow system was placed in a tissue culture incubator providing 5% CO2 as well as physiological temperature (37°C) and pH (7.4). Nodule deformation and destabilization in response to shear stress were captured with a JENOPTIK Progres C3 digital camera under an Olympus IX70 microscope. The total numbers of nodules on the glass slides were quantified under microscope before and after PSS exposure. Nodule destabilization is expressed in terms of percentage of nodules detached from the glass slides for quantitative purposes.
Computational fluid dynamic simulation.
Computational fluid dynamic (CFD) code was developed to recapitulate shear force distribution (3) on the calcific nodules. Generation of three-dimensional (3-D) geometries and meshes was reconstructed in Solidworks (Concord, MA) based on the measured dimensions. To characterize the fluid velocity gradients near the substrate and nodules, we constructed fine-mesh sizes immediately adjacent to the channel wall and nodular contours. The flow field was modeled by applying the 3-D Navier-Stokes equations. The governing equations, including mass and momentum equations, were solved for laminar, unsteady, incompressible, and non-Newtonian flow. The mass flux, calculated based on the steady flow rates, was applied as the inlet boundary condition and implemented in Solidworks Flow Simulation. The inlet Reynolds number (Re) was calculated from the mean velocity at the inlet.
MMP expression and activity assays.
CVC nodules grown in six-well plates were incubated in the presence or absence of monocytic THP-1 cells (5 × 105/ml) or oxLDL (50 μg/ml) for 1–3 days in DMEM-5% FBS. Media supernatants were collected for MMP activity assays and zymogram. The cells were lysed in RNA isolation lysis buffer for measurement of MMP expression by quantitative RT-PCR. MMP activity was measured using the Sensolyte Generic MMP Assay Kit (AnaSpec), and the arbitrary units of MMP activity were calculated, per the manufacturer's instructions, using reference positive controls.
Zymogram.
The supernatant of medium (15 μl) was mixed with 2× loading buffer (Bio-Rad) and loaded into 4–16% Blue Casein zymogram gel (Invitrogen). After running at 125 V for 1.5 h at room temperature, the gel was incubated with zymogram renaturing buffer and rinsed with zymogram developing buffer. The gel was then incubated at 37°C for 36 h. After drying, the gel was scanned into image and the band intensity as indication of MMP activity was quantified with ImageJ software (National Institutes of Health, Bethesda, MD).
Overexpression of MMP-9.
CVC were grown for 3 days on glass slides and then infected with control (Adv-DL) or recombinant MMP-9 adenoviruses (Adv-MMP-9) at multiplicity of infection (MOI) of 1:100. The cells were allowed to grow for additional 3 days before shear stress exposure as described above.
Measurement of MMP expression.
MMP expression was measured by quantitative RT-PCR as previously described (26). Total RNA was isolated using the Aurum Total RNA Isolation Kit (Bio-Rad) following the manufacturer's instructions. Potential genomic DNA contamination was removed with on-column DNase I digestion. Approximately 0.5–1 μg of total RNA was reverse-transcribed with Bio-Rad's iScript cDNA synthesis kit. The primers were designed on the basis of bovine MMP sequences, and thus preferentially detected MMP expression in CVC. The primer sequences were as follows: bMMP1: forward, ATGGGAAGAAAGTTGAAAGGCAGAGAAACG and reverse, GGGCTGCTTCATCACATTCAGGGTTTC; bMMP2: forward, CGCCGTCGCCCATCATCAAATTT and reverse, TTGGGGCAGCCGTAGAAGGTGTTT; bMMP3: forward, CCTTCCGATTCTGCTGTTGCTATGTGTG and reverse, TGTCTTCCTTGTCCCTTGCAGCCC; bMMP9: forward, GCACCACCACAACATCACCTACTGGATC and reverse, ATGTCAGCTTCGGGGCCGTACACT; and bGAPDH: forward, ATGGTGAAGGTCGGAGTGAACGGATT and reverse, GCGACGATGTCCACTTTGCCAGAA.
Statistical analysis.
Data are expressed as means ± SD. Multiple comparisons were made by one-way analysis of variance (ANOVA), and statistical significance for pairwise comparison was determined using the Tukey test. Values of P < 0.05 were considered statistically significant.
RESULTS
CVC-derived calcifying nodules resembled calcified atherosclerotic plaque in vivo.
