

Abstract
Hyperandrogenism and chronic low-grade inflammation are related in polycystic ovary syndrome (PCOS), but it is unknown whether hyperandrogenemia can activate inflammation. We determined the effect of oral androgen administration on fasting and glucose-stimulated nuclear factor-κB (NF-κB) activation and expression and related markers of inflammation in mononuclear cells (MNC) of lean reproductive-age women. Sixteen lean, ovulatory reproductive-age women were treated with 130 mg of DHEA or placebo (n = 8 each) for 5 days in a randomized, controlled, double-blind fashion. Nuclear activation of NF-κB, p65 and p105 NF-κB subunit RNA, TNFα and IL-1β mRNA, and NF-κB p65 and inhibitory-κB (IκB) protein were quantified from MNC obtained while fasting and 2 h after glucose ingestion, before and after DHEA or placebo administration. Before treatment, subjects receiving DHEA or placebo exhibited no differences in androgens or any inflammatory markers while fasting and after glucose ingestion. Compared with placebo, DHEA administration raised levels of testosterone, androstenedione, and DHEA-S, increased the percent change in fasting and glucose-challenged activated NF-κB, p65, p105, TNFα, and IL-1β RNA and p65 protein, and decreased the percent change in fasting and glucose-challenged IκB protein. We conclude that elevation of circulating androgens to the range observed in PCOS upregulates the NF-κB inflammation pathway in lean reproductive-age women. Thus, hyperandrogenemia activates and sensitizes MNC to glucose in this population.
Keywords: androgens, nuclear factor-κB, hyperglycemia
the polycystic ovary syndrome (PCOS) is the most common female endocrinopathy, affecting between 8 and 12% of reproductive-age women (37). The disorder is characterized by hyperandrogenism, chronic oligo- or anovulation, and polycystic ovaries, with two out of these three findings required to diagnose PCOS (43a). Circulating androgen levels in PCOS are typically above the normal premenopausal female range based on common upper limit values [testosterone >70 ng/dl, dehydroepiandrosterone sulfate (DHEA-S) >300 μg/dl], which may vary depending on the assay used for measurement (10, 34). Whereas testosterone levels in the disorder are usually below the male range (testosterone >300 ng/dl), androgen levels near the androgen-producing tumor range (testosterone >200 ng/dl, DHEA-S >700 μg/dl) have been reported in severe cases (49, 50). As many as 70% of women with PCOS exhibit insulin resistance, with the compensatory hyperinsulinemia considered to be a promoter of the hyperandrogenism (9, 40). In addition, women with PCOS are often obese, which is strongly associated with insulin resistance and hyperglycemia (12, 33).
Hyperglycemia is proinflammatory due to its ability to generate reactive oxygen species (ROS) from circulating mononuclear cells (MNC). ROS-induced oxidative stress activates nuclear factor-κB (NF-κB), a transcription factor heterodimer typically consisting of the DNA-binding subunits p50 and p65 (6, 45). The p50 subunit in particular is derived from p105, a larger precursor that is transcribed and translated before being processed to yield p50 (18). In resting MNC, NF-κB is present in the cytoplasm complexed to its inhibitory protein, known as inhibitory-κB (IκB). Following activation, IκB dissociates from NF-κB and undergoes ubiquitination and degradation. NF-κB is freed to undergo nuclear translocation and subsequent DNA binding to promote transcription of TNFα, a known mediator of insulin resistance (8, 15, 28, 44). We have shown previously that in PCOS there is increased NF-κB activation and altered TNFα release from MNC following oral glucose ingestion independent of obesity (21, 22). Thus, nuclear translocation of activated NF-κB within MNC in response to hyperglycemia may serve as a cardinal inflammatory signal in the induction of insulin resistance in PCOS.
The cause of increased MNC sensitivity to hyperglycemia in PCOS is unknown. Our previous reports have demonstrated that, in PCOS, fasting and hyperglycemia-induced NF-κB activation and TNFα release from MNC are highly correlated with circulating androgens (23, 24, 27). These findings raise the possibility that hyperandrogenemia is capable of activating MNC of women with PCOS. In contrast, MNC of normal, lean, reproductive-age women are not sensitive to hyperglycemia and do not exhibit an inflammatory response to glucose challenge (21, 23, 24, 27).
We embarked on a double-blinded, placebo-controlled study to examine the effect of oral androgen administration on NF-κB nuclear activation in MNC of lean reproductive-age women in the fasting state and following glucose challenge. We also examined this effect on the mRNA content of NF-κB heterodimer subunits TNFα and IL-1β and the protein content of NF-κB p65 and IκB. We hypothesized that oral androgen administration increases NF-κB activation and NF-κB and cytokine gene expression and decreases IκB protein in MNC of lean reproductive-age women.
MATERIALS AND METHODS
Participants.
Sixteen lean, healthy women between 20 and 40 yr of age volunteered to participate in the study. All subjects had a body mass index (BMI) between 18 and 25 kg/m2 and were ovulatory, as evidenced by regular menses and a luteal-phase serum progesterone level >5 ng/ml. All subjects exhibited normal circulating androgen levels and had no evidence of hyperandrogenic skin manifestations or polycystic ovaries on ultrasound.
All subjects were screened for diabetes or inflammatory illnesses, and none were smokers or taking medications that would affect carbohydrate metabolism or immune function for ≥6 wk prior to study participation. None of the subjects were involved in any regular exercise program for ≥6 mo before study participation. The study was approved by the Mayo Clinic Institutional Review Board (IRB), and all subjects provided written informed consent in accordance with IRB guidelines for the protection of human subjects.
Study design.
Subjects were randomly assigned by a research pharmacist to receive a single, oral, daily 130-mg dose of micronized DHEA (n = 8; Spectrum Chemical and Laboratory Products, Gardena, CA) or identical placebo (n = 8) at 9 PM for 5 days. Participants and study personnel were blinded to the treatment assignment. All subjects underwent an oral glucose tolerance test (OGTT) beginning at 9 AM after an overnight fast of ∼12 h before and after treatment. The pretreatment OGTT was performed between days 5 and 8 after the onset of menses on the morning before DHEA or placebo was begun, and the posttreatment OGTT was performed on the morning after the 5 days of assigned treatment were completed. The women were provided with a healthy diet consisting of 50% carbohydrate, 35% fat, and 15% protein for 3 consecutive days before each OGTT. Body composition was assessed immediately before the initial OGTT.
OGTT.
All subjects ingested a 75-g glucose beverage. Blood samples were drawn while the subjects were fasting and 10, 20, 30, 60, 90, 120, and 180 min after glucose ingestion. Androgen trough levels were measured from the fasting samples. Glucose tolerance was assessed by World Heath Organization criteria (39). Area under the curve (AUC) for glucose and insulin was calculated using the trapezoidal rule (29). Insulin sensitivity was derived from glucose and insulin measurements throughout the entire 180 min of the OGTT by insulin sensitivity OGTT (ISOGTT) using the following formula: 10,000 ÷ (fasting glucose × fasting insulin) × (mean glucose × mean insulin) (38).
