

Abstract
The molecular mechanisms of acute lung injury are incompletely understood. MicroRNAs (miRNAs) are crucial biological regulators that act by suppressing their target genes and are involved in a variety of pathophysiologic processes. miR-127 appears to be down-regulated during lung injury. We set out to investigate the role of miR-127 in lung injury and inflammation. Expression of miR-127 significantly reduced cytokine release by macrophages. Looking into the mechanisms of regulation of inflammation by miR-127, we found that IgG Fcγ Receptor I (FcγRI/CD64) was a target of miR-127, as evidenced by reduced CD64 protein expression in macrophages over-expressing miR-127. Furthermore, miR-127 significantly reduced the luciferase activity with a reporter construct containing the native 3′-UTR of CD64. Importantly, we demonstrated that miR-127 attenuated lung inflammation in an IgG immune complex (IgG IC) model in vivo. Collectively, these data show that miR-127 targets macrophage CD64 expression and promotes the reduction of lung inflammation. Understanding how miRNAs regulate lung inflammation may represent an attractive way to control inflammation induced by infectious or non-infectious lung injury.
Introduction
Acute lung injury (ALI) is characterized by hypoxemia, pulmonary edema, reduced lung compliance, and impaired gas exchange (1). Severe lung injury leads to acute respiratory distress syndrome, characterized by severe lung inflammation and profound hypoxemia, and frequently results in multiple organ failure (2). Both ALI and ARDS are major causes of morbidity and mortality. The molecular and immunological mechanisms of acute lung injury remain incompletely understood.
When the lung encounters an exogenous insult, epithelial cells and macrophages are the primary lines of defense. The injured cells trigger a cascade of events including acute inflammatory response, recruitment of immune cells such as monocytes/macrophages, and release of cytokines (IL-1, IL-6, and TNF-α), chemokines, growth factors, and prostaglandins (3). Through innate immunity, the structures of invading microorganisms, including lipids, carbohydrates, peptides, and nucleic acids, are first recognized by pattern-recognition receptors (PRRs) (4). PRRs include the Toll-like receptor family (TLR), including TLR4 (4). TLR4 is essential for responses to bacterial lipopolysaccharide (LPS) as well as to various endogenous ligands, such as hyaluronan (HA) fragments (5, 6). Engagement of the TLR4 receptor triggers the activation of an intracellular signaling pathway, resulting in subsequent cytokine/chemokine production and release (6). Through the adaptive immune system, Fc receptors recognize the Fc domain of immunoglobulin (Ig) and thereby link the antibody-mediated immune response to cellular effector functions including phagocytosis, release of inflammatory mediators, and clearance of immune complexes. Fc gamma receptors (FcγRs) belong to the Ig superfamily and are the most important Fc receptors for phagocytosis of opsonized microbes, mainly including FcγRI, FcγRII, FcγRIII, and FcγRIV (7). Whereas FcγRI (CD64) is constitutively present on only monocytes and macrophages, FcγRIII is expressed in many tissues, but absent in lymphocytes. FcγRII is present on almost all hematopoietic cells. FcγRI, FcγRIII and FcγRIV function as activating receptors, where FcγRII acts as a negative regulator. Alveolar macrophages express FcγRI, FcγRII, and FcγRIII (8). FcγRI deficient mice showed impaired cytokine release, phagocytosis, and cellular cytotoxicity in IC-induced inflammation, suggesting a critical role for FcγRI in IgG2a-IC-dependent immune functions (9).
Numerous human diseases are thought to result from the failure to regulate the production and clearance of immune complexes. Circulating immune complexes were found in patients with systemic lupus erythematosus (10), rheumatoid arthritis (11), Goodpasture syndrome (12), and nephritis (13). In the respiratory system, basement membrane destruction by immune complexes are found in patients with ARDS (14), idiopathic interstitial pneumonias (15), and hypersensitivity pneumonitis/alveolitis (16). Studies suggest that the anti-IL-8 autoantibody:IL-8 immune complexes were found in lung fluids from patients with ALI/ARDS and correlated both with the development and outcome of ARDS (14, 17). Anti-KC:KC complexes induced lung inflammation in mice and were associated with the development of severe pulmonary inflammation (18). A recent study suggested that anti-chemokine autoantibody:chemokine immune complexes may contribute to the pathogenesis of lung inflammation by inducing activation of endothelial cells through engagement of the IgG receptor FcγRIIa (14). The molecular mechanisms by which the immune complex regulate inflammatory responses are largely unclear.
