

Abstract
We explored the application of electron detachment dissociation (EDD) and infrared multiphoton dissociation (IRMPD) tandem mass spectrometry to fluorescently labeled sialylated oligosaccharides. Standard sialylated oligosaccharides and a sialylated N-linked glycan released from human transferrin were investigated. EDD yielded extensive glycosidic cleavages and cross-ring cleavages in all cases studied, consistently providing complementary structural information compared to IRMPD. Neutral losses and satellite ions such as C – 2H ions were also observed following EDD. In addition, we examined the influence of different fluorescent labels. The acidic label 2-aminobenzoic acid (2-AA) enhanced signal abundance in negative-ion mode. However, few cross-ring fragments were observed for 2-AA labeled oligosaccharides. The neutral label 2-aminobenzamide (2-AB) resulted in more cross-ring cleavages compared to 2-AA labeled species, but not as extensive fragmentation as for native oligosaccharides, likely resulting from altered negative charge locations from introduction of the fluorescent tag.
Keywords: derivatized oligosaccharides, Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR), tandem mass spectrometry, high resolution, structural characterization
1 Introduction
Glycosylation plays essential roles in a variety of cellular processes, including tumor growth and metastasis, immune response, and cell-cell communication [1–6]. Sialic acids (e.g., N-acetyl neuraminic acid, NeuAc) are an important family of sugars that contains a carboxylic acid at the C-1 position of the six-member sugar ring. Sialic acids are often found at the terminal positions of glycans and glycoconjugates and are involved in a large number of protein-glycan and glycan-glycan interactions in cellular processes, including intercellular adhesion, signaling, and microbial attachment [7]. Compared to proteomics and genomics, glycomics analysis faces unique challenges due to the non-template driven biosynthesis and the highly diverse structures of glycans. In order to achieve thorough structural elucidation of glycans, monosaccharide composition, degree of branching (for branched glycans), linkage type, and anomeric configuration all need to be determined.
Mass spectrometry (MS)-based structural characterization of glycans is gaining popularity due to its high sensitivity and ability to perform high-throughput analysis [8–11]. Fourier transform ion cyclotron resonance (FT-ICR) MS is a powerful tool for the structural analysis of glycans benefiting from ultra-high mass accuracy, high resolution, and compatibility with various tandem mass spectrometry (MS/MS) techniques [12–14]. While mass profiling of glycans can be obtained by MS, detailed structure elucidation requires MS/MS. Collision activated dissociation (CAD) [15–17], infrared multiphoton dissociation (IRMPD) [18–20], high-energy CAD (heCAD) [21–23], electron capture dissociation (ECD) [24–28], electron detachment dissociation (EDD) [29–34], negative electron transfer dissociation (NETD) [35], 157 nm photodissociation [36–38], and electron induced dissociation (EID) [39] have all been applied for glycan structural characterization.
During IRMPD, precursor ions are irradiated by an IR laser (typically 10.6 μm CO2 laser). Multiple photons are absorbed and the corresponding energy is redistributed over all precursor ion vibrational modes [40, 41]. Compared to CAD, IRMPD is more advantageous for glycan structural analysis because of its ability to readily generate secondary fragmentation, which provides higher fragmentation efficiency, particularly for large glycans [18]. EDD was first introduced in 2001 for characterization of peptide anions [42]. During EDD, polyanions are irradiated with >10 eV energy electrons. Electron detachment occurs to generate charge-reduced species, followed by subsequent radical-driven fragmentation [43]. Our lab previously showed that EDD provides complementary structural information for model oligosaccharides and sialylated N-glycans compared to IRMPD [34, 44]. Amster and co-workers investigated the fragmentation behavior in EDD of glycosaminoglycans, and found that EDD yielded extensive glycosidic and cross-ring cleavages, whereas CAD and IRMPD mostly resulted in glycosidic cleavages [29–33, 45]. EDD could also be utilized to differentiate the isomers glucuronic acid and iduronic acid [29]. The combination of EDD and IRMPD/CAD is a highly valuable tool for structural characterization of carbohydrates, particularly acidic carbohydrates in negative-ion mode, due to the ability to provide complementary structural information.
Fluorescent labeling of glycans is frequently employed prior to MS for several reasons. First, introduction of a hydrophobic label to hydrophilic glycans improves ionization efficiency [46]. Second, fluorescent labels enable UV- or fluorescence-based detection in high-performance liquid chromatography (HPLC), and also enhance glycan retention on reverse phase (e.g., C18) columns [47–50]. Native glycans often exhibit poor retention, while derivatized glycans can be separated and identified with C18 columns [51]. In addition, recent studies have demonstrated that quantitation of glycans can be obtained by utilizing stable isotopic labeling [52–54]. The most commonly used labeling method is reductive amination. The primary amine of the label reacts with the aldehyde group of the glycan to generate a Schiff base intermediate, which is then stabilized through reduction to form a secondary amine [55]. 2-aminobenzoic acid (2-AA), 2-aminobenzamide (2-AB), 2-aminopyridine (2-AP), and 5-amino-2-naphthalenesulfonic acid (ANSA) are some of the commonly utilized fluorescent labels [56–68].