Body composition assessment.
Height without shoes and body weight were measured to the nearest 1.0 cm and 0.1 kg, respectively. Percent (%) total body fat, %truncal fat, and the ratio of truncal fat to leg fat were determined by dual-energy X-ray absorptiometry using a Prodigy model scanner (General Electric Healthcare, Little Chalfont, Buckinghamshire, UK). Truncal fat content was defined as the area between the dome of the diaphragm (cephalad limit) and the top of the greater trochanter (caudal limit) (46).
Molecular assays.
MNC were isolated from blood samples obtained during the OGTT at 0 and 120 min (2 h). Nuclear-bound NF-κB was quantified by electrophoretic mobility shift assay (EMSA), as described previously (26). The mRNA content of p50, p105, TNFα, and IL-1β was quantified by RT-PCR, as described previously (5). Primer Express software (PE Biosystems, Foster City, CA) was used to select the following primer and probe sequences: p65 (GenBank NM_021975): forward primer CATGCGCTTCCGCTACAAGT, reverse primer TGTGTAGCCATTGATCTTGATGGT, and probe CCAGGCGAGAGGAG; p105 (GenBank NM_003998): forward primer GTCACTCTAACGTATGCAACAGGAA, reverse primer TTGCAAGCTGCATAGCCTTCT, and probe AGCCTCTATCCATGGCTA; TNFα (GenBank NM_000594): forward primer CCCAGGCAGTCAGATCATCTTC, reverse primer GTTTGCTACAACATGGGCTACA, and probe CGAACCCCGAGTGACAA; and IL-1β (GenBank NM_000576): forward primer CCACCTCCAGGGACAGGAT, reverse primer TTGTCATTACTTTCTTCTCCTTGTACAAA, and probe AACAAGTGGTGTTCTCCA. A 28S rRNA signal was used to normalize against differences in RNA isolation and degradation and remained constant with DHEA treatment.
The protein content of p65, IκB, and actin was quantified by Western blotting (WB), as described previously (1), using a 1:1,000 dilution of a monoclonal antibody against p65 (Transduction Laboratories, San Diego, CA), IκB (Transduction Laboratories), or actin (Santa Cruz Biotechnology, Santa Cruz, CA). Densitometry following EMSA or WB was performed on scanned films using Carestream Molecular Imaging software version 5.0.2.30 (Rochester, NY), and all values for p65 and IκB were corrected for loading using those obtained for actin.
Plasma and serum measurements.
Plasma glucose was measured using a hexokinase reagent on a Roche Cobas c311 instrument (Roche Diagnostics, Indianapolis, IN). Plasma insulin and serum testosterone were measured by a two-site immunoenzymatic assay and a competitive chemiluminescent immunoassay, respectively, on a DxI 800 automated system (Beckman Instruments, Chaska, MN). Serum androstenedione was measured by liquid chromatography-triple quad mass spectrometry on an API 5000 system (Applied Biosystems, Foster City, CA). Serum DHEA-S was measured by a solid-phase, competitive, chemiluminescent immunoenzymatic assay on a Siemens Immulite 2000 instrument (Siemens Healthcare Diagnostics, Deerfield, IL). Plasma TNFα was measured by high-sensitivity ELISA (Quantikine; R & D Systems, Minneapolis, MN). Serum cortisol was measured by ELISA (R & D Systems). All samples from each subject were measured in duplicate in the same assay at the end of the study. The inter- and intra-assay coefficients of variation for all assays were ≤7.4 and 8%, respectively.
Statistics.
The StatView software package (SAS Institute, Cary, NC) was used for statistical analysis. Sample size was calculated based on an expected difference of ≥30% between groups in activated NF-κB with a SD of 22% and desired power of 80%, using our previous study as a reference (23). The primary end point was change from baseline in molecular markers of inflammation between DHEA and placebo groups. Secondary outcomes were also assessed as change from baseline within group. Data are presented as means ± SE. Unpaired (DHEA vs. placebo) and paired (before vs. after treatment) Student's t-tests were performed. In view of interindividual variability, %change from baseline to assess the response to DHEA or placebo was calculated for each participant from fasting values (0) before and after either treatment using the following formula: {[before (0) − after (0)] ÷ before (0)} × 100. %Change from baseline to assess the response to glucose ingestion was calculated from the fasting value (0) and the 2-h post-glucose ingestion value (2) for each OGTT before and after DHEA or placebo administration using the following formulas: {[before (0) − before (2)] ÷ before (0)} × 100; and {[after (0) − after (2)] ÷ after (0)} × 100. Correlation analyses were performed by Pearson linear regression using the method of least squares. Results were considered significant at a two-tailed α-level of 0.05.
RESULTS
Body composition, glycemic status, and insulin sensitivity.
Age, height, weight, BMI, %total body fat, %truncal fat, and the ratio of truncal fat to leg fat (<1.0) were similar in the group treated with DHEA compared with the placebo group (Table 1). All subjects had a normal glucose response during the OGTT, with fasting glucose levels <100 mg/dl and 2-h glucose levels ranging between 95 and 134 mg/dl, before and after treatment. Fasting levels and AUC for glucose and insulin and ISOGTT were similar in both groups and remained unchanged before and after DHEA or placebo administration. Glucose levels 2 h after glucose ingestion were similar in both groups before DHEA or placebo administration and exhibited a modest but significant (P < 0.04) decline after placebo compared with DHEA administration.
Table 1.