MicroRNAs (miRNAs) are critical regulators of gene expression. Mature miRNAs bind target mRNAs at complementary sites in 3′-untranslated regions (3′-UTRs) or coding sequences and thereby trigger down-regulation, suppression of target gene expression (19). Emerging evidence also shows that miRNAs play an important role in both adaptive and innate immunity (20). miRNAs are involved in innate immunity through the regulation of Toll-like receptor signaling and cytokine responses. For example, reports showed that miR-146, miR-155, and miR-132 are strongly up-regulated after LPS treatment in human monocyte THP1 (21-23). miR-146 may target IRAK-1 and TRAF6, two essential components of the TLR signaling pathways, thereby acting as a negative regulator of the inflammatory response (21). miR-147 is induced by toll-like receptor stimulation and can regulate macrophage inflammatory response by decreasing LPS-induced TNF-α and IL-6 production (24). miRNAs also regulate adaptive immune responses. For example, silencing of miR-126 by antagomir impairs Th2 responses in the lung (25). miR-181a appears to act as a negative regulator of T cell receptor signaling, by targeting Bcl-2 and CD69, mediating positive selection (26, 27). Furthermore, miR-155 has a role in regulating T helper cell differentiation and the germinal center reaction to produce an optimal T cell-dependent antibody response (28).
We have recently analyzed miRNA expression after bleomycin-induced lung injury and repair (29) and identified that miR-127 expression was decreased one day after bleomycin treatment. In this report, we examined miR-127 expression in both non-infectious and infectious lung injury. In order to examine the role of miR-127 in macrophages, we established a miR-127 stable over-expression of MH.S cells. We demonstrated that miR-127 directly down-regulates CD64 and attenuated lung inflammation in vivo. Taken together, these data suggest that miR-127 plays a previously unrecognized role in preventing exaggerated inflammatory responses to complement injury by negatively regulating FcγRI.
Materials and Methods
Reagents
Peptidoglycan (PGN) from Bacillus subtilis and LPS from Escherichia coli 0111:B4 were purchased from Sigma-Aldrich. Hyaluronan (HA) fragments (about 200,000 kDa in molecular weight, LPS free) were from ICN. Immune complexes were prepared according to previous reports (30). Briefly, IgG-BSA immune complexes were produced by slowly adding 4 volumes of IgG anti-BSA (5 mg/ml, MP Biomedicals) to 1 volume of BSA (10 mg/ml, Sigma) and incubating at 37°C for 30 minutes. The complexes were recovered by centrifugation and resuspended in RPMI-1640 medium. Macrophage cell lines MH.S and RAW246.7, as well as human monocyte cell line U937, were stimulated with IgG-BSA immune complexes (100 μg/ml) or RPMI-1640 medium as control.
Cells
The mouse macrophage cell line RAW264.7, alveolar macrophage cell line MH.S, and human monocyte cell line U937 were purchased from the American Type Culture Collection (ATCC). Cells were cultured in RPMI1640 plus 10% FBS at 37°C and 5% CO2. U937 were differentiated and activated with phorbol 12-myristate 13-acetate (PMA) (100 nM, Sigma) before other treatment.
miRNA oligonucleotides
2′-OME modified miRNAs were chemically synthesized by IDT. All bases were 2′-O-methyl (2′-OME) modified and a cholesterol moiety was linked to 3′ terminus. miR-127: 5′- mUmCmG mGmAmU mCmCmG mUmCmU mGmAmG mCmUmU mGmGmC mU/3CholTEG/ -3′, where m denotes 2′-OME modification. CEMR67: 5′-mUmCmA mCmAmA mCmCmUm CmCmUm AmGmAm AmAmG mAmG/3CholTEG/ -3′.