CVC is a subpopulation of smooth muscle cells from the medial layer of the artery wall producing raised nodules in tissue culture over a 1- to 3-wk period. These nodules ranged from 20 to 2,000 μm in diameter (with average size in the 100- to 200-μm range) and were positively stained by the von Kossa technique demonstrating calcification in the shoulder regions (27). When CVC, within diffusion chambers, were subcutaneously implanted in ApoE-null mice, they produced atherosclerotic plaque-like structures (Fig. 1, A and B). These CVC-derived structures included fibrous caps and cholesterol clefts (Fig. 1B). The friable shoulder regions revealed an osteoblast-like differentiation of CVC (Fig. 1C) as demonstrated by von Kossa staining (Fig. 1D). These structures were positively stained for collagen type II (Fig. 2A), alkaline phosphatase (Fig. 2B), smooth muscle α-actin (Fig. 2C), and lipids (Fig. 2D). Thus, the CVC-derived calcific nodules harbored histological features of calcific atherosclerotic plaque.
Fig. 1.
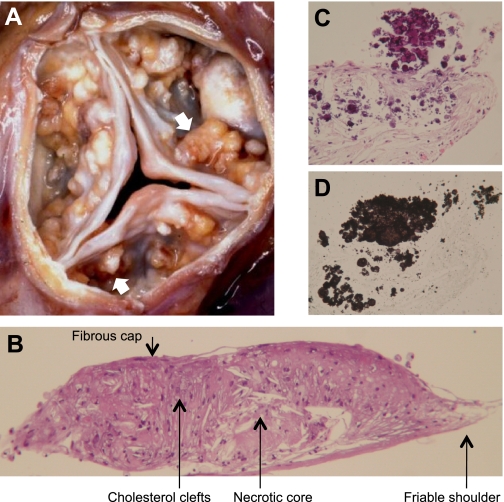
Calcific atherosclerotic nodules. A: gross pathology of human calcific aortic valve stenosis, showing numerous calcific nodules (arrows) on the aortic face of the valve leaflets (kindly provided by Michael Fishbein, UCLA School of Medicine). B: histochemical staining of calcifying vascular cell (CVC) nodule produced in vivo within a subcutaneous diffusion chamber implanted in a hyperlipidemic ApoE-deficient mouse. C and D: high-magnification images of serial sections from the shoulder region revealing calcium mineral deposits by hematoxylin and eosin (C) and von Kossa staining (D). Nodules measure approximately 0.5–2 mm in diameter.
Fig. 2.
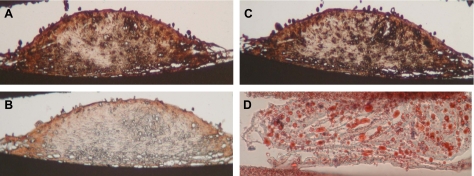
Characterization of CVC-derived calcific nodules. A–D: immunohistochemical and histochemical staining of cryosections of in vivo CVC nodules using antibodies for collagen type II (A), alkaline phosphatase (B), smooth muscle α-actin (C), and Oil Red O for lipids (D).
CVC-derived nodules deformed and destabilized under shear stress.
CVC nodules deformed in response to PSS (mean τave = 23 dyn/cm2; temporal gradient ∂τ/∂t = 71 dyn·cm·−2·s−1 at 1 Hz) (Supplemental Video S1; Supplemental Material for this article is available online at the Journal website). Time-lapse microvideography showed nodules undergoing deformation over 0.5-s to 260-s periods (Fig. 3). Extended exposure of shear stress led to destabilization of the nodules, culminating in detachment from the substrate (Supplemental Video S2).
Fig. 3.
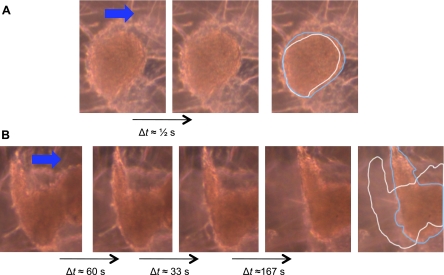
Videomicrographic sequences of CVC nodule deformation in response to pulsatile shear stress. CVC were cultured for 6 days and exposed to pulsatile shear stress simulating flow in the common carotid artery [time-averaged shear stress (τave) = 23 dyn/cm2; temporal gradient (∂τ/∂t) = 71 dyn·cm−2·s−1]. A: deformation of nodule over ∼0.5 s. B: destabilization of nodule over ∼260 s. The nodule eventually detached from the slide (Supplemental Video S2). The rightmost panel of each sequence is a schematic of the morphological deformation showing outlines of the original (white line) and final (blue line) nodule morphology.