It has been previously reported that labels may change mass spectrometric fragmentation behaviors of glycans [48, 50, 69]. For neutral glycans and labeled glycans which do not contain acidic groups, positive-ion mode analysis is frequently employed. 2-AB-tagged oligosaccharides were examined in both protonated and sodiated forms by CAD, heCAD, post-source decay (PSD), and ultraviolet photodissociation (UVPD) [59–61, 64, 70, 71]. Harvey reported that 2-AB labeling at the reducing end did not significantly alter fragmentation behavior of N-glycans in CAD [59]. Protonated species mostly produced B- and Y-type glycosidic cleavages, whereas sodiated species generated additional C-, and Y-type as well as cross-ring fragments [60, 71]. 355 nm UVPD of fluorescently labeled sodiated oligosaccharides resulted in efficient fragmentation, generating series of A- and C-ions, complementary to CAD [70]. 2-AB labeled oligosaccharides were also analyzed in negative-ion mode MALDI-TOF/TOF-MS, demonstrating considerably different fragmentation patterns compared to positive-ion mode [63]. Various cross-ring fragments such as 1,3A-type ions were observed, providing linkage information. In addition, fucose residues were stabilized in negative-ion mode analysis, which allowed improved fucosylation site determination [63]. Acidic glycans (e.g., sialylated glycans) or glycans with acidic labels (e.g., 2-AA) tend to produce abundant signals in negative-ion mode, which makes negative-ion mode analysis a highly favorable choice. 2-AA labeled N-glycans mostly generate Y-type ions containing the reducing end and also some A-type cross-ring cleavages in negative-ion mode CAD [15]. Here, we investigate the fragmentation behaviors of fluorescently labeled sialylated oligosaccharides by EDD and IRMPD, and compare those to the fragmentation patterns of the corresponding underivatized species.
2 Materials and methods
2.1 Reagents
Disialyl-lacto-N-tetraose (DSLNT) and LS-tetrasaccharide b (LSTb) were purchased from V-labs Inc (Covington, LA). Human apo-transferrin, 1,4-dithio-DL-threitol (DTT), iodoacetamide, 2-AA, 2-AB, boric acid, sodium acetate, and sodium cyanoborohydride (NaCNBH3) were obtained from Sigma Chemical Co. (St. Louis, MO). NH4HCO3, NH4OH, methanol, acetonitrile, glacial acetic acid, dimethyl sulfoxide (DMSO), and formic acid were obtained from Fisher (Fair Lawn, NJ). Peptide-N-glycosidase F (PNGase F) was purchased from Calbiochem (Gibbstown, NJ).
2.2 Preparation of N-linked glycans
The glycoprotein was reduced in 5 mM DTT at 56 °C for 45 min, alkylated by 15 mM iodoacetamide in the dark at room temperature for 1 h, and digested with PNGase F (2 U) in 50 mM NH4HCO3 (pH 8) overnight at 37 °C.
2.3 Fluorescent labeling of oligosaccharides
Oligosaccharides (2 nmol) were dried down, reconstituted in 60 μL freshly prepared labeling reagent, and incubated at 80 °C for 1h (2-AA) or 65 °C for 2h (2-AB). For 2-AA labeling, labeling reagent was prepared by dissolving 30 mg 2-AA and 30 mg NaCNBH3 in 1 mL methanol containing 4% sodium acetate (w/v) and 2% boric acid (w/v). For 2-AB labeling, labeling reagent was 0.35 M 2-AB and 1 M NaCNBH3 in a DMSO/glacial acetic acid mixture (7:3 (v/v)).
2.4 Purification of oligosaccharides
The fluorescently labeled oligosaccharides were purified by SPE graphitized carbon cartridge (Alltech Associates Inc., Deerfield, IL). For each sample, a carbon cartridge was washed with 0.1% (v/v) formic acid in 80% acetonitrile/H2O (v/v), followed by deionized water. The solution containing labeled oligosaccharides was loaded and the cartridge was then washed by deionized water to remove salts and other contaminants. The glycans were eluted with 0.1% formic acid (v/v) in 20% or 40% acetonitrile/H2O (v/v). The solution was then dried down in a vacuum concentrator (Eppendorf, Hamberg, Germany) and reconstituted in 50% methanol, 0.1% NH4OH (v/v) solution for MS analysis.
2.5 Mass spectrometry
All mass spectra were collected with an actively shielded 7 T FT-ICR mass spectrometer with a quadrupole front-end (APEX-Q, Bruker Daltonics, Billerica, MA), as previously described [72]. An indirectly heated hollow dispenser cathode was used to perform EDD [73]. IRMPD was performed with a vertically mounted 25 W, 10.6 μM CO2 laser (Synrad, Mukilteo, WA). Samples were infused via an Apollo II electrospray ion source at a flow rate of 70 μL/h with the assistance of N2 nebulizing gas. Following ion accumulation in the first hexapole for 50 ms, ions were mass selectively accumulated in the second hexapole for 1–6 s. Ions were then transferred through high voltage ion optics and captured with dynamic trapping in an Infinity ICR cell [74]. The accumulation sequence up to the ICR cell fill was looped 3 times to optimize precursor ion signal to noise (S/N) ratio. For EDD, the cathode heating current was kept at 2.0 A and the cathode voltage was pulsed to a bias voltage of − 30 to − 35 V [34] for 1 s. An extraction lens located in between the cathode and the ICR cell was kept − 0.8 to − 1.0 V higher than the cathode bias voltage. An electron current of a few μA is optimum for EDD [75]. IRMPD was performed with a laser power of 10 W with firing times ranging from 0.25–1s.
2.6 Data analysis
All mass spectra were acquired with XMASS software (Bruker Daltonics) with 256 data points from m/z 100 to 2000 and summed over 60–100 scans. Data processing was performed with MIDAS software [76]. Data were zero filled once, Hanning apodized, and exported to Microsoft Excel for internal frequency-to-mass calibration with a two-term calibration equation [77]. Product ion spectra were interpreted with the aid of the web application GlycoFragment (http://www.glycosciences.de/tools/GlycoFragments/) [78]. Product ions were not assigned unless they were at least 3 times the noise level.