Custom locked nucleic acid (LNA) oligonucleotides were synthesized by Exiqon, LNA-miR127: 5′-+T+CGG+ATC+CGT+CTG+AGC+TTG+G+C-3′, LNA-anti-miR127: 5′-+G+CCA+AGC+TCA+GAC+GGA+TCC+G+A-3′, control probe (CEMR67): 5′-+T+CACA+ACCT+CCT+AGA+AAG+A+G-3′, where the plus (+) sign denotes the LNA modification.
MiR-127 precursor, inhibitor, and control probe were purchased from Ambion.
Expression constructs and stable transduction
MiR-127 lentiviral construct was generated as follows: a region of 101 nucleotides containing pre-miR-127 was amplified from mouse genomic DNA by using the following primers, 5′MIR127: GATC ACTG TTAA CCAG CCTG CT and 3′MIR127: TTAA CTCG AGGG AGGC AGAT GA. They were then cloned into the HpaI/XhoI sites of a pSico vector (31), named pSico-miR-127. Viral particles were produced by calcium phosphate–mediated transfection into 293T cells as described (32). Lentiviral supernatants were collected 48 hours after transfection, passed through a 0.22-μm filter, and used directly to infect MH.S cells. GFP-positive cells were sorted 3-4 days after infection and resulted in MH.S-SicoGFP and MH.S-miR127GFP cells. Recombinant adenoviral stocks expressing Cre recombinase were purchased from the Gene Transfer Vector Core facility of University of Iowa College of Medicine (Iowa City, IA). Infections of GFP-positive cells were performed by using 100 plaque-forming units of virus per cell. Four days after adenovirus infections, the GPF-negative cells were sorted from GFPpos infected with adeno-Cre as MH.S-Sico and MH.S-miR127 cells. Cre recombination was discriminated by PCR amplification of genomic DNA with the following primers, pSico-Cre-UP: GGGA CAGC AGAG ATCC AGTT and pSico-Cre-DN: ACCG GAAC CCTT AAAC ATGA. The unrecombined viral DNA results in a 1921 bp band, and the recombination viral DNA results in a 495 bp band.
Animals, IgG immune complex-induced lung injury, bleomycin-induced lung injury, and BAL analysis
All experiments were carried out using 8- to 12-week-old C57Bl/6 female mice (Jackson Laboratory). All studies were conducted in accordance with NIH guidelines for the care and use of animals, and with approval from the Duke University Animal Care and Use Committee.
Rabbit anti-BSA IgG (MP Biomedicals catalog 55275; 10 μg in 20 μl of PBS) was administered intranasally, and 5 mg/kg of BSA (Sigma A8412) in 200 μl of PBS was injected intravenously immediately thereafter. Control mice received BSA intravenously in the absence of an intranasal dose of anti-BSA. To assess permeability, 0.25% Evans blue (Alfa Aesar, catalog A16774) was administrated with BSA intravenously. Since neither CEMR67 nor PBS had any effect, either CEMR67 or PBS was used as control in certain experiments. When miR-127 and control probe CEMR67 were used, 100 μg in 40 μl PBS was instilled intranasally 2 hours before anti-BSA IgG administration.
Animals were sacrificed 4 h after IgG IC-induced alveolitis according to reports (33). The trachea was catheterized and lungs were lavaged with 0.8 ml of ice-cold PBS three times. The first 0.8 ml of BALF was collected for ELISA, and the total and differential cells in BALF were counted by use of a hemocytometer. Cell differentials were performed on cytospin preparations stained with protocol Hema3 stain set (Fisher Healthcare). Evans blue concentration in BALF was determined by ELISA reader under 405 nm for lung permeability analysis. Total protein level was determined by bicinchoninic acid protein assay.
Under anesthesia, 2.5 U/kg bleomycin (Blenoxane from Mayne Pharma Inc., Paramus, New Jersey) dissolved in sterile PBS was administered via trachea as previously described (6, 34, 35). Lung tissues were harvested one day post bleomycin challenges or from untreated mice.