Computational fluid dynamic reconstruction of the pulsatile flow field illustrated a stream of velocity vectors with a range of magnitudes and directions (Fig. 4A). Mean shear forces were accentuated upstream of the nodules corresponding with the shoulder regions where nodular deformation and detachment were initialized (Fig. 4B, Supplemental Video S2). Instantaneous shear stress profiles for an individual nodule further revealed shear stress mismatch at the interface between the shoulders and substrates (Fig. 4C). A plot depicting the magnitude of shear stress over a given nodule showed the variations in stress in relation to the aspect ratio of the nodule (Fig. 4D). This analysis provides an in silico model of calcific atherosclerotic plaque disruption under pulsatile flow.
Fig. 4.
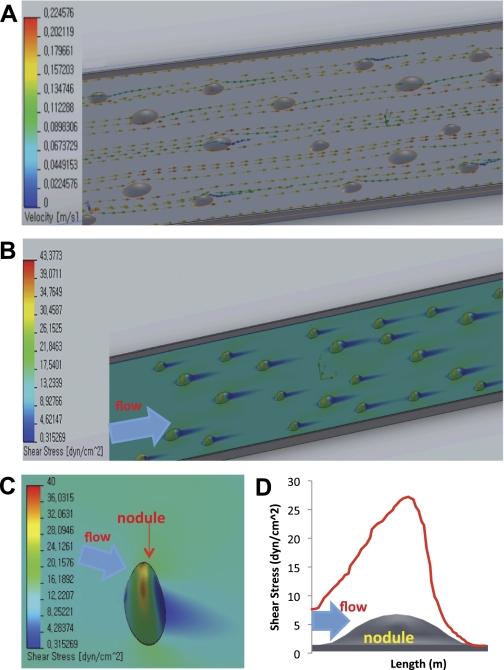
Computer fluid dynamical (CFD) simulation of shear force distribution on CVC nodules under pulsatile shear stress. A: instantaneous stream of velocity vectors in the flow field in relation to the CVC nodules. B: shear stress concentration on the shoulder regions of nodules. C: instantaneous distribution of shear stress on an individual nodule in relation to the substrate. D: the distribution of mean shear stress on the single nodule surface area of an individual nodule. Mean shear stress on individual nodule is 16.6 dyn/cm2.
OxLDL and monocytic THP-1 cells promoted the destabilization of CVC-derived nodules.
Metabolically active components of thin cap fibrous atheroma include biologically active lipoprotein particles and monocytes/macrophages, both of which impart oxidative stress in rupture-prone lesions (4, 15, 32). Here, we sought to quantify the destabilization of calcified CVC nodules by a well-defined PSS in the presence of oxLDL or THP-1 monocytes. Coculture of CVC nodules with THP-1 cells significantly increased the numbers of nodules detached by pulsatile flow from 27 ± 6% to 56 ± 6%, P < 0.05, n = 3 (see Fig. 7A). Similarly, oxLDL treatment also increased nodule detachment from 17 ± 4% to 28 ± 2%, P < 0.05, n = 3 (see Fig. 7B). These findings suggest that active lipids and monocytes/macrophages promote destabilization of calcified nodules.
Fig. 7.
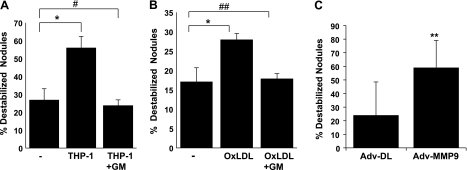
MMPs mediated THP-1 cells and oxLDL-induced CVC nodule instability. CVC nodules grown on glass slides were cocultured with or without THP-1 cells (A) or treated with or without 50 μg/ml of oxLDL for 3 days in the presence or absence of an MMP inhibitor, GM6001 (GM) (B), or infected with recombinant MMP-9 adenovirus (Adv-MMP-9) or control adenovirus (Adv-DL) at multiplicity of 100 for 3 days (C). The nodules were then exposed to pulsatile shear stress. Numbers of detached nodules were calculated as percentage of total nodules (*P < 0.05, n = 3; **P < 0.05, n = 5; #P = 0.42 vs. control, n = 3; ##P = 0.80 vs. control, n = 3).
OxLDL and THP-1 upregulated MMP activity in CVC.