3 Results
MS/MS spectra of oligosaccharides mainly contain two types of bond cleavages: glycosidic cleavages which occur between monosaccharide residues, and cross-ring cleavages occurring across sugar rings [79]. Glycosidic cleavages provide structural information regarding monosaccharide composition, whereas cross-ring cleavages aid the determination of linkage type. EDD and IRMPD fragmentation patterns of the 2-AA and 2-AB labeled sialylated oligosaccharides DSLNT and LSTb, and an N-glycan released from human transferrin were investigated. Structures of 2-AA and 2-AB are shown in Figure 1.
Figure 1.
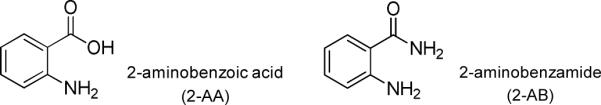
Structures of 2-aminobenzoic acid (2-AA) and 2-aminobenzamide (2-AB).
3.1 DSLNT
DSLNT is a branched di-sialylated oligosaccharide with the composition Neu5Acβ3Galβ3(Neu5Acβ6)GlcNAcβ3Galβ4Glc. Negative-ion mode IRMPD of 2-AB-labeled DSLNT resulted in an almost complete series of Y-ions, all containing the fluorescent label (Figure 2a), similar to the fragmentation patterns from MALDI-PSD of 2-AB labeled oligosaccharides [61]. Y4α(3β) (m/z = 1117.4), corresponding to loss of either sialic acid, was the most abundant species among the product ions, illustrating the gas-phase lability of sialic acids. No cross-ring fragments were found, thus precluding acquisition of linkage information. EDD of doubly deprotonated 2-AB-labeled DSLNT resulted in extensive fragmentation (Figure 2b). All product ions were singly charged, which may arise from two different fragmentation mechanisms: either via direct fragmentation of activated precursor ions, or via the charged reduced radical ions generated after electron detachment [32]. Glycosidic cleavages between every neighboring monosaccharide were observed, along with five cross-ring cleavages, including 0,2A-, 1,5A-, 0,2X-, and 1,5X-type ions. 0,2X- and 1,5X-type ions are generally not present in CAD/IRMPD spectra, but they have been observed from high-energy CAD, laser induced dissociation (LID) of sodiated fluorescently labeled oligosaccharides [63, 69], and from EDD of underivatized oligosaccharides and GAGs [30, 32, 34]. Among all the product ions, C1α(1β) (m/z = 308.1), corresponding to loss of one sialic acid, was the most abundant species. CO2 loss from the charge reduced species, glycosidic fragments, and from cross-ring fragments was frequently observed, presumably originating from the carboxylic acid of the sialic acids. This neutral loss complicates the spectral appearance. Several satellite peaks, such as C − 2H, Y − 2H, and Y − 16 Da (or Z + 2H) [34] were also found in the spectra. Such ions were only observed in EDD, indicating that they may arise from radical driven fragmentation pathways [30]. Compared to IRMPD, EDD produced additional structural information including both glycosidic and cross-ring cleavages, rendering EDD a highly valuable and promising tool for structural characterization of fluorescently labeled oligosaccharides in negative-ion mode.
Figure 2.

(a) IRMPD (80 scans, 0.8 s at 10 W) and (b) EDD (80 scans, 1s irradiation, cathode bias – 30 V) spectra of doubly deprotonated 2-AB-labeled DSLNT (m/z = 703.75). Cross-ring fragments are highlighted in bold and reducing end product ions lacking 2-AB are underlined in the spectra. All product ions are singly charged. Fragmentation patterns from IRMPD and EDD are summarized in Figure 2c.
MS/MS of 2-AA labeled DSLNT is shown in Figure 3. Compared to 2-AB-labeled species, 2-AA-labeled DSLNT readily generated abundant signal in negative-ion mode. In order to compare the fragmentation behaviors, shorter ion accumulation time was chosen for 2-AA-tagged species to yield the same precursor ion abundance. IRMPD of 2-AA-labeled DSLNT mostly resulted in Y-ions containing the fluorescent tag, which could aid determination of monosaccharide composition and “sequence” of the oligosaccharide (Figure 3a). Again, no cross-ring cleavages were produced. However, in contrast to 2-AB-labeled DSLNT, the Y4α(3β) product ion, corresponding to loss of one sialic acid, does not dominate the IRMPD spectrum of 2-AA-labeled DSLNT. Instead, Y4α/Y3β, corresponding to loss of both sialic acids, is dominant and Y3α/Y3β product ions, which also have lost both sialic acids and an additional galactose residue, are observed. The latter product was not detected in IRMPD of 2-AB-labeled DSLNT. The higher abundance of these two product ions for the 2-AA-labeled species is likely due to the higher probability of retaining charge on the reducing end due to the acidic label.
Figure 3.

(a) IRMPD (80 scans, 1 s at 10 W) and (b) EDD (80 scans, 1s irradiation, cathode bias – 30 V) spectra of doubly deprotonated 2-AA-labeled DSLNT (m/z = 704.24). Cross-ring fragments are highlighted in bold and reducing end fragment ions lacking 2-AA are underlined in the spectra. All product ions are singly charged. Fragmentation patterns from IRMPD and EDD are summarized in Figure 3c.