RNA analysis
RNA was isolated from cells with TRIzol reagent (Invitrogen) or mirVana™ miRNA Isolation Kit (Ambion). The expression of miR-127 was normalized to sno202 and analyzed using Taqman miRNA assays (Applied Biosystems).
Luciferase assay
Construct generation. The full length FcγRI 3′-UTR was amplified by PCR from mouse genomic DNA with the following primers: FcγRI-Forward 5′-GGT GAC TAG TGG ACC CGA GCA GCC-3′ and FcγRI-Reverse 5′-AAG TGA GCT CAG GTC CTC TGG AAG-3′. The amplified 1404 bp product was inserted into the SpeI/SacI sites of the luciferase expression vector pMIR-REPORT (Applied Biosystems) immediately downstream of the luciferase gene and termed pMIR-REPORT[FcγRI-3′-UTR]. A mutant construct was generated with two primers, FcγRI-3m-Forward 5′-GAC TGG CCG CGG CTG AGA CAA GCT GGG TAA TCA GAC-3′ and FcγRI-3m-Reverse 5′-GCC GCG GCC AGT CTG TAT ATT TGC TTT ATT TAA GAG TTG CAT GCC-3′ cloned into pMIR-REPORT, resulting in pMIR-REPORT[mFcγRI-3′-UTR].
Transfection. RAW264.7 cells were co-transfected with 0.4 μg of firefly luciferase report vector and 0.08 μg of the control vector containing Renilla luciferase, pRL-TK (Promega) in 24-well plates by using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. For each well, 10 nM miR-127 precursor and miR-127 inhibitor (Ambion) were used. For luciferase assay, Firefly and Renilla luciferase activities were measured consecutively by using dual-luciferase assays (Promega) 48 h after transfection according to manufacturer's suggestions.
Cytokine and complement production
TNF-α, IL-6, IL-1β, MIP-1β, MIP-2, MCP-1, KC, and C5a protein levels in supernatant collected from cell culture or in BALF of injured mice were measured immunologically using commercial ELISA kits (R&D Systems). Complement C5a in BAL of injured mice were determined with ELISA with antibodies from BD Pharmingen (Capture antibody: purified rat anti-mouse C5a, catalog 558027; standard: purified recombinant mouse C5a, catalog 622597; detect antibody: biotin rat anti-mouse C5a, catalog 558028).
Protein analysis
Protein extracts were isolated from cells or lung tissues. CD64, STAT3 and Phospho-Stat3 (Tyr705) levels were determined using goat polyclonal anti-CD64 (R&D systems), mouse monoclonal anti-Stat3 (Cell signaling), or rabbit monoclonal anti-phospho-Stat3 (Tyr705) (Cell signaling) and normalized to GAPDH (Cell signaling).
Microarray analysis
Microarray analysis was performed using the Affymetrix Mouse Genome 430 2.0 Array (Affymetrix). Gene chips were scanned by the GenePix 4000B scanner (Molecular Devices, Sunnyvale, CA). Data and images were obtained using GenePix Pro 6.1 (Molecular Devices, Sunnyvale, CA) according to the manufacturer's instructions. RNAs extracted from MH.S-Sico cells were hybridized on Cy3, and RNAs extracted from MH.S-miR127 cells were hybridized on Cy5. Data were background subtracted and normalized within the array using the LOESS normalization by Genespring GX 11 software. The complete microarray data set is available at National Center for Biotechnology Information Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/), under accession number GSE27642.
Flow cytometry
RAW264.7 and MH.S cells were stained with PE conjugated anti- mouse CD64 antibody (BD Pharmingen, catalog 558455) and flow cytometry was used to determine the CD64 staining positive cells in MH.S-Sico and MH.S-miR127 cells, to determine the CD64 staining positive cells in miR-127 precursor, inhibitor and control probe transfected Raw 264.7 cell, and to select top 1% of highest CD64 expressing cells, as CD64hi, and bottom 1% of lowest CD64 expressing cells, as CD64lo.