To test whether induction of MMPs contribute to the mechanism of destabilization, we analyzed MMP activity. Both oxLDL treatment and THP-1 cell treatment significantly induced MMP activity compared with control (CVC = 4.9 ± 0.5, THP-1 = 4.5 ± 2.1, CVC + THP-1 = 13.8 ± 4.1, P < 0.01 vs. CVC; CVC + oxLDL = 9.7 ± 2.6, *P < 0.05 vs. CVC, n = 4) (Fig. 5A). To verify changes in MMP activity by THP-1 and oxLDL treatment, we performed casein zymogram assay as a sensitive measurement for stromelysin such as MMP-3 (stromelysin 1) by using the media supernatants of treatments. A single band of 50- to 60-kDa range, presumably MMP-3, was shown in the zymogram. In agreement with the MMP activity assay, THP-1 and oxLDL increased MMP-3 activity in the zymogram (Fig. 5B). Furthermore, coculture of CVC with THP-1 cells significantly increased MMP-1, MMP-3, and MMP-9, but decreased MMP-2 mRNA expression in a time-dependent manner (Fig. 6A, P < 0.05, n = 3). OxLDL treatment also regulated MMP expression to a similar extent (Fig. 6B, P < 0.05, n = 3), suggesting that active lipids and monocytes destabilize calcific nodules via MMPs.
Fig. 5.

Matrix metalloproteinase (MMP) activity in CVC exposed to THP-1 cells or oxidized LDL (oxLDL). CVC grown in 6-well plates were cocultured with THP-1 cells or treated with 50 μg/ml of oxLDL for 3 days. A: MMP activity was assayed in the supernatant as described in materials and methods (#P < 0.01, *P < 0.05, n = 3). B: media supernatants were used to run casein zymogram. OxLDL and THP-1 increased casein-degrading activity in zymogram.
Fig. 6.

Effects of THP-1 cells and oxLDL on MMP expression in CVC. CVC grown in 6-well plates were cocultured with or without THP-1 cells for 1, 2, or 3 days (A) or treated with or without 50 μg/ml of oxLDL for 3 days (B). Expression of MMPs was measured by quantitative RT-PCR (*P < 0.05, vs. control; n = 3).
OxLDL- and THP-1-induced MMP activity mediated the destabilization of CVC nodules.
To corroborate the role of MMPs in nodule destabilization, we cocultured CVC nodules with THP-1 cells or treated the nodules with oxLDL in the presence or absence of an MMP inhibitor, GM6001 (10 μM), and quantified nodule destabilization under PSS. MMP inhibition completely abrogated THP-1-induced CVC nodule destabilization (Fig. 7A; nodules detached: control = 27 ± 6%, THP-1 = 56 ± 6%, P < 0.05 vs. control; THP-1 + GM6001 = 24 ± 3%, P = 0.42 vs. control, n = 3). MMP inhibitors also reversed oxLDL-mediated nodule destabilization to a similar extent (control = 17 ± 4%, oxLDL = 28 ± 2%, P < 0.05 vs. control, oxLDL + GM6001 = 18 ± 1%, P = 0.80 vs. control, n = 3) (Fig. 7B). Hence, monocytes and oxLDL promoted destabilization of calcified nodules via an elevated MMP activity. To further demonstrate the destabilizing role of MMP, CVC nodules were infected with MMP-9 adenovirus (Adv-MMP-9) for overexpression of MMP-9 or with control adenovirus (Adv-DL). MMP-9 overexpression significantly increased nodule destabilization as compared with control (Fig. 7C, nodule detached: Adv-DL = 24 ± 24.5%, Adv-MMP-9 = 59.1 ± 19.9%, P < 0.05, n = 5).
DISCUSSION
This study provides a novel in vitro model of calcific nodule destabilization and characterizes its mechanical vulnerability to programmed fluid shear stress in response to metabolically active lipids and macrophages. This model, consisting of CVC-derived nodules, when grown in vivo, demonstrated histological features characteristic of calcific atherosclerotic plaque, including a fibrous cap, cholesterol clefts, foam cells, thin shoulder, lipids, collagen II, alkaline phosphatase, and calcium mineral deposits. Computational fluid dynamic (CFD) reconstruction further provided hemodynamic profiles for the individual nodules, demonstrating shear stress mismatch at the interface between the nodules and substrates. While both THP-1 cells and oxLDL upregulated MMP expression and activity in CVC nodules, blocking of MMP activity with GM6001 completely reversed nodule destabilization. Artificially overexpressing MMP-9 in CVC further accentuated nodule destabilization. Taken together, these findings provide new biomechanical insights into the interplay between active metabolic factors and calcification in a dynamic environment leading to calcific plaque destabilization.