Age, body composition, glucose, insulin, and cortisol levels, and insulin sensitivity
| Placebo | DHEA | |
|---|---|---|
| Age, yr | 28 ± 2 | 28 ± 3 |
| Height, cm | 162.1 ± 2.7 | 164.2 ± 1.8 |
| Body weight, kg | 58.4 ± 2.9 | 62.0 ± 2.0 |
| Body mass index, kg/m2 | 23.0 ± 0.5 | 22.1 ± 0.4 |
| Total body fat, % | 35.8 ± 1.9 | 31.0 ± 2.7 |
| Truncal fat, % | 33.2 ± 2.3 | 29.0 ± 2.9 |
| Truncal fat/leg fat ratio | 0.97 ± 0.08 | 0.90 ± 0.03 |
| Pretreatment | ||
| Fasting glucose, mg/dl | 90 ± 2.1 | 85 ± 1.7 |
| 2-h Glucose, mg/dl | 118 ± 3 | 121 ± 5 |
| Glucose AUC, mg/dl × 180 min | 21,676 ± 510 | 21,849 ± 416 |
| Fasting insulin, μiU/ml | 4.2 ± 0.4 | 3.8 ± 0.2 |
| Insulin AUC, μiU/ml × 180 min | 4,664 ± 572 | 5,104 ± 485 |
| ISOGTT | 10.3 ± 0.9 | 10.6 ± 0.9 |
| Cortisol, μg/dl | 22.0 ± 2.2 | 21.8 ± 2.5 |
| Posttreatment | ||
| Fasting glucose, mg/dl | 89 ± 2 | 84 ± 2 |
| 2-h Glucose, mg/dl | 107 ± 4* | 119 ± 4 |
| Glucose AUC, mg/dl × 180 min | 21,866 ± 846 | 22,218 ± 610 |
| Fasting insulin, μiU/ml | 5.5 ± 0.9 | 3.9 ± 0.3 |
| Insulin AUC, μiU/ml × 180 min | 6,210 ± 945 | 5,141 ± 464 |
| ISOGTT | 8.4 ± 1.2 | 10.6 ± 1.0 |
| Cortisol, μg/dl | 22.8 ± 2.2 | 22.3 ± 3.1 |
Values are expressed as means ± SE; n = 8. DHEA, dehydroepiandrosterone; ISOGTT, insulin sensitivity oral glucose tolerance test; AUC, area under the curve. Conversion factors to Systeme International units: glucose × 0.0551 (mmol/l), insulin × 7.175 (pmol/l), cortisol × 27.59.
P < 0.05.
Serum androgen levels.
Before DHEA or placebo administration, serum levels of testosterone, androstenedione, and DHEA-S were similar in both groups (Fig. 1). After DHEA or placebo administration, all three of these androgen levels were significantly (P < 0.002) higher in the group treated with DHEA compared with the placebo group.
Fig. 1.
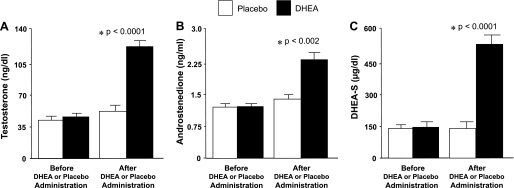
Serum levels of testosterone (A), androstenedione (B), and dehydroepiandrosterone sulfate (DHEA-S; C) before and after administration of DHEA or placebo. Compared with placebo, DHEA administration raised the levels of testosterone (P < 0.0001), androstenedione (P < 0.002), and DHEA-S (P < 0.0001).
Nuclear activation of NF-κB.
Supershift and cold competition experiments verified the specificity of the EMSA bands representing NF-κB nuclear activation (Fig. 2A). The %change in activated NF-κB from MNC obtained while fasting was significantly (P < 0.04) higher after DHEA compared with placebo (Fig. 2, B and C). Before DHEA or placebo administration, the %change in activated NF-κB decreased and was not significantly different between groups following oral glucose ingestion. After DHEA or placebo administration, the %change in activated NF-κB decreased once again following oral glucose ingestion in the placebo group but increased in the DHEA group and was significantly (P < 0.005) different between groups. The within-group analysis revealed a significant increase in the %change in activated NF-κB (−6 ± 6 vs. 16 ± 5, P < 0.03) after DHEA administration and no change after placebo.
Fig. 2.
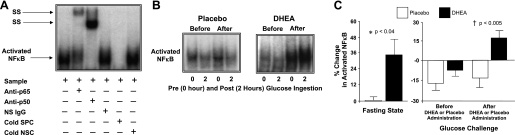
A: electrophoretic mobility shift assay (EMSA) showing nuclear factor-κB (NF-κB) in nuclear extracts from mononuclear cells (MNC). A supershift (SS) of the NF-κB band occurred during incubation with specific antibodies against NF-κB subunits but not during incubation with nonspecific (NS) IgG. Neutralization of the NF-κB band occurred during incubation with a specific cold competitor (SPC) of the oligonucleotide consensus sequence but not during incubation with a nonspecific cold competitor (NSC). B: representative EMSA bands from the 2 study groups showing the quantity of NF-κB in nuclear extracts from MNC in samples collected in the fasting state (0) and 2 h post-glucose ingestion (2), before and after treatment with DHEA or placebo. The samples used to quantify NF-κB from both study groups were run on the same gel. C: densitometric quantitative analysis comparing the change from baseline (%) in MNC-derived activated NF-κB between the 2 study groups for fasting samples before and after (before vs. after, 0) DHEA or placebo administration (left) and for fasting and 2 h post-glucose ingestion samples for each oral glucose tolerance test (OGTT; before, 0 vs. 2; after, 0 vs. 2) as a measurement of the response to glucose challenge before and after DHEA or placebo administration (right). After DHEA administration, the %change in activated NF-κB was significantly greater compared with placebo in the fasting state (P < 0.04) and in response to glucose ingestion (P < 0.005).
NF-κB subunits p65 and p105 and TNFα and IL-1β mRNA content.
The %change in mRNA content of MNC-derived p65, p105, TNFα, and IL-1β in the fasting state was significantly (P < 0.05) higher after DHEA compared with placebo (Fig. 3). Before DHEA or placebo administration, there was no significant difference between groups in the %change in p65, p105, TNFα, and IL-1β mRNA content following oral glucose ingestion. After DHEA or placebo administration, the %change in p65, p105, TNFα, and IL-1β mRNA content was significantly (P < 0.05) higher in the DHEA group compared with the placebo group. The within-group analysis revealed no significant changes in the mRNA content of either NF-κB subunit or either cytokine after administration of DHEA or placebo.
Fig. 3.
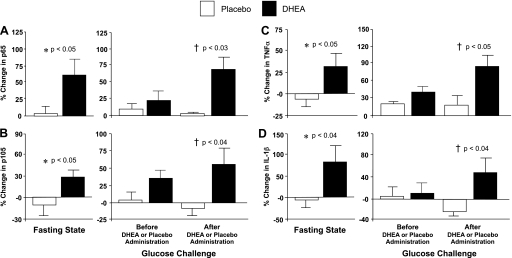
Comparison between groups of the change from baseline (%) in mRNA content of NF-κB subunits p65 (A) and p105 (B), the translated precursor of p50, and the proinflammatory cytokines TNFα (C) and IL-1β (D) in MNC for fasting samples before and after (before vs. after, 0) DHEA or placebo administration (left) and for fasting and 2 h post-glucose ingestion samples for each OGTT (before, 0 vs. 2; after, 0 vs. 2) as a measurement of the response to glucose challenge before and after DHEA or placebo administration (right). Values are normalized to 28S rRNA expression. After DHEA administration, the %change in p65, p105, TNFα, and IL-1β mRNA transcripts increased significantly compared with placebo in the fasting state (P < 0.05) and in response to glucose ingestion (P < 0.05).