Synthetic 2′-O-methyl (2′-OME)- and cholesterol-modified, and Fam (5′)-labeled miR-127 oligonucleotide or control oligonucleotide without Fam labeling (100 μg in 40 μl PBS) were intranasally administered to C57BL/6J mice. BAL cells were isolated 2 hours later and stained with CD64-PE (BD Biosciences, Cat# 558592) and F4/80-APC (AbD Serotec, Cat# MCA497APCT) antibodies. Macrophages uptaking Fam-labeled miR-127 oligonucleotide, and CD64 expression on BAL cells isolated from miR-127 and control oligonucleotide treated mice were analyzed with flow cytometry.
Histology
To morphologically assess lung injury 4 h after IgG IC deposition, the lungs were inflated with 10% formalin via a tracheal cannula and removed from the thoracic cavity. Tissue was fixed overnight, embedded in paraffin, and sectioned for staining with hematoxylin-eosin.
Statistics
Data are expressed as the mean ± SEM where applicable. We assessed differences in measured variables using the unpaired two-sided Student t-test or Wilcoxon rank-sum test. Differences between multiple groups were calculated using one-way ANOVA with Tukey-Kramer post test or two-way ANOVA with Bonferroni multiple comparisons. Statistical difference was accepted at P < 0.05. Prism5.0 software was used to perform statistical analysis.
Results
Mature miR-127 is regulated during inflammation
There are limited reports suggesting that miR-127 may play a role in development (36) and tumor progression (37). However, there is no report on miR-127 targets or on its role in inflammation. We recently analyzed miRNA expression after bleomycin-induced lung injury with a miRNA array study (29) and identified that miR-127 expression was regulated after injury. Bleomycin treatment led to down-regulation of miR-127 at day 1 after bleomycin (Fig. 1A). We also found that miR-127 was down-regulated after IgG IC-induced lung injury (Fig. 1B). We reasoned that miR-127 may play a role in regulating lung injury and inflammatory response. When we measured the expression of miR-127 in murine RAW264.7 macrophages and MH.S alveolar macrophages upon LPS stimulation, we found that overnight treatment with LPS showed a marginal down-regulation of miR-127 expression in both MH.S (Fig. S1A) and RAW264.7 macrophages (Fig. S1B).
Figure 1. Lung injury regulated miR-127 expression.
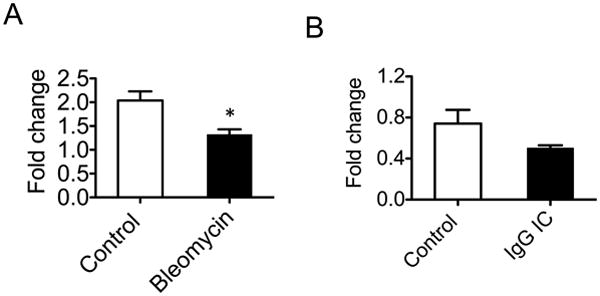
A. Total RNA was isolated from normal lungs and lungs one day after intratracheal bleomycin instillation (2.5U/kg). miR-127 expression was determined with TaqMan microRNA assay. B. miR-127 was decreased after IgG IC-induced lung injury.
Over-expression of miR-127 decreased cytokine expression
We next determined the ability of miR-127 to modulate the inflammatory response in macrophages in vitro. To accomplish this, we transduced MH.S cells with a lentivirus encoding miR-127 (pSico-miR127) or an empty lentivirus (pSico) as control (31). Pre-miR-127 gene was cloned into a pSico vector in which CMV-GFP cassette is located downstream of the U6 promoter and is flanked by loxP sites such that Cre-mediated recombination is expected to result in the loss of GFP expression and the activation of miRNA expression (Fig. S2A). PCR confirmation of Cre-mediated recombination was performed to amplify the recombined and un-recombined viral DNA (Fig. S2B). High-efficiency transduction by the vector was achieved as indicated by uniform GFP expression in infected cells (Fig. S2C). After super-infection with a Cre-expressing recombinant adenovirus, near complete recombination with concomitant loss of GFP fluorescence was observed (Fig. S1C). One week after Cre expression, more than 2 fold higher level of miR-127 expression was detected in cells infected with pSico-miR-127 versus control cells (Fig. S1D). This suggested we had successfully established the miR-127 over-expressed MH.S cell line.