EDD of 2-AA-labeled DSLNT demonstrated similar fragmentation compared to the 2-AB-labeled species (Figure 3b). Thirteen glycosidic and four cross-ring cleavages were observed, including 1,5A-, 0,2X-, 1,3X- and 1,5X-type ions. All product ions were singly charged. Extensive neutral loss (CO2, CH3O, and H2O) from the charged reduced species and from glycosidic fragments, and internal fragments were observed in the EDD spectrum. Similar to Figure 2b, satellite ions such as C4 − 2H were found. Their absence in the IRMPD spectrum of the same species suggests that such ions may originate from radical-driven fragmentation, or from electronic excitation. Compared to 2-AB-labeled species, 2-AA-labeled DSLNT generated fewer cross-ring fragments (four compared to five), suggesting that the nature of the reducing end substitute affects EDD fragmentation behavior.
3.2 Sialylated N-glycan
IRMPD and EDD of the fluorescently labeled di-sialylated N-glycan from human transferrin were also examined (see Figure 4 and Supplementary Figure S1). Most product ions from IRMPD of the 2-AB-labeled glycan (Figure 4a) were singly charged, while one product from cross-ring cleavage (m/z = 1130.9) was doubly charged. Most glycosidic fragments were B- and C-type ions contained the non-reducing end. EDD of the 2-AB-labeled N-glycan produced a large number of glycosidic and cross-ring fragments, which provided rich structural information (Figure 4b). In addition to glycosidic cleavages between every neighboring monosaccharide, three cross-ring cleavages were observed, including 1,3A-, 1,5A-, and 0,2A-type ions. 0,2A-, 1,3A-, and 1,5A-type ions have been previously reported from MS/MS of deprotonated 2-AB labeled neutral N-glycans in MALDI-TOF/TOF analysis [63], and such product ions are also commonly observed in negative-ion mode CAD of glycans [80–82]. The 1,5A5 ion (m/z = 1769.6) at the branching mannose of the chitobiose core aided determination of the positions of the two antenna. Unlike EDD of 2-AA- and 2-AB-labeled DSLNT, no X-type ions were found.
Figure 4.

MS/MS of a 2-AA- (a), (b) and 2-AB- (c), (d) derivatized N-linked glycan released from human transferrin. Fragmentation patterns from IRMPD (a), (c) and EDD (b), (d) are shown.
MS/MS of the 2-AA-tagged N-glycan showed somewhat different fragmentation behavior compared to the 2-AB-tagged species. IRMPD of the 2-AA-labeled N-glycan resulted in efficient fragmentation, generating eleven glycosidic and two cross-ring cleavages (Figure 4c). It is interesting to note that 2,4X1 containing the fluorescent tag (m/z = 484.2) was observed. Typically, X-type ions are absent in CAD/IRMPD of N-glycans [18, 83]. The observation of X-type ions following derivatization may indicate that the charge locations are different in the 2-AA-labeled N-glycan compared to the underivatized glycan. Introduction of the acidic tag 2-AA adds an additional likely site for deprotonation. EDD of the 2-AA-labeled N-glycan resulted in only glycosidic cleavages (Figure 4d). All observed Y- and Z-type ions contained the fluorescent label 2-AA, suggesting that one negative charge is located on 2-AA. Compared to EDD of the 2-AB-labeled N-glycan, the most distinct difference was the absence of cross-ring fragments. This difference may be explained by the altered charge location (as further discussed below).
3.3 LSTb
LSTb is a branched mono-sialylated oligosaccharide with the composition Galβ3(Neu5Acβ6)GlcNAcβ3Galβ4Glc. Negative-ion mode IRMPD and EDD of 2-AA-labeled LSTb were investigated and the results are shown in Figure 5. IRMPD of 2-AA-labeled LSTb (Figure 5a) yielded a complete series of Y- and Z-ions and also one cross-ring fragment (0,2X3α). Again, all the Y- and Z-type ions contained the fluorescent tag. EDD of 2-AA-labeled LSTb (Figure 5b) produced six additional glycosidic and two additional cross-ring fragments, including 1,5X2, and 1,5X3β ions. 0,2X- and 1,5X-type ions have also been reported in positive-ion mode high-energy CAD, LID of sodiated 2-AB-labeled oligosaccharides, and EDD of sialylated oligosaccharides [34, 63, 69]. Similar to EDD of 2-AA-labeled DSLNT and the N-glycan, unique product ions such as neutral loss (H2O and CH3O) from the charge reduced species and C – 2H type satellite ions were observed in the EDD spectrum.
Figure 5.


(a) IRMPD (80 scans, 1 s at 10 W) and (b) EDD (80 scans, 1s irradiation, cathode bias – 30 V) spectra of doubly deprotonated 2-AA-labeled LSTb (m/z = 558.69). Cross-ring fragments are highlighted in bold, reducing end fragment ions lacking 2-AA are underlined in the spectra, and electronic noise peaks are indicated by asterisks. All product ions are singly charged. Fragmentation patterns from IRMPD and EDD are summarized in Figure 5c.
2-AB labeling of LSTb was also conducted and the labeling reaction was successful, however, even with careful tuning of the instrument parameters, the signal abundance of doubly charged species was too low for EDD. Therefore, we were not able to investigate EDD of 2-AB-labeled LSTb. In EDD, at least two precursor ion charges are required. In addition, due to the low efficiency of EDD [27], abundant precursor ion signal is a prerequisite to observe product ions. LSTb has only one acidic site, which is the carboxylic acid on the sialic acid and 2-AB is a neutral tag that does not enhance ionization in negative-ion mode. In contrast, 2-AA-labeled LSTb readily generated abundant signal in MS due to the acidity of 2-AA, thus EDD fragmentation could easily be achieved. These observations suggest that the chosen fluorescent tag for derivatization has a strong influence on whether EDD will be successful.