NF-κB p65 subunit and IκB protein content.
The %change in p65 protein content from MNC obtained while fasting was significantly higher after DHEA compared with placebo (Fig. 4, A and B). Before DHEA or placebo administration, the %change in p65 protein content decreased and was similar in both groups following oral glucose ingestion. After DHEA or placebo administration, the %change in p65 protein content decreased following oral glucose ingestion in the placebo group but increased in the DHEA group and was significantly different between groups. The within-group analysis revealed no significant changes in p65 protein content after administration of DHEA or placebo.
Fig. 4.

Representative Western blots from the 2 study groups showing the protein content of p65 (A) and actin (C) and inhibitory-κB (IκB) and actin in MNC homogenates in samples collected in the fasting state (0) and 2 h post-glucose ingestion (2) before and after treatment with DHEA or placebo. The samples used to quantify proteins from both study groups were run on the same gel. Densitometric quantitative analysis comparing the change from baseline (%) in MNC-derived p65 (B) and IκB protein content (D) between the 2 study groups for fasting samples before and after (before vs. after, 0) DHEA or placebo administration (B and D, left) and for fasting and 2 h post-glucose ingestion samples for each OGTT (before, 0 vs. 2; after, 0 vs. 2) as a measurement of the response to glucose challenge before and after DHEA or placebo administration (B and D, right). After DHEA administration, the %change in p65 was significantly greater, and the %change in IκB was reduced significantly compared with placebo in the fasting state (p65: P < 0.002; IκB: P < 0.05) and in response to glucose ingestion (p65: P < 0.01; IκB: P < 0.03).
In contrast, the %change in IκB protein content from MNC obtained while fasting decreased significantly after DHEA compared with placebo (Fig. 4, C and D). Before DHEA or placebo administration, the %change in IκB protein content increased and was not significantly different between groups following oral glucose ingestion. After DHEA or placebo administration, the %change in IκB protein content increased following oral glucose ingestion in the placebo group but decreased in the DHEA group and was significantly different between groups. The within-group analysis revealed no significant changes in IκB protein expression after administration of DHEA or placebo.
Circulating TNFα and cortisol levels.
The %change in fasting TNFα levels was significantly higher after DHEA compared placebo (Fig. 5). Before DHEA or placebo administration, there was no significant difference between groups in the %change in TNFα levels following oral glucose ingestion. After DHEA or placebo administration, the %change in TNFα levels decreased following oral glucose ingestion in the placebo group but increased in the DHEA group and was significantly different between groups. The within-group analysis revealed no significant changes in TNFα levels after administration of DHEA or placebo. Fasting cortisol levels were similar in both groups and remained unchanged before and after DHEA or placebo administration.
Fig. 5.
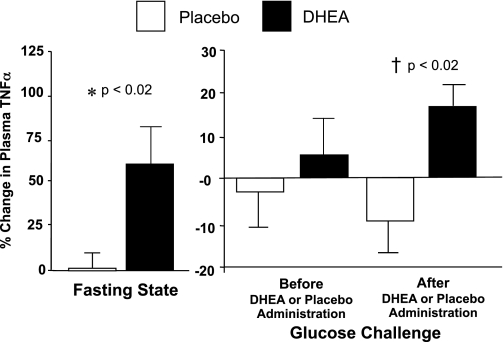
Comparison between groups of the change from baseline (%) in plasma levels of the proinflammatory cytokine TNFα for fasting samples before and after (before vs. after, 0) DHEA or placebo administration (left) and for fasting and 2 h post-glucose ingestion samples for each OGTT (before, 0 vs. 2; after, 0 vs. 2) as a measurement of the response to glucose challenge before and after DHEA or placebo administration (right). After DHEA administration, the %change in plasma TNFα increased significantly compared with placebo in the fasting state (P < 0.02), and in response to glucose ingestion (P < 0.02).
Correlations.
Measurements of body composition were not correlated with any inflammatory markers or with insulin sensitivity in the fasting state or in response to glucose ingestion (data not shown).
Serum testosterone and DHEA-S levels after DHEA or placebo administration were positively correlated with the %change in MNC-derived activated NF-κB and p65 protein content in the fasting state for the combined groups (Table 2). Testosterone levels after DHEA or placebo administration were positively correlated with the %change in fasting IL-1β RNA content and negatively correlated with the %change in fasting IκB protein content. Androstenedione levels after DHEA or placebo administration were positively correlated with the %change in fasting p65 protein content. All three posttreatment androgen levels were positively correlated with the %change in fasting plasma TNFα levels.
Table 2.
Pearson correlations for the combined groups of circulating androgen levels after DHEA or placebo administration vs. change from baseline (%) in the fasting state of inflammation markers that signifies the treatment response to DHEA and placebo
| RNA Content (%Change) |
Protein Content (%Change) |
|||||||
|---|---|---|---|---|---|---|---|---|
| Intranuclear NF-κB (%Change) | p65 | p105 | TNFα | IL-1β | p65 | IκB | Plasma TNFα (%Change) | |
| Testosterone, ng/dl | ||||||||
| r | 0.613 | 0.128 | 0.354 | 0.243 | 0.661 | 0.613 | −0.497 | 0.774 |
| P | 0.012* | 0.637 | 0.179 | 0.364 | 0.005* | 0.012* | 0.049* | 0.001* |
| Androstenedione, ng/dl | ||||||||
| r | 0.408 | 0.246 | 0.262 | 0.294 | 0.340 | 0.535 | −0.390 | 0.614 |
| P | 0.117 | 0.358 | 0.327 | 0.269 | 0.198 | 0.033* | 0.136 | 0.019* |
| DHEA-S, μg/dl | ||||||||
| r | 0.590 | 0.252 | 0.276 | 0.285 | 0.532 | 0.743 | −0.413 | 0.682 |
| P | 0.016* | 0.346 | 0.300 | 0.284 | 0.034 | 0.001* | 0.112 | 0.007* |
p65, NF-κB p65 subunit; p105, NF-κB p105 subunit; r, correlation coefficient; P, level of significance; DHEA-S, dehydroepiandrosterone sulfate.
P < 0.05.
After DHEA or placebo administration, all three androgen levels were positively correlated with the %change in activated NF-κB and p65 protein content and negatively correlated with the %change in IκB protein content in response to glucose ingestion (Table 3). After DHEA or placebo administration, testosterone and DHEA-S levels were also positively correlated with the %change in plasma TNFα in response to glucose ingestion.
Table 3.