To test the hypothesis that the gain of miR-127 function plays a role in the release of cytokines during inflammation, MH.S-miR127 and control cells were treated with LPS. Over-expression of miR-127 in MH.S cells significantly decreased production of pro-inflammatory cytokines induced by LPS, such as TNF-α, IL-1β, and IL-6 (Fig. 2A-C). These data suggest that miR-127 functions as a negative regulator of inflammatory response. Given the ability of miR-127 to down-regulate TLR4 induced inflammatory responses, we next investigated if miR-127 also modulates activation of peptidoglycan (PGN), hyaluronan fragments, and IgG immune complex (IgG IC) stimulated cells. As shown in Fig. 2, miR-127 significantly attenuated TNF-α and IL-1β production induced by PGN and hyaluronan fragments (Fig. 2A-B), as well as IL-6 production induced by PGN and IgG IC (Fig. 2C).
Figure 2. Stable expression of miR-127 in MH.S cells attenuated inflammatory response.
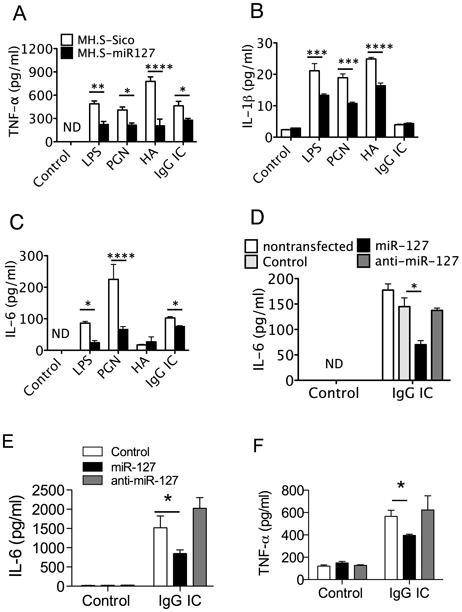
A-C. MH.S-miR127 and control cells were treated with LPS, hyaluronan (HA), PGN, and IgG IC for 4 h. The levels of TNF-α (A), IL-1β (B), and IL-6 (C) in the supernatants were determined by ELISA. D-E. miR-127 precursor attenuated IgG IC-induced inflammatory response in RAW264.7 cells and MH.S cells. RAW264.7 cells (D) and MH.S cells (E) were transiently transfected with 10 nM control probes, miR-127 precursor, and miR-127 inhibitor. At 48 h after transfection, the cells were cultured with or without IgG IC for 4 h. The levels of IL-6 in the supernatants were then determined. F. Human monocyte U937 cells were activated with PMA at 100 nM for 24 hours, and the cells were washed and then cultured in regular medium for 24 hours before transfection. The cells were transiently transfected with 10 nM control probes, miR-127 precursor, and miR-127 inhibitor. At 48 h after transfection, the cells were cultured without or with IgG IC for 4 h. The levels of IL-6 in the supernatants were then determined. miR-127 precursor attenuated IgG IC-induced TNF-α production human U937 cells. Values are presented as mean ± SEM. n = 3-4, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 compared to control group. The experiments were repeated 3 times.
To further address this question, RAW264.7 cells, MH.S cells, and PMA treated human monocyte U937 cells were transiently transfected with a specific miR-127 precursor, miR-127 inhibitor or control probes. miR-127 precursor markedly inhibited IgG IC-induced IL-6 by RAW264.7 cells (Fig. 2D), TNF-α by MH.S cells (Fig. 2E), and human TNF-α by U937 cells (Fig. 2F). However, miR-127 precursor, miR-127 inhibitor or the control probes alone did not alter cytokine production by RAW264.7, MH.S cells, or U937 cells (Fig. 2D-F). These results demonstrated that miR-127 possesses the ability to suppress cytokine releases in various inflammation models in vitro.