Vascular cells implanted into mice via the diffusion chamber technique engendered organized mesenchymal tissue including bone and cartilage (10). Our in vitro model of atherosclerotic plaque was generated by CVC, also known as vascular mesenchymal cells, a subpopulation of smooth muscle cells from the medial layer of the aortic wall (5, 43). In addition to showing the histological features of atherosclerotic plaques, CVC-derived nodules also expressed a wide range of genes and gene products typical of calcified atherosclerotic plaque, including osteopontin, fibronectin, α-actin, caldesmon, bone morphogenetic protein-2, osteonectin, osteocalcin, collagen types I and III, laminin, matrix GLA protein, and cytokines. Previously, the cells were termed calcifying vascular cells (CVC) because of their ability to calcify spontaneously, a process requiring about 6–10 days and accelerated by treatment with β-glycerophosphate (5 mM).
It has been suggested that plaque instability is caused by a dynamic imbalance in oxidative stress (17), inflammation (18, 45), and proteolytic activity (11). Fluid shear stress promotes mechanical failure when plaque is raised, focal, and eccentric (16, 18). We previously showed that CVC-derived calcific nodules detach in response to a ramping magnitude in shear stress (27). In the present study, we used videomicroscopy to capture real-time nodule deformation and destabilization in response to PSS at a constant time-averaged shear stress. We applied CFD to demonstrate shear force distribution, highlighting the shear mismatch between the shoulder regions of the nodule and the substrate [extracellular matrix (ECM)].
These vascular mesenchymal cells, originally termed calcifying vascular cells (CVC), undergo osteoblastic differentiation, express bone-specific proteins, and produce ECM that incorporates hydroxyapatite mineral under regulation of developmental genes. We also demonstrated that these cells are also multipotent, with the capacity for chondrogenic, leiomyogenic (smooth muscle), and stromogenic (marrow stromal) lineage acquisition. Given their substantial capacity for self-renewal, we proposed that they are closely related to mesenchymal stem cells (7, 33). Interestingly, these cells self-organize into patterns of regularly spaced nodular aggregates or ridges in a reaction-diffusion process governed by morphogens: bone morphogenetic protein-2 and its inhibitor (12). These findings suggest that the artery wall contains mesenchymal stem cells with lineage plasticity and the capacity for self-organization, thus accounting for both ectopic tissues in atherosclerotic arteries as well as the long-known patchy distribution of atherosclerosis. In the present study, we also provide evidence that CVC-derived nodules are a relevant model for calcified atherosclerotic plaque.
Oxidized LDL and monocyte-macrophages are metabolically active components of mechanically unstable plaque (4, 15, 32). Here, we showed that monocytes/macrophages and oxLDL promote nodule destabilization under physiological shear stress. Despite interexperimental variations due to passage number or degree of calcification, both THP-1 monocytic cells and oxLDL consistently destabilized the calcified nodules. Thus, the CVC nodules responded to the metabolically active factors in a similar manner as in vivo atherosclerotic plaque would.
MMPs, proteolytic enzymes that break down extracellular matrix, such as that produced by CVC nodules on a glass substrate (27), are implicated in tissue remodeling, angiogenesis, and tumor metastasis. They are produced by CVC and play an important role in plaque stability (22, 31). Dual roles of MMP in initial thickening and late-stage rupture of atherosclerotic plaque have been documented (23, 30, 31). Increased MMP activity in the advanced plaque may be responsible for the thinning of the fibrous cap that renders the necrotic core prone to rupture (13, 15, 25). In our in vitro model, oxLDL and monocyte treatment increased overall MMP activity and increased the expression of MMP-1 (collagenase 1), MMP-3 (stromelysin 1), and MMP-9 (gelatinase B), but decreased MMP-2 (gelatinase A) expression. The latter is consistent with the finding of Wilson et al. (44) that mildly oxidized LDL decreased MMP-2 expression. However, Haug et al. (19) found the opposite with oxidized LDL in vascular smooth muscle cells. In macrophages derived from a U937 cell line, oxLDL was reported to upregulate MMP-2 expression, suggesting that the effects of oxLDL on MMP-2 expression may be cell-type and cell-stage specific. Despite a decrease in MMP-2 expression, the total MMP activity increased in response to oxLDL and THP-1, at least in part because of increased expression by CVC. The mechanism of how THP-1 and oxLDL regulate the expression of MMP in CVC remains an interesting question for future study. While MMP traditionally have been thought to function primarily in the degradation of extracellular matrix molecules, recent data suggest that MMPs may also function as important regulators of matrix biology, inflammation, and osteogenesis including in the control of arterial calcification (21). Mechanical interactions of cells with their matrix and environment, including cell-generated traction and matrix stiffness, strongly influence vascular pathology, including the development of calcification. Thus, MMPs effects on the mechanical strength of extracellular matrix may influence vascular cell calcification, and conversely, calcification may alter cell-generated traction and MMP expression (8).