Pearson correlations for the combined groups of circulating androgen levels after DHEA or placebo administration vs. change in baseline (%) during posttreatment glucose challenge of inflammation markers that signifies the posttreatment response to glucose ingestion
| RNA Content (%Change) |
Protein Content (%Change) |
|||||||
|---|---|---|---|---|---|---|---|---|
| Intranuclear NF-κB (%Change) | p65 | p105 | TNFα | IL-1β | p65 | IκB | Plasma TNFα (%Change) | |
| Testosterone, ng/dl | ||||||||
| r | 0.656 | 0.285 | 0.251 | 0.353 | 0.326 | 0.596 | −0.629 | 0.554 |
| P | 0.006* | 0.303 | 0.349 | 0.180 | 0.217 | 0.015* | 0.009* | 0.040* |
| Androstenedione, ng/dl | ||||||||
| r | 0.568 | 0.069 | 0.006 | 0.235 | 0.166 | 0.735 | −0.490 | 0.337 |
| P | 0.022* | 0.807 | 0.982 | 0.382 | 0.538 | 0.001* | 0.049* | 0.238 |
| DHEA-S, μg/dl | ||||||||
| r | 0.639 | 0.334 | 0.355 | 0.461 | 0.383 | 0.617 | −0.527 | 0.588 |
| P | 0.008* | 0.223 | 0.178 | 0.072 | 0.143 | 0.011* | 0.036* | 0.027* |
P < 0.05.
After DHEA or placebo administration, glucose levels 2 h post-glucose ingestion were positively correlated with the %change in MNC-derived activated NF-κB (r = 0.68, P < 0.005) and negatively correlated with IκB protein content (r = −0.53, P < 0.04) in response to glucose ingestion for the combined groups (data not shown).
The %change in fasting-activated NF-κB was positively correlated with the %change in IL-1β and plasma TNFα in the fasting state for the combined groups (Table 4). The %change in the fasting mRNA and protein contents of p65 were positively correlated with each other. The %change in fasting p65 mRNA content was also positively correlated with the %change in fasting mRNA contents of p105 and TNFα. The %change in fasting TNFα mRNA content was positively correlated with the %change in fasting contents of p105 mRNA and p65 protein. The %change in fasting IL-1β was positively correlated with the %change in fasting plasma TNFα.
Table 4.
Pearson correlations for the combined groups between the change in baseline (%) of inflammation markers in the fasting state to each other that signify the treatment response to DHEA or placebo
| RNA Content (%Change) |
Protein Content (%Change) |
||||||
|---|---|---|---|---|---|---|---|
| p65 | p105 | TNFα | IL-1β | p65 | IκB | Plasma TNFα (% Change) | |
| Intranuclear | |||||||
| NF-κB (%Change) | |||||||
| r | 0.234 | 0.018 | 0.018 | 0.330 | 0.237 | 0.071 | 0.770 |
| P | 0.383 | 0.621 | 0.619 | 0.019* | 0.376 | 0.793 | 0.001* |
| p65 | |||||||
| r | 0.597 | 0.764 | 0.012 | 0.156 | 0.118 | 0.118 | |
| P | 0.015* | 0.0006* | 0.966 | 0.564 | 0.686 | 0.686 | |
| p105 | |||||||
| r | 0.833 | 0.385 | 0.176 | 0.153 | 0.153 | ||
| P | 0.0001* | 0.141 | 0.514 | 0.600 | 0.600 | ||
| TNFα | |||||||
| r | 0.211 | −0.077 | 0.086 | 0.086 | |||
| P | 0.433 | 0.778 | 0.771 | 0.771 | |||
| IL-1β | |||||||
| r | 0.150 | 0.287 | 0.609 | ||||
| P | 0.578 | 0.281 | 0.021* | ||||
P < 0.05.
With regard to the response to glucose ingestion after DHEA or placebo administration, the %change in activated NF-κB was positively correlated with the %change in mRNA and protein contents of p65 and negatively correlated with the %change in IκB protein content (Table 5). The %change in p65 mRNA content was also positively correlated with the %change in mRNA contents of p105 and TNFα. The %change in p105 mRNA content was positively correlated with the %change in mRNA contents of TNFα and IL-1β. The %change in TNFα mRNA content was positively correlated with the %change in p65 protein content.
Table 5.
Pearson correlations for the combined groups between the change in baseline (%) of inflammation markers during glucose challenge after DHEA or placebo administration to each other that signify the posttreatment response to glucose ingestion
| RNA Content (%Change) |
Protein Content (%Change) |
||||||
|---|---|---|---|---|---|---|---|
| p65 | p105 | TNFα | IL-1β | p65 | IκB | Plasma TNFα (%Change) | |
| Intranuclear | |||||||
| NF-κB NF-κB (%Change) | |||||||
| r | 0.582 | 0.253 | 0.383 | 0.320 | 0.615 | −0.524 | 0.215 |
| P | 0.023* | 0.345 | 0.144 | 0.267 | 0.011* | 0.037* | 0.460 |
| p65 | |||||||
| r | 0.729 | 0.758 | 0.457 | 0.368 | 0.303 | 0.303 | |
| P | 0.002* | 0.001* | 0.087 | 0.177 | 0.314 | 0.314 | |
| p105 | |||||||
| r | 0.573 | 0.554 | 0.145 | 0.393 | 0.393 | ||
| P | 0.020* | 0.026* | 0.592 | 0.165 | 0.165 | ||
| TNFα | |||||||
| r | 0.177 | 0.652 | 0.504 | 0.504 | |||
| P | 0.513 | 0.006* | 0.066 | 0.066 | |||
| IL-1β | |||||||
| r | 0.206 | 0.352 | 0.137 | ||||
| P | 0.444 | 0.181 | 0.640 | ||||
P < 0.05.
DISCUSSION
Our data clearly show for the first time that MNC of lean reproductive-age women become activated and sensitized to physiological hyperglycemia after circulating androgens are raised to levels observed in PCOS and that this generates an inflammatory response in this healthy, previously uninflamed population. Oral androgen administration for 5 days activates NF-κB, the cardinal signal of inflammation, in the fasting state with further activation following glucose ingestion. These same conditions also progressively increase NF-κB gene expression along with transcribed and circulating cytokines and decrease the protein content of IκB, the cytoplasmic inhibitor of NF-κB. Androgen levels after DHEA or placebo administration are directly related to changes in activated NF-κB, p65 protein content, and transcribed or circulating cytokines and are inversely related to the changes in IκB protein content. Furthermore, the selected study population is lean with normal adiposity. These findings provide support for the concept that, in PCOS, hyperandrogenism may be the progenitor of diet-induced inflammation independent of obesity or excess abdominal adiposity.