3.4 Influence of reducing end derivatization on EDD fragmentation
EDD fragmentation patterns of DSLNT, LSTb, and the N-linked glycan released from human transferrin with and without fluorescent labels are summarized in Table 1. Although product ions resulting from glycosidic and cross-ring cleavages were observed in almost all cases, the degree of fragmentation was different, particularly for the number of observed cross-ring cleavages. Underivatized oligosaccharides provided the most cross-ring fragments in all three sialylated oligosaccharides investigated, followed by 2-AB labeled oligosaccharides. This result is similar to previous studies reported by Harvey and co-workers in positive-ion mode [69]. The latter authors compared the effects of reducing end substituent in high-energy CAD of N-linked oligosaccharides, including underivatized and 2-AB-labeled oligosaccharides, and N-glycopeptides with one and four amino acids. Compared to 2-AB-derivatized oligosaccharides, the native species generated a more complete series of glycosidic fragments and also more cross-ring fragments due to the open nature of the reducing terminus ring [69].
Table 1.
EDD fragmentation summary of DSLNT, LSTb, and an N-glycan released from transferrin with or without fluorescent labels.
| EDD FRAGMENTATION | DSLNT | LSTb | N-glycan from transferrin |
|---|---|---|---|
| Unlabeled | 10 glycosidic 6 cross-ring |
11 glycosidic 7 cross-ring |
16 glycosidic 5 cross-ring |
| 2-AA labeled | 13 glycosidic 4 cross-ring |
13 glycosidic 3 cross-ring |
10 glycosidic 0 cross-ring |
| 2-AB labeled | 11 glycosidic 5 cross-ring |
N/A | 11 glycosidic 3 cross-ring |
2-AA labeling enhances signal abundance of oligosaccharides in negative-ion mode, however, the 2-AA-derivatized oligosaccharides generated the smallest number of cross-ring cleavages. Particularly, no cross-ring fragments were observed for the 2-AA-labeled N-glycan (compared to three from the 2-AB-labeled species and five from the native glycan), which makes 2-AA labeling less favorable in EDD of derivatized oligosaccharides. One possible explanation of this observation is that the introduction of a fluorescent tag at the reducing end of oligosaccharides alters the locations of the negative charges. It has been previously reported that deprotonated hydroxyl is required to obtain cross-ring cleavages in negative-ion mode [80–82]. Without derivatization, it is most favorable for the negative charges to be located on the sialic acids, but also possible to have deprotonated hydroxyl groups. When 2-AA is introduced at the reducing end, the carboxylic acid on 2-AA is preferably deprotonated, which competes with deprotonation of hydroxyl groups, thus making cross-ring cleavages less likely to occur. 2-AB is a neutral fluorescent label; therefore introduction of 2-AB does not alter the location of negative charges. However, for less acidic oligosaccharides, which are difficult to doubly deprotonate, 2-AB is less favorable due to the lack of an acidic moiety.
4 Concluding remarks
We demonstrate that EDD of fluorescently labeled sialylated oligosaccharides results in extensive fragmentation, providing rich glycosidic and cross-ring fragments. We also show that complementary structural information can be obtained from EDD compared to IRMPD of the same species. When investigating the influence of introducing different fluorescent labels at the reducing end of oligosaccharides, we found that not only does the labels affect signal abundance, but they also have a strong influence on the EDD fragmentation behavior. Native oligosaccharides showed the most extensive fragmentation compared to their 2-AA- and 2-AB-labeled counterparts. The acidic tag 2-AA promotes precursor ion signal abundance in negative-ion mode. However, it introduces another deprotonation site to compete with the sialic acids and hydroxyl groups, thus suggesting that deprotonated hydroxyls are important for cross-ring fragmentation in EDD, as in CAD [80–82]. The neutral label 2-AB does not significantly impede generation of cross-ring cleavages, but for small and less acidic glycans, which are difficult to doubly deprotonate, 2-AB may not be the best choice.
Supplementary Material
MS/MS spectra of a 2-AA- (a), (b) and 2-AB- (c), (d) derivatized N-linked glycan released from human transferrin. Spectra from IRMPD (a), (c) and EDD (b), (d) are shown.
Acknowledgments
This work was supported by Award Number R21CA138331 from the National Cancer Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health. W.Z. was partially supported by a George Ashworth Analytical Chemistry Fellowship from the University of Michigan.