While numerous in vivo studies have revealed a dual role for MMP in atherogenesis and plaque rupture, preclinical and clinical studies of plaque stability have demonstrated mixed results for modulating MMP expression and activities (30). In our studies, inhibition of MMP activities completely blocked oxLDL- and monocyte-induced nodule instability, whereas overexpression of MMP-9 accentuated its instability. Thus, MMP may predominately destabilize advanced calcific plaques. In sum, the phenotypic and biomechanical similarities between CVC-derived nodules and calcific atherosclerotic plaque provide a novel in vitro model to assess the interplay between metabolically active stressors and calcific atherosclerotic plaque. This well-defined hemodynamic system further reveals that oxLDL and monocytes may simulate clinical plaque instability via increased expression of matrix remodeling metalloproteinases.
GRANTS
This project was supported by grants from the National Institutes of Health National Heart Lung and Blood Institute HL-091302 (T. K. Hsiai), HL-083015 (T. K. Hsiai), and HL-081202 (L. L. Demer) and the National Institute of Diabetes and Digestive and Kidney Diseases DK-081346 (Y. Tintut).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: R.L., Y.T., L.L.D., and T.H. conception and design of research; R.L., D.M., K.F., R.M., and Y.T. performed experiments; R.L., D.M., Y.T., L.L.D., and T.H. analyzed data; R.L., J.L., Y.T., L.L.D., and T.H. interpreted results of experiments; R.L., J.L., and Y.T. prepared figures; R.L. drafted manuscript; R.L., D.M., K.F., Y.T., L.L.D., and T.H. edited and revised manuscript; R.L., L.L.D., and T.H. approved final version of manuscript.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Jack Gauldie (McMaster Univ., Hamilton, ON, Canada) for providing us with the control and human MMP-9 adenovirus, and Dr. Jennifer Robert for amplifying and purifying the adenoviruses.
REFERENCES
- 1.Abedin M, Tintut Y, Demer LL. Vascular calcification: mechanisms and clinical ramifications. Arterioscler Thromb Vasc Biol 24: 1161–1170, 2004 [DOI] [PubMed] [Google Scholar]
- 2.Ai L, Rouhanizadeh M, Wu JC, Takabe W, Yu H, Alavi M, Li R, Chu Y, Miller J, Heistad DD, Hsiai TK. Shear stress influences spatial variations in vascular Mn-SOD expression: implication for LDL nitration. Am J Physiol Cell Physiol 294: C1576–C1585, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ai L, Zhang L, Dai W, Hu C, Shung KK, Hsiai TK. Real-time assessment of flow reversal in an eccentric arterial stenotic model. J Biomech 43: 2678–2683, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barascuk N, Skjøt-Arkil H, Register TC, Larsen L, Byrjalsen I, Christiansen C, Karsdal MA. Human macrophage foam cells degrade atherosclerotic plaques through cathepsin K mediated processes. BMC Cardiovasc Disord 10: 19, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bostrom K, Watson KE, Horn S, Wortham C, Herman IM, Demer LL. Bone morphogenetic protein expression in human atherosclerotic lesions. J Clin Invest 91: 1800–1809, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Byon CH, Javed A, Dai Q, Kappes JC, Clemens TL, Darley-Usmar VM, McDonald JM, Chen Y. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J Biol Chem 283: 15319–15327, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Campagnoli C, Roberts IA, Kumar S, Bennett PR, Bellantuono I, Fisk NM. Identification of mesenchymal stem/progenitor cells in human first-trimester fetal blood, liver, and bone marrow. Blood 98: 2396–2402, 2001 [DOI] [PubMed] [Google Scholar]
- 8.Chen JH, Simmons CA. Cell-matrix interactions in the pathobiology of calcific aortic valve disease: critical roles for matricellular, matricrine, and matrix mechanics cues. Circ Res 108: 1510–1524, 2011 [DOI] [PubMed] [Google Scholar]