As expected, there is no evidence of baseline inflammation in the lean, healthy reproductive-age women selected for study. Before DHEA or placebo administration, NF-κB gene expression and activation is suppressed following glucose ingestion, thereby limiting nuclear translocation and cytokine transcription. This may be the normal in vivo response to physiological hyperglycemia in young, healthy, lean women and is consistent with our previous reports that hyperglycemia suppresses MNC-derived NF-κB activation and TNFα release along with associated mediators of inflammation in young, healthy, lean men and women (21, 22, 23, 25, 32). The placebo group also exhibits minimal posttreatment alteration in the parameters evaluated while fasting and reproduction of the typical response to glucose ingestion. Suppression of NF-κB activation may be a physiological benefit in the presence of hyperglycemia when there is a need to increase glucose disposal. MNC are capable of infiltrating muscle and adipose tissue, culminating in paracrine interactions between MNC-derived macrophages and these insulin-sensitive tissues (41, 48, 51). In fact, ablation of MNC-derived macrophages in muscle of insulin-resistant animals has been shown to improve insulin sensitivity (42). Thus, increased TNFα release from MNC-derived macrophages following NF-κB activation may inhibit insulin signaling and impair glucose uptake. Conversely, optimal insulin signaling to facilitate glucose disposal in young healthy lean women may be due to control of NF-κB activation by limiting the amount of total and intranuclear NF-κB and increasing the amount of IκB in the postprandial state.
The acute elevations in circulating androgens induced by oral androgen administration activate previously resting MNC in the fasting state and subsequently increase MNC sensitivity to glucose ingestion in the selected population lacking baseline inflammation. Indeed, there is increased NF-κB gene expression and activation and cytokine transcription along with decreased IκB protein after DHEA treatment in the fasting state and in response to glucose ingestion. These key findings showcase the ability of elevated circulating androgens to upregulate the NF-κB inflammation pathway, similar to what occurs in chronically inflamed, insulin-resistant populations (13, 19). This effect is corroborated by the direct relationship of activated NF-κB, p65 protein, and circulating TNFα and the inverse relationship of IκB protein with androgen levels after DHEA or placebo administration under the same conditions. The direct relationship of activated NF-κB and the inverse relationship of IκB protein with glucose levels measured 2 h post-glucose ingestion after DHEA or placebo administration highlight the impact of androgen-induced MNC sensitization to hyperglycemia on NF-κB activation. Further evidence is provided by the direct relationships between the expression of inflammation markers at the level of mRNA and protein. Our current data mimic those observed in our previous studies of hyperglycemia-induced inflammation in women with PCOS regardless of body mass status and in obese reproductive-age women without PCOS (21, 22, 23, 24, 26). Direct binding of androgen to its receptor present in MNC has been shown to upregulate the NF-κB inflammation pathway and may be responsible for the proinflammatory mechanism of hyperandrogenemia observed in the current study (4, 43). Performance of in vitro experiments that expose MNC of lean, healthy reproductive-age women to androgen in the presence and absence of androgen receptor-blocking agents is merited to confirm this mechanism.
Insulin sensitivity is normal and unaltered after DHEA treatment in the lean, healthy reproductive-age women in our study despite androgen-induced upregulation of the NF-κB inflammation pathway. This is in contrast to the insulin resistance in PCOS that may be promoted by chronic low-grade inflammation (14, 20, 23, 44). Anti-inflammatory, insulin-sensitizing, glucose-lowering responses have been described following DHEA administration partially in relation to the first-pass liver effect associated with oral administration (3, 30, 54). However, these responses attributed to DHEA have been described mostly in animals with a fundamentally different physiology compared with humans and primarily under conditions associated with severe immune challenges such as genetically induced obesity, sepsis, trauma, and hemorrhage (3, 7). It is paradoxical with limited clinical relevance that glucose levels 2 h post-glucose ingestion are lower after placebo compared with treatment with DHEA. Thus, it is unlikely that potential ameliorating effects of DHEA administration have prevented a decline in insulin sensitivity by counteracting the proinflammatory response elicited in the lean, healthy reproductive women in our study.
Body composition characteristics of our study population may account for the unaltered insulin sensitivity. The subjects chosen are lean and have a truncal fat/leg fat ratio below 1.0, indicative of normal abdominal adiposity. In contrast, women with PCOS are often obese, and lean women with PCOS often have increased abdominal adiposity (11, 21, 31, 53). This is important because it is now clear that excess adipose tissue is a reservoir for MNC-derived macrophages, which are the major source of adipose tissue TNFα and also activate adipocyte TNFα production in a paracrine fashion (16, 17, 51, 52). Thus, the chronic low-grade inflammation present in PCOS may be related to excess adiposity, particularly abdominal adiposity. Inflamed adipose tissue in PCOS may perpetuate chronic NF-κB activation, thereby promoting more sustained excess TNFα release from circulating MNC and resident macrophages to mediate insulin resistance. In fact, previous observations in young adults demonstrate that changes in insulin sensitivity are a function of abdominal adiposity (35, 36). We have reported previously that, in PCOS, abdominal adiposity is directly related to hyperglycemia-induced NF-κB activation and inversely related to insulin sensitivity (21, 23). In contrast, measurements of abdominal adiposity are not associated with any inflammatory markers or insulin sensitivity in our study population. Finally, our small sample size powered primarily for comparing the inflammatory response between groups or the short duration of treatment may contribute to the inability to observe a change in insulin sensitivity or across-the-board within-group significant differences. It remains to be determined whether chronic androgen administration in a larger sample of lean reproductive-age women can optimally alter MNC for induction of measurable downstream metabolic aberrations.
In conclusion, androgen-induced hyperandrogenemia comparable with what is observed in PCOS activates and sensitizes MNC to physiological hyperglycemia in lean reproductive-age women. This is manifested by increases in the gene expression and activation of NF-κB and decreases in IκB protein expression in the fasting state and in response to glucose ingestion. These unique observations support the contention that hyperandrogenism, the hallmark feature of PCOS, may be the progenitor of diet-induced inflammation in the disorder.
GRANTS
This research was supported by Grants HD-048535 (to F. González) and DK-41973 (to K. S. Nair) from the National Institutes of Health and RR-024150 to the CTSA from the National Center for Research Resources.
DISCLOSURES
The authors have nothing to disclose.
AUTHOR CONTRIBUTIONS
F.G. and K.S.N. did the conception and design of the research; F.G., J.K.D., E.B., and J.M.S. performed the experiments; F.G. analyzed the data; F.G. interpreted the results of the experiments; F.G. prepared the figures; F.G. drafted the manuscript; F.G. and K.S.N. edited and revised the manuscript; F.G., K.S.N., J.K.D., E.B., and J.M.S. approved the final version of the manuscript.
ACKNOWLEDGMENTS
We thank the nursing staff of the Mayo Clinic Center for Translational Science Activities' (CTSA) Clinical Research Unit for supporting the implementation of the study and assisting with data collection.