List of abbreviations
- 2-AA
2-aminobenzoic acid
- 2-AB
2-aminobenzamide
- 2-AP
2-aminopyridine
- ANSA
5-amino-2-naphthalenesulfonic acid
- ACD
Collision activated dissociation
- DMSO
Dimethyl sulfoxide
- DSLNT
Disialyl-lacto-N-tetraose
- DTT
1,4-dithio-DL-threitol
- ECD
Electron capture dissociation
- EDD
Electron detachment dissociation
- EID
Electron induced dissociation
- ETD
Electron transfer dissociation
- FTICR-MS
Fourier transform ion cyclotron resonance mass spectrometry
- HeCAD
High-energy CAD
- IRMPD
Infrared multiphoton dissociation
- LID
Laser induced dissociation
- LSTb
LS-tetrasaccharide b
- PNGase F
Peptide-N-glycosidase F
- SPE
Solid phase extraction
- UVPD
Ultraviolet photodissociation
5 References
- [1].Varki A. Glycobiol. 1993;3:97–130. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kim YJ, Varki A. Glycoconj. J. 1997;14:569–576. doi: 10.1023/a:1018580324971. [DOI] [PubMed] [Google Scholar]
- [3].Raman R, Raguram S, Venkataraman G, Paulson JC, Sasisekharan R. Nat. Methods. 2005;2:817–824. doi: 10.1038/nmeth807. [DOI] [PubMed] [Google Scholar]
- [4].Dube DH, Bertozzi CR. Nat. Rev. Drug Discov. 2005;4:477–488. doi: 10.1038/nrd1751. [DOI] [PubMed] [Google Scholar]
- [5].Bertozzi CR, Kiessling LL. Science. 2001;291:2357–2364. doi: 10.1126/science.1059820. [DOI] [PubMed] [Google Scholar]
- [6].Finkelstein J. Nature. 2007;446:999–999. [Google Scholar]
- [7].Varki A. Nature. 2007;446:1023–1029. doi: 10.1038/nature05816. [DOI] [PubMed] [Google Scholar]
- [8].Morelle W, Michalski JC. Curr. Pharm. Des. 2005;11:2615–2645. doi: 10.2174/1381612054546897. [DOI] [PubMed] [Google Scholar]
- [9].Zaia J. Chem. Biol. 2008;15:881–892. doi: 10.1016/j.chembiol.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Krishnamoorthy L, Mahal LK. ACS Chem. Biol. 2009;4:715–732. doi: 10.1021/cb900103n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Mechref Y, Novotny MV. Chem. Rev. 2002;102:321–369. doi: 10.1021/cr0103017. [DOI] [PubMed] [Google Scholar]
- [12].Marshall AG, Hendrickson CL, Jackson GS. Mass Spectrom. Rev. 1998;17:1–35. doi: 10.1002/(SICI)1098-2787(1998)17:1<1::AID-MAS1>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- [13].Park YM, Lebrilla CB. Mass Spectrom. Rev. 2005;24:232–264. doi: 10.1002/mas.20010. [DOI] [PubMed] [Google Scholar]
- [14].Marshall AG, Hendrickson CL. Annu. Rev. Anal. Chem. 2008;1:579–599. doi: 10.1146/annurev.anchem.1.031207.112945. [DOI] [PubMed] [Google Scholar]
- [15].Harvey DJ. J. Mass Spectrom. 2005;40:642–653. doi: 10.1002/jms.836. [DOI] [PubMed] [Google Scholar]
- [16].Sagi D, Peter-Katalinic J, Conradt HS, Nimtz M. J. Am. Soc. Mass. Spectrom. 2002;13:1138–1148. doi: 10.1016/S1044-0305(02)00412-9. [DOI] [PubMed] [Google Scholar]
- [17].Zaia J, Miller MJC, Seymour JL, Costello CE. J. Am. Soc. Mass. Spectrom. 2007;18:952–960. doi: 10.1016/j.jasms.2007.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lancaster KS, An HJ, Li BS, Lebrilla CB. Anal. Chem. 2006;78:4990–4997. doi: 10.1021/ac0600656. [DOI] [PubMed] [Google Scholar]
- [19].Xie YM, Lebrilla CB. Anal. Chem. 2003;75:1590–1598. doi: 10.1021/ac026009w. [DOI] [PubMed] [Google Scholar]
- [20].Zhang JH, Schubothe K, Li BS, Russell S, Lebrilla CB. Anal. Chem. 2005;77:208–214. doi: 10.1021/ac0489824. [DOI] [PubMed] [Google Scholar]
- [21].Harvey DJ, Naven TJP, Kuster B, Bateman RH, et al. Rapid Commun. Mass Spectrom. 1995;9:1556–1561. doi: 10.1002/rcm.1290091517. [DOI] [PubMed] [Google Scholar]
- [22].Harvey DJ, Bateman RH, Green MR. J. Mass Spectrom. 1997;32:167–187. doi: 10.1002/(SICI)1096-9888(199702)32:2<167::AID-JMS472>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- [23].Lewandrowski U, Resemann A, Sickmann A. Anal. Chem. 2005;77:3274–3283. doi: 10.1021/ac048399n. [DOI] [PubMed] [Google Scholar]
- [24].Zhao C, Xie B, Chan SY, Costello CE, O'Connor PB. J. Am. Soc. Mass. Spectrom. 2008;19:138–150. doi: 10.1016/j.jasms.2007.10.022. [DOI] [PubMed] [Google Scholar]
- [25].Adamson JT, Hakansson K. Anal. Chem. 2007;79:2901–2910. doi: 10.1021/ac0621423. [DOI] [PubMed] [Google Scholar]
- [26].Budnik BA, Haselmann KF, Elkin YN, Gorbach VI, Zubarev RA. Anal. Chem. 2003;75:5994–6001. doi: 10.1021/ac034477f. [DOI] [PubMed] [Google Scholar]