- 9.Demer LL, Tintut Y. Vascular calcification: pathobiology of a multifaceted disease. Circulation 117: 2938–2948, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farrington-Rock C, Crofts NJ, Doherty MJ, Ashton BA, Griffin-Jones C, Canfield AE. Chondrogenic and adipogenic potential of microvascular pericytes. Circulation 110: 2226–2232, 2004 [DOI] [PubMed] [Google Scholar]
- 11.Galis ZS, Khatri JJ. Matrix metalloproteinases in vascular remodeling and atherogenesis: the good, the bad, and the ugly. Circ Res 90: 251–262, 2002 [PubMed] [Google Scholar]
- 12.Garfinkel A, Tintut Y, Petrasek D, Bostrom K, Demer LL. Pattern formation by vascular mesenchymal cells. Proc Natl Acad Sci USA 101: 9247–9250, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.George SJ, Dwivedi A. MMPs, cadherins, and cell proliferation. Trends Cardiovasc Med 14: 100–105, 2004 [DOI] [PubMed] [Google Scholar]
- 14.Gimbrone MA, Jr, Cotran RS. Human vascular smooth muscle in culture. Growth and ultrastructure. Lab Invest 33: 16–27, 1975 [PubMed] [Google Scholar]
- 15.Gough PJ, Gomez IG, Wille PT, Raines EW. Macrophage expression of active MMP-9 induces acute plaque disruption in apoE-deficient mice. J Clin Invest 116: 59–69, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Griendling KK, FitzGerald GA. Oxidative stress and cardiovascular injury: Part II: animal and human studies. Circulation 108: 2034–2040, 2003 [DOI] [PubMed] [Google Scholar]
- 17.Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res 86: 494–501, 2000 [DOI] [PubMed] [Google Scholar]
- 18.Harrison DGK, Landmesser U, Hornig B, Drexler H. Role of oxidative stress in atherosclerosis. Am J Cardiol 91: 7A–11A, 2003 [DOI] [PubMed] [Google Scholar]
- 19.Haug C, Lenz C, Diaz F, Bachem MG. Oxidized low-density lipoproteins stimulate extracellular matrix metalloproteinase Inducer (EMMPRIN) release by coronary smooth muscle cells. Arterioscler Thromb Vasc Biol 24: 1823–1829, 2004 [DOI] [PubMed] [Google Scholar]
- 20.Hwang J, Ing MH, Salazar A, Lassegue B, Griendling K, Navab M, Sevanian A, Hsiai TK. Pulsatile versus oscillatory shear stress regulates NADPH oxidase subunit expression: implication for native LDL oxidation. Circ Res 93: 1225–1232, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Irwin CL, Guzman RJ. Matrix metalloproteinases in medial arterial calcification: potential mechanisms and actions. Vascular 17 Suppl 1: S40–S44, 2009 [DOI] [PubMed] [Google Scholar]
- 22.Johnson JL. Matrix metalloproteinases: influence on smooth muscle cells and atherosclerotic plaque stability. Expert Rev Cardiovasc Ther 5: 265–282, 2007 [DOI] [PubMed] [Google Scholar]
- 23.Johnson JL, George SJ, Newby AC, Jackson CL. Divergent effects of matrix metalloproteinases 3, 7, 9, and 12 on atherosclerotic plaque stability in mouse brachiocephalic arteries. Proc Natl Acad Sci USA 102: 15575–15580, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kolodgie FD, Katocs AS, Jr, Largis EE, Wrenn SM, Cornhill JF, Herderick EE, Lee SJ, Virmani R. Hypercholesterolemia in the rabbit induced by feeding graded amounts of low-level cholesterol. Methodological considerations regarding individual variability in response to dietary cholesterol and development of lesion type. Arterioscler Thromb Vasc Biol 16: 1454–1464, 1996 [DOI] [PubMed] [Google Scholar]
- 25.Lherke MGM, Broedl UC, Lebherz C, Laubender RP, Becker A, von Ziegler F, Tittus J, Reiser M, Becker C, Göke Steinback G, Leber AW, Parhofer KG. MMP-1 serum levels predict coronary atherosclerosis in humans. Cardiovasc Diabetol 8: 50, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li R, Ning Z, Cui J, Khalsa B, Ai L, Takabe W, Beebe T, Majumdar R, Sioutas C, Hsiai T. Ultrafine particles from diesel engines induce vascular oxidative stress via JNK activation. Free Radic Biol Med 46: 775–782, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin TC, Tintut Y, Lyman A, Mack W, Demer LL, Hsiai TK. Mechanical response of a calcified plaque model to fluid shear force. Ann Biomed Eng 34: 1535–1541, 2006 [DOI] [PubMed] [Google Scholar]