REFERENCES
- 1. Aljada A, Ghanim H, Dandona P. Translocation of p47phox and activation of NADPH oxidase in mononuclear cells. Methods Molec Biol 196: 99–103, 2002 [DOI] [PubMed] [Google Scholar]
- 2. Andrews NC, Faller DV. A rapid micropreparation technique for extraction of DNA-binding proteins from limited numbers of mammalian cells. Nucleic Acids Res 19: 2499, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aoki K, Saito T, Satoh S, Mukasa K, Kaneshiro M, Kawasaki S, Okamura A, Sehihara H. Dehydroepiandrosterone suppresses the elevated hepatic glucose-6-phospatase and fructose-1,6-biphosphatase activities in C57BL/Ksj-db/db mice. Diabetes 48: 1579–1585, 1999 [DOI] [PubMed] [Google Scholar]
- 4. Ashcroft GS, Mills SL. Androgen receptor-mediated inhibition of cutaneous wound healing. J Clin Invest 110: 615–624, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Balagopal P, Schimke JC, Ades P, Adey D, Nair KS. Age effect on transcript levels and synthesis rate of muscle MHC and response to resistance exercise. Am J Physiol Endocrinol Metab 280: E203–E208, 2001 [DOI] [PubMed] [Google Scholar]
- 6. Baldwin AS., Jr Series introduction: the transcription factor NF-kappaB and human disease. J Clin Invest 107: 3–6, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Barkhausen T, Hildebrand F, Krettek C, van Griensven M. DHEA-dependent and organ-specific regulation of TNF-alpha mRNA expression in a murine polymicrobial sepsis and trauma model. Critical Care 13: R114, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med 336: 1066–1071, 1997 [DOI] [PubMed] [Google Scholar]
- 9. Burghen GA, Givens JR, Kitabachi AE. Correlation of hyperandrogenism with hyperinsulinemia in polycystic ovarian disease. J Clin Endocrinol Metab 50: 113–116, 1980 [DOI] [PubMed] [Google Scholar]
- 10. Carmina E, Rosato F, Jannμ A, Rizzo M, Longo RA. Extensive clinical experience: relative prevalence of different androgen excess disorders in 950 women referred because of clinical hyperandrogenism. J Clin Endocrinol Metab 91: 2–6, 2006 [DOI] [PubMed] [Google Scholar]
- 11. Carmina E, Bucchieri S, Esposito A, Del Puente A, Mansueto P, Orio F, Di Fede G, Rini G. Abdominal fat quantity and distribution in women with polycystic ovary syndrome and extent of its relation to insulin resistance. J Clin Endocrinol Metab 92: 2500–2505, 2007 [DOI] [PubMed] [Google Scholar]
- 12. Ciaraldi TP, Kolterman OG, Olefsky JM. Mechanism for the postreceptor defect in insulin action in human obesity. Decrease in glucose transport system activity. J Clin Invest 68: 875–878, 1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dandona P, Aljada A, Mohanty P, Ghanim H, Hamouda W, Assian E, Ahmad S. Insulin inhibits intranuclear nuclear factor kappaB and stimulates IkappaB in mononuclear cells in obese subjects: evidence for an anti-inflammatory effect? J Clin Endocrinol Metab 86: 3257–3265, 2001 [DOI] [PubMed] [Google Scholar]
- 14. Escobar-Morreale HF, Luque-Ramírez M, González F. Circulating inflammatory markers in polycystic ovary syndrome: a systematic review and metaanalysis. Fertil Steril 95: 1048–1058, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr Rev 23: 599–622, 2002 [DOI] [PubMed] [Google Scholar]
- 16. Fain JN, Bahouth SW, Madan AK. TNFalpha release by the nonfat cells of human adipose tissue. Int J Obes 28: 616–622, 2004 [DOI] [PubMed] [Google Scholar]
- 17. Fain JN, Madan AK, Hiler ML, Cheema P, Bahouth SW. Comparison of the release of adipokines by adipose tissue, adipose tissue matrix, and adipocytes from visceral and subcutaneous abdominal adipose tissues of obese humans. Endocrinology 145: 2273–2282, 2004 [DOI] [PubMed] [Google Scholar]
- 18. Fan CM, Maniatis T. Generation of p50 subunit of NF-kappa B by processing of p105 through an ATP-dependent pathway. Nature 354: 395–398, 1991 [DOI] [PubMed] [Google Scholar]
- 19. Ghanim H, Aljada A, Daoud N, Deopurkar R, Chaudhuri A, Dandona P. Role of inflammatory mediators in the suppression of insulin receptor phosphorylation in circulating mononuclear cells of obese subjects. Diabetologia 50: 278–285, 2007 [DOI] [PubMed] [Google Scholar]
- 20. Ghanim H, Aljada A, Hofmeyer D, Syed T, Mohanty P, Dandona P. Circulating mononuclear cells in the obese are in a proinflammatory state. Circulation 110: 1564–1571, 2004 [DOI] [PubMed] [Google Scholar]
- 21. González F, Minium J, Rote NS, Kirwan JP. Hyperglycemia alters tumor necrosis factor-alpha release from mononuclear cells in women with polycystic ovary syndrome. J Clin Endocrinol Metab 90: 5336–5342, 2005 [DOI] [PubMed] [Google Scholar]
- 22. González F, Rote NS, Minium J, Kirwan JP. Reactive oxygen species-induced oxidative stress in the development of insulin resistance and hyperandrogenism in polycystic ovary syndrome. J Clin Endocrinol Metab 91: 336–340, 2006 [DOI] [PubMed] [Google Scholar]
- 23. González F, Rote NS, Minium J, Kirwan JP. Increased activation of nuclear factor κB triggers inflammation and insulin resistance in polycystic ovary syndrome. J Clin Endocrinol Metab 91: 1508–1512, 2006 [DOI] [PubMed] [Google Scholar]
- 24. González F, Rote NS, Minium J, Kirwan JP. In vitro evidence that hyperglycemia stimulates tumor necrosis factor-α release in obese women with polycystic ovary syndrome. J Endocrinol 188: 521–529, 2006 [DOI] [PubMed] [Google Scholar]
- 25. González F, Minium J, Rote NS, Kirwan JP. Altered tumor necrosis factor-α release from mononuclear cells of obese reproductive age women during hyperglycemia. Metabolism 55: 271–277, 2006 [DOI] [PubMed] [Google Scholar]
- 26. González F, Rote NS, Minium J, O'Leary VB, Kirwan JP. Obese reproductive age women exhibit a proatherogenic inflammatory response during hyperglycemia. Obesity 15: 2436–2444, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. González F, Rote NS, Minium J, Kirwan JP. Insulin sensitivity and hyperandrogenism in polycystic ovary syndrome are related to activated nuclear factor κB from mononuclear cells in the fasting state. Program of the 89th Annual Meeting of the Endocrine Society, Toronto, ON, Canada, 2007, p. 142 [Google Scholar]
- 28. Hotamisligil GS, Murray DL, Choy LN, Spiegelman BM. Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc Natl Acad Sci USA 91: 4854–4858, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jones NH. Finding the area under the curve using jmp and a trapezoidal rule [Online]. SAS Institute. http://www.jmp.com/about/newsletters/jmpercable/pdf/05_fall_1997.pdf [1997].