- [27].McFarland MA, Marshall AG, Hendrickson CL, Nilsson CL, et al. J. Am. Soc. Mass. Spectrom. 2005;16:752–762. doi: 10.1016/j.jasms.2005.02.001. [DOI] [PubMed] [Google Scholar]
- [28].Liu H, Hakansson K. Int. J. Mass Spectrom. 2011 in press, doi: 10.1016/j.ijms.2010.10.030. [Google Scholar]
- [29].Wolff JJ, Chi LL, Linhardt RJ, Amster IJ. Anal. Chem. 2007;79:2015–2022. doi: 10.1021/ac061636x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wolff JJ, Laremore TN, Busch AM, Linhardt RJ, Amster IJ. J. Am. Soc. Mass. Spectrom. 2008;19:294–304. doi: 10.1016/j.jasms.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wolff JJ, Laremore TN, Busch AM, Linhardt RJ, Amster IJ. J. Am. Soc. Mass. Spectrom. 2008;19:790–798. doi: 10.1016/j.jasms.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wolff JJ, Amster IJ, Chi LL, Linhardt RJ. J. Am. Soc. Mass. Spectrom. 2007;18:234–244. doi: 10.1016/j.jasms.2006.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wolff JJ, Laremore TN, Leach FE, Linhardt RJ, Amster IJ. Eur. J. Mass Spectrom. 2009;15:275–281. doi: 10.1255/ejms.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Adamson JT, Hakansson K. J. Am. Soc. Mass. Spectrom. 2007;18:2162–2172. doi: 10.1016/j.jasms.2007.09.007. [DOI] [PubMed] [Google Scholar]
- [35].Wolff JJ, Leach FE, Laremore TN, Kaplan DA, et al. Anal. Chem. 2010;82:3460–3466. doi: 10.1021/ac100554a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Devakumar A, Thompson MS, Reilly JP. Rapid Commun. Mass Spectrom. 2005;19:2313–2320. doi: 10.1002/rcm.2058. [DOI] [PubMed] [Google Scholar]
- [37].Devakumar A, Mechref Y, Kang P, Novotny MV, Reilly JP. J. Am. Soc. Mass. Spectrom. 2008;19:1027–1040. doi: 10.1016/j.jasms.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Devakumar A, Mechref Y, Kang P, Novotny MV, Reilly JP. Rapid Commun. Mass Spectrom. 2007;21:1452–1460. doi: 10.1002/rcm.2981. [DOI] [PubMed] [Google Scholar]
- [39].Gao D, Zhou W, Hakansson K. 58th ASMS Conference on Mass Spectrometry and Allied Topics; Salt Lake City, UT. 2010. [Google Scholar]
- [40].Brodbelt JS, Wilson JJ. Mass Spectrom. Rev. 2009;28:390–424. doi: 10.1002/mas.20216. [DOI] [PubMed] [Google Scholar]
- [41].Eyler JR. Mass Spectrom. Rev. 2009;28:448–467. doi: 10.1002/mas.20217. [DOI] [PubMed] [Google Scholar]
- [42].Budnik BA, Haselmann KF, Zubarev RA. Chem. Phys. Lett. 2001;342:299–302. [Google Scholar]
- [43].Zubarev RA. Mass Spectrom. Rev. 2003;22:57–77. doi: 10.1002/mas.10042. [DOI] [PubMed] [Google Scholar]
- [44].Zhou W, Håkansson K. 57th ASMS Conference on Mass Spectrometry and Allied Topics; Philadelphia, PA. 2009. [Google Scholar]
- [45].Wolff JJ, Laremore TN, Aslam H, Linhardt RJ, Amster IJ. J. Am. Soc. Mass. Spectrom. 2008;19:1449–1458. doi: 10.1016/j.jasms.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Yoshino K, Takao T, Murata H, Shimonishi Y. Anal. Chem. 1995;67:4028–4031. doi: 10.1021/ac00117a034. [DOI] [PubMed] [Google Scholar]
- [47].Lamari FN, Kuhn R, Karamanos NK. J. Chromatogr. B. 2003;793:15–36. doi: 10.1016/s1570-0232(03)00362-3. [DOI] [PubMed] [Google Scholar]
- [48].Lattova E, Snovida S, Perreault H, Krokhin O. J. Am. Soc. Mass. Spectrom. 2005;16:683–696. doi: 10.1016/j.jasms.2005.01.021. [DOI] [PubMed] [Google Scholar]
- [49].Anumula KR. Anal. Biochem. 2006;350:1–23. doi: 10.1016/j.ab.2005.09.037. [DOI] [PubMed] [Google Scholar]
- [50].Ruhaak LR, Zauner G, Huhn C, Bruggink C, et al. Anal. Bioanal. Chem. 2010;397:3457–3481. doi: 10.1007/s00216-010-3532-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wuhrer M, Deelder AM, Hokke CH. J. Chromatogr. B. 2005;825:124–133. doi: 10.1016/j.jchromb.2005.01.030. [DOI] [PubMed] [Google Scholar]
- [52].Bowman MJ, Zaia J. Anal. Chem. 2007;79:5777–5784. doi: 10.1021/ac070581b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Xia BY, Feasley CL, Sachdev GP, Smith DF, Cummings RD. Anal. Biochem. 2009;387:162–170. doi: 10.1016/j.ab.2009.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Prien JM, Prater BD, Qin Q, Cockrill SL. Anal. Chem. 2010;82:1498–1508. doi: 10.1021/ac902617t. [DOI] [PubMed] [Google Scholar]