CD64 is a target of miR-127
We next focused our study on miR-127 regulation in IgG IC induced inflammation because putative mRNA targets predicted in silico [MicroCosm Targets Version 5 (http://www.ebi.ac.uk/enright-srv/microcosm/htdocs/targets/v5/)] were members of pathways implicated in FcγR mediated immune response in macrophages, including FcγRI, P13K cat class IA, VAV-1, PIP5KI, and Myosin II. Notably, CD64 was predicted to have a strong miR-127 binding site (Fig. 3A) and showed a minimum free energy of –26.24 kcal/mol.
Figure 3. FcγRI (CD64) is a target of miR-127.
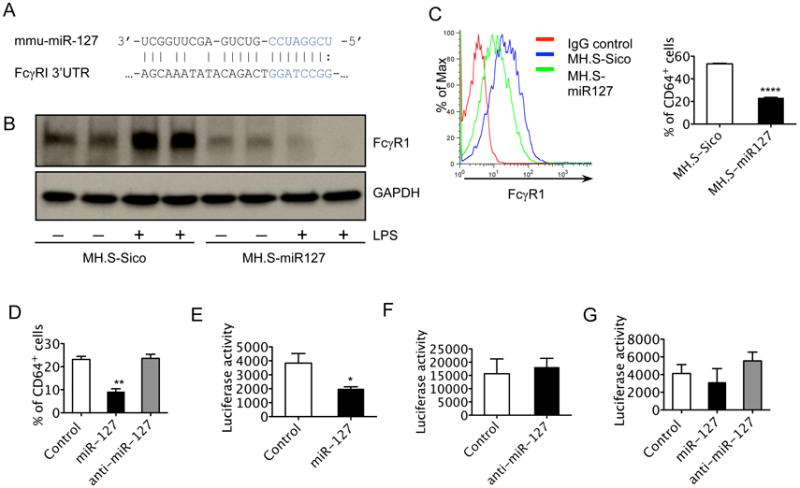
A. Predicted binding site for miR-127 in 3′-UTR of CD64. miR-127 seed sequence was in blue. B. Western blot analysis was performed to determine the levels of CD64 protein in MH.S-miR127 and control cells without or with LPS (100 ng/ml) treatment for 4h. C. Flow cytometry was performed to determine the percent of CD64 staining positive cells in MH.S-miR127 and control cells. D. RAW264.7 cells were transfected with 10nM control probes, LNA-miR127, and LNA-anti-miR127. 48 h after transfection, the cells were stained with CD64 antibody. The percent of CD64 staining positive cells were then determined. E-F. Luciferase activity of miR-127 precursor (E), miR-127 inhibitor (F) or control probe and pMIR-REPORT[FcγRI-3′-UTR] co-transfection. G. Luciferase activity of miR-127 precursor, miR-127 inhibitor or control probe and pMIR-REPORT[mFcγRI-3′-UTR] (containing a mutated 3′UTR of FcγRI) co-transfection (n = 3-4, *p < 0.05, **p < 0.01, ****p < 0.0001). The experiments were repeated 3 times.
We then performed cDNA microarray analysis to determine if CD64 expression is different between MH.S-miR127 and control MH.S-Sico cells. The microarray analysis of RNA isolated from MH.S-miR127 and MH.S-Sico cells revealed a down-regulation of FcγRI transcripts in MH.S-miR127 cells compared to MH.S-Sico cells (Fig. S3). These data display an inverse relationship between CD64 expression and miR-127 expression, indicating that CD64 is a putative target of miR-127.