- 30. Kimura M, Tanaka S, Yamada Y, Kiuchi Y, Yamakawa T, Sekihara H. Dehydroepiandrosterone decreases serum tumor necrosis factor-alpha and restores insulin sensitivity: independent effect from secondary weight reduction in genetically obese Zucker fatty rats. Endocrinology 139: 3249–3253, 1998 [DOI] [PubMed] [Google Scholar]
- 31. Kirchengast S, Huber J. Body composition characteristics and body fat distribution in lean women with polycystic ovary syndrome. Hum Reprod 16: 1255–1260, 2001 [DOI] [PubMed] [Google Scholar]
- 32. Kirwan JP, Krishnan RK, Weaver JA, Del Aguila LF, Evans WJ. Human aging is associated with altered TNF-α production during hyperglycemia and hyperinsulinemia. Am J Physiol Endocrinol Metab 281: E1137–E1143, 2001 [DOI] [PubMed] [Google Scholar]
- 33. Kolterman OG, Insel J, Saekow M, Olefsky JM. Mechanisms of insulin resistance in human obesity. Evidence for receptor and post receptor defects. J Clin Invest 65: 1272–1284, 1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Knochenhauer ES, Key TJ, Kahser-Miller M, Waggoner W, Boots LR, Azziz R. Prevalence of the polycystic ovary syndrome in unselected black and white women of the southeastern Unites States: a prospective study. J Clin Endocrinol Metab 83: 3078–3082, 1998 [DOI] [PubMed] [Google Scholar]
- 35. Kriketos AD, Greenfield JR, Peake PW, Furler SM, Denyer GS, Charlesworth JA, Campbell LV. Inflammation, insulin resistance, and adiposity: a study of first-degree relatives of type 2 diabetic subjects. Diabetes Care 27: 2033–2040, 2004 [DOI] [PubMed] [Google Scholar]
- 36. Linné Y. Effects of obesity on women's reproduction and complications during pregnancy. Obes Rev 5: 137–143, 2004 [DOI] [PubMed] [Google Scholar]
- 37. March WA, Moore VM, Willson KJ, Phillips DI, Norman RJ, Davies MJ. The prevalence of polycystic ovary syndrome in a community sample assessed under contrasting diagnostic criteria. Hum Reprod 25: 544–551, 2010 [DOI] [PubMed] [Google Scholar]
- 38. Matsuda M, DeFronzo R. Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care 22: 1462–1470, 1999 [DOI] [PubMed] [Google Scholar]
- 39. Modan M, Harris MI, Halkin H. Evaluation of WHO and NDDG criteria for impaired glucose tolerance. Results from two national samples. Diabetes 38: 1630–1635, 1989 [DOI] [PubMed] [Google Scholar]
- 40. Nestler JE, Jakubowicz DJ, de Vargas AF, Brik C, Quintero N, Medina F. Insulin stimulates testosterone biosynthesis by human thecal cells from women with polycystic ovary syndrome by activating its own receptor and using inositolglycan mediators as the signal transduction system. J Clin Endocrinol Metab 83: 2001–2005, 1998 [DOI] [PubMed] [Google Scholar]
- 41. Nguyen MT, Favelyukis S, Nguyen AK, Reichart D, Scott PA, Jenn A, Liu-Bryan R, Glass CK, Neels JG, Olefsky JM. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via toll-like receptors 2 and 4 and JNK-dependent pathways. J Biol Chem 282: 35279–35292, 2007 [DOI] [PubMed] [Google Scholar]
- 42. Patsouris D, Li PP, Thapar D, Chapman J, Olefsky JM, Neels JG. Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab 8: 301–309, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ripple MO, Henry WF, Schwarze SR, Wilding G, Weindruch R. Effect of antioxidants on androgen-induced AP-1 and NF-kappaB DNA-binding activity in prostate carcinoma cells. J Natl Cancer Inst 14: 1227–1232, 1999 [DOI] [PubMed] [Google Scholar]
- 43a. Rotterdam ESHRE/ASRM-Sponsored PCOS Consensus Workshop Group Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome. Fertil Steril 81: 19–25, 2004 [DOI] [PubMed] [Google Scholar]
- 44. Rui L, Aguirre V, Kim JK, Shulman GI, Lee A, Corbould A, Dunaif A, White MF. Insulin/IGF-1 and TNF-alpha stimulate phosphorylation of IRS-1 at inhibitory Ser307 via distinct pathways. J Clin Invest 107: 181–189, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest 107: 7–11, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Taylor RW, Keil D, Gold EJ, Williams SM, Goulding A. Body mass index, waist girth, and waist-to-hip ratio as indexes of total and regional adiposity in women: evaluation using receiver operating characteristic curves. Am J Clin Nutr 67: 44–49, 1998 [DOI] [PubMed] [Google Scholar]
- 48. Varma V, Yao-Borengasser A, Rasouli N, Nolen GT, Phanavanh B, Starks T, Gurley C, Simpson P, McGehee RE, Jr, Kern PA, Peterson CA. Muscle inflammatory response and insulin resistance: synergistic interaction between macrophages and fatty acids leads to impaired insulin action. Am J Physiol Endocrinol Metab 296: E1300–E1310, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Waggoner W, Boots LR, Azziz R. Total testosterone and DHEAS levels as predictors of androgen-secreting neoplasms: a populational study. Gynecol Endocrinol 13: 394–400, 1999 [DOI] [PubMed] [Google Scholar]
- 50. Webster RA. Reproductive function and pregnancy. In: Henry's Clinical Diagnosis and Management by Laboratory Methods (21st ed.), edited by McPherson RA, Pincus MR. Philadelphia, PA: Saunders Elsevier, 2006, chapt. 25, p. 364–378 [Google Scholar]
- 51. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112: 1796–1808, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 112: 1821–1830, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yildirim B, Sabir N, Kaleli B. Relation of intra-abdominal fat distribution to metabolic disorders in nonobese patients with polycystic ovary syndrome. Fertil Steril 79: 1358–1364, 2003 [DOI] [PubMed] [Google Scholar]
- 54. Zhang Z, Araghi-Niknam M, Liang B, Inserra P, Ardestani SK, Jiang S, Chow S, Watson RR. Prevention of immune dysfunction and vitamin E loss by dehydroepiandrosterone and melatonin supplementation during murine retrovirus infection. Immunology 96: 291–297, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]