- [55].Bigge JC, Patel TP, Bruce JA, Goulding PN, et al. Anal. Biochem. 1995;230:229–238. doi: 10.1006/abio.1995.1468. [DOI] [PubMed] [Google Scholar]
- [56].Maslen S, Sadowski P, Adam A, Lilley K, Stephens E. Anal. Chem. 2006;78:8491–8498. doi: 10.1021/ac0614137. [DOI] [PubMed] [Google Scholar]
- [57].Maslen SL, Goubet F, Adam A, Dupree P, Stephens E. Carbohydr. Res. 2007;342:724–735. doi: 10.1016/j.carres.2006.12.007. [DOI] [PubMed] [Google Scholar]
- [58].Wuhrer M, Koeleman CAM, Deelder AM. Anal. Chem. 2009;81:4422–4432. doi: 10.1021/ac900278q. [DOI] [PubMed] [Google Scholar]
- [59].Harvey DJ. Analyst. 2000;125:609–617. [Google Scholar]
- [60].Harvey DJ. J. Am. Soc. Mass. Spectrom. 2000;11:900–915. doi: 10.1016/S1044-0305(00)00156-2. [DOI] [PubMed] [Google Scholar]
- [61].Sato Y, Suzuki M, Nirasawa T, Suzuki A, Endo T. Anal. Chem. 2000;72:1207–1216. doi: 10.1021/ac9908750. [DOI] [PubMed] [Google Scholar]
- [62].Wuhrer M, Koeleman CAM, Hokke CH, Deelder AM. Int. J. Mass Spectrom. 2004;232:51–57. [Google Scholar]
- [63].Wuhrer M, Deelder AM. Anal. Chem. 2005;77:6954–6959. doi: 10.1021/ac051117e. [DOI] [PubMed] [Google Scholar]
- [64].Morelle W, Slomianny MC, Diemer H, Schaeffer C, et al. Rapid Commun. Mass Spectrom. 2005;19:2075–2084. doi: 10.1002/rcm.2033. [DOI] [PubMed] [Google Scholar]
- [65].Chen XY, Flynn GC. J. Am. Soc. Mass. Spectrom. 2009;20:1821–1833. doi: 10.1016/j.jasms.2009.06.003. [DOI] [PubMed] [Google Scholar]
- [66].Takegawa Y, Deguchi K, Ito S, Yoshioka S, et al. Rapid Commun. Mass Spectrom. 2005;19:937–946. doi: 10.1002/rcm.1872. [DOI] [PubMed] [Google Scholar]
- [67].Deguchi K, Takegawa Y, Ito H, Miura N, et al. Rapid Commun. Mass Spectrom. 2006;20:412–418. doi: 10.1002/rcm.2320. [DOI] [PubMed] [Google Scholar]
- [68].Briggs JB, Keck RG, Ma S, Lau WD, Jones AJS. Anal. Biochem. 2009;389:40–51. doi: 10.1016/j.ab.2009.03.006. [DOI] [PubMed] [Google Scholar]
- [69].Kuster B, Naven TJP, Harvey DJ. Rapid Commun. Mass Spectrom. 1996;10:1645–1651. doi: 10.1002/(SICI)1097-0231(199610)10:13<1645::AID-RCM664>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- [70].Wilson JJ, Brodbelt JS. Anal. Chem. 2008;80:5186–5196. doi: 10.1021/ac800315k. [DOI] [PubMed] [Google Scholar]
- [71].Morelle W, Page A, Michalski JC. Rapid Commun. Mass Spectrom. 2005;19:1145–1158. doi: 10.1002/rcm.1900. [DOI] [PubMed] [Google Scholar]
- [72].Yang J, Mo JJ, Adamson JT, Hakansson K. Anal. Chem. 2005;77:1876–1882. doi: 10.1021/ac048415g. [DOI] [PubMed] [Google Scholar]
- [73].Tsybin YO, Witt M, Baykut G, Kjeldsen F, Hakansson P. Rapid Commun. Mass Spectrom. 2003;17:1759–1768. doi: 10.1002/rcm.1118. [DOI] [PubMed] [Google Scholar]
- [74].Caravatti P, Allemann M. Org. Mass Spectrom. 1991;26:514–518. [Google Scholar]
- [75].Yang J, Hakansson K. Int. J. Mass Spectrom. 2008;276:144–148. [Google Scholar]
- [76].Senko MW, Canterbury JD, Guan SH, Marshall AG. Rapid Commun. Mass Spectrom. 1996;10:1839–1844. doi: 10.1002/(SICI)1097-0231(199611)10:14<1839::AID-RCM718>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- [77].Ledford EB, Rempel DL, Gross ML. Anal. Chem. 1984;56:2744–2748. doi: 10.1021/ac00278a027. [DOI] [PubMed] [Google Scholar]
- [78].Lohmann KK, von der Lieth CW. Nucleic Acids Res. 2004;32:W261–W266. doi: 10.1093/nar/gkh392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Domon B, Costello CE. Glycoconj. J. 1988;5:397–409. [Google Scholar]
- [80].Harvey DJ. J. Am. Soc. Mass. Spectrom. 2005;16:622–630. doi: 10.1016/j.jasms.2005.01.004. [DOI] [PubMed] [Google Scholar]
- [81].Harvey DJ. J. Am. Soc. Mass. Spectrom. 2005;16:631–646. doi: 10.1016/j.jasms.2005.01.005. [DOI] [PubMed] [Google Scholar]
- [82].Harvey DJ. J. Am. Soc. Mass. Spectrom. 2005;16:647–659. doi: 10.1016/j.jasms.2005.01.006. [DOI] [PubMed] [Google Scholar]
- [83].Pikulski M, Hargrove A, Shabbir SH, Anslyn EV, Brodbelt JS. J. Am. Soc. Mass. Spectrom. 2007;18:2094–2106. doi: 10.1016/j.jasms.2007.09.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
MS/MS spectra of a 2-AA- (a), (b) and 2-AB- (c), (d) derivatized N-linked glycan released from human transferrin. Spectra from IRMPD (a), (c) and EDD (b), (d) are shown.