- 28.Miller JD, Chu Y, Brooks RM, Richenbacher WE, Pena-Silva R, Heistad DD. Dysregulation of antioxidant mechanisms contributes to increased oxidative stress in calcific aortic valvular stenosis in humans. J Am Coll Cardiol 52: 843–850, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mody N, Parhami F, Sarafian TA, Demer LL. Oxidative stress modulates osteoblastic differentiation of vascular and bone cells. Free Radic Biol Med 31: 509–519, 2001 [DOI] [PubMed] [Google Scholar]
- 30.Newby AC. Dual role of matrix metalloproteinases (matrixins) in intimal thickening and atherosclerotic plaque rupture. Physiol Rev 85: 1–31, 2005 [DOI] [PubMed] [Google Scholar]
- 31.Newby AC. Metalloproteinases and vulnerable atherosclerotic plaques. Trends Cardiovasc Med 17: 253–258, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nishi KIH, Uno M, Kitzato KT, Horiguchi H, Shinno K, Nagahiro S. Oxidized LDL in carotid plaques and plasma associates with plaque instability. Arterioscler Thromb Vasc Biol 22: 1649–1654, 2002 [DOI] [PubMed] [Google Scholar]
- 33.Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR. Multilineage potential of adult human mesenchymal stem cells. Science 284: 143–147, 1999 [DOI] [PubMed] [Google Scholar]
- 34.Rajamannan NM, Subramaniam M, Rickard D, Stock SR, Donovan J, Springett M, Orszulak T, Fullerton DA, Tajik AJ, Bonow RO, Spelsberg T. Human aortic valve calcification is associated with an osteoblast phenotype. Circulation 107: 2181–2184, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rouhanizadeh M, Soundararajan G, Lo R, Arcas D, Browand FK, Hsiai TK. MEMS sensors to resolve spatial variations in shear stress in a 3-D blood vessel bifurcation model. IEEE Sens J 6: 78–88, 2006 [Google Scholar]
- 36.Shao JS, Aly ZA, Lai CF, Cheng SL, Cai J, Huang E, Behrmann A, Towler DA. Vascular Bmp Msx2 Wnt signaling and oxidative stress in arterial calcification. Ann NY Acad Sci 1117: 40–50, 2007 [DOI] [PubMed] [Google Scholar]
- 37.Shao JS, Cai J, Towler DA. Molecular mechanisms of vascular calcification: lessons learned from the aorta. Arterioscler Thromb Vasc Biol 26: 1423–1430, 2006 [DOI] [PubMed] [Google Scholar]
- 38.Speer MY, Giachelli CM. Regulation of cardiovascular calcification. Cardiovasc Pathol 13: 63–70, 2004 [DOI] [PubMed] [Google Scholar]
- 39.Stary HC. The sequence of cell and matrix changes in atherosclerotic lesions of coronary arteries in the first forty years of life. Eur Heart J 11, Suppl E: 3–19, 1990 [DOI] [PubMed] [Google Scholar]
- 40.Takabe W, Li R, Ai L, Yu F, Berliner JA, Hsiai TK. Oxidized low-density lipoprotein-activated c-Jun NH2-terminal kinase regulates manganese superoxide dismutase ubiquitination: implication for mitochondrial redox status and apoptosis. Arterioscler Thromb Vasc Biol 30: 436–441, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tintut Y, Alfonso Z, Saini T, Radcliff K, Watson K, Bostrom K, Demer LL. Multilineage potential of cells from the artery wall. Circulation 108: 2505–2510, 2003 [DOI] [PubMed] [Google Scholar]
- 42.Vattikuti R, Towler DA. Osteogenic regulation of vascular calcification: an early perspective. Am J Physiol Endocrinol Metab 286: E686–E696, 2004 [DOI] [PubMed] [Google Scholar]
- 43.Watson KE, Bostrom K, Ravindranath R, Lam T, Norton B, Demer LL. TGF-beta 1 and 25-hydroxycholesterol stimulate osteoblast-like vascular cells to calcify. J Clin Invest 93: 2106–2113, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wilson D, Massaeli H, Pierce GN, Zahradka P. Native and minimally oxidized low density lipoprotein depress smooth muscle matrix metalloproteinase levels. Mol Cell Biochem 249: 141–149, 2003 [PubMed] [Google Scholar]
- 45.Witztum JR. The oxidation hypothesis of atherosclerosis. Lancet 344: 793–795, 1994 [DOI] [PubMed] [Google Scholar]
- 46.Wolf RL, Wehrli SL, Popescu AM, Woo JH, Song HK, Wright AC, Mohler ER, 3rd, Harding JD, Zager EL, Fairman RM, Golden MA, Velazquez OC, Carpenter JP, Wehrli FW. Mineral volume and morphology in carotid plaque specimens using high-resolution MRI and CT. Arterioscler Thromb Vasc Biol 25: 1729–1735, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.