Next, Western blot analysis of CD64 was performed in MH.S-miR127 and control cells with and without LPS treatment. CD64 protein expression was induced by LPS in control cells, but protein expression of CD64 in MH.S-miR127 cells was significantly inhibited in response to LPS stimulation (Fig. 3B). Since CD64 is an integral membrane glycoprotein, we performed fluorescence-activated cell sorting (9) to investigate if membrane CD64 is regulated by miR-127. As expected, flow cytometry analysis showed a decrease in CD64 positive staining on MH.S-miR127 cells when compared with control cells (Fig. 3C). We also found decreased CD64 positive staining in miR-127 precursor transfected RAW 264.7 cells compared with the transfection with miR127 inhibitor or control probe (Fig. 3D). Together, these data demonstrate that CD64 is a potential target of miR-127 and is down regulated by miR-127 over-expression.
To further obtain direct evidence that CD64 is a target of miR-127, and to determine whether miR-127 targets and represses CD64 directly through a 3′-UTR interaction, we constructed a luciferase reporter vector containing the full-length CD64 3′-UTR (pMIR-REPORT[FcγRI-3′-UTR]) and analyzed the effect of miR-127 on luciferase expression. As anticipated, co-transfection of miR-127 attenuated expression of luciferase activity from the pMIR-REPORT[FcγRI-3′-UTR] reporter (Fig. 3E), whereas the inhibition was not observed when either control miRNA or miR-127 inhibitor were used (Fig. 3F). Correspondingly, we also generated a mutant reporter (pMIR-REPORT[mFcγRI-3′-UTR]) in which the seed binding sequence in 3′-UTR of CD64 was mutated. The luciferase activity in pMIR-REPORT[mFcγRI-3′-UTR] and miR-127 co-transfected cells showed no differences when compared with pMIR-REPORT[mFcγRI-3′-UTR] and control miRNA or miR-127 inhibitor co-transfection (Fig. 3G). These suggest that miR-127 reduced the luciferase activity by binding to CD64-3′UTR, but had no effect on CD64 mutant 3′-UTR. Therefore, we concluded that CD64 is a bona fide target of miR-127.
Expression of CD64 is positively correlated with IgG IC-induced inflammatory response
To test whether CD64 functionally regulates IgG IC-induced cytokine expression, RAW264.7 and MH.S cells were stained with anti-CD64 antibody and flow cytometry analysis was used to sort out the top 1% of the highest CD64 expressing cells, as CD64hi, and the bottom 1% of the lowest CD64 expressing cells, as CD64lo. Next, the highest and lowest CD64 expressed RAW264.7 and MH.S cells were treated with IgG IC and cytokine expression in the conditioned media were measured. RAW264.7-CD64lo cells showed a decrease in TNF-α, IL-1β and IL-6 release after IgG IC stimulation as compared to RAW264.7-CD64hi cells (Fig. 4A). Similarly, MH.S-CD64lo cells showed a decrease in pro-inflammatory cytokines such as IL-6, after IgG IC stimulation, as compared to MH.S-CD64hi cells (Fig. 4B). RAW264.7-CD64lo cells also showed a decrease in IL-1β release after LPS stimulation as compared to RAW264.7-CD64hi cells (Fig. 4C). These data demonstrate that expression of CD64 is closely associated with the inflammatory response of macrophages activated by IgG IC.
Figure 4. Expression of CD64 correlated with IgG IC induced inflammatory response.
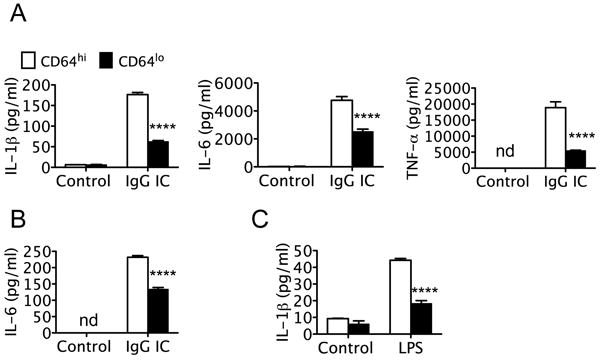
A. RAW264.7 cells were sorted for the highest or lowest 1% cells expressing CD64. CD64hi and CD64lo were treated without or with IgG IC for 4 h. The levels of IL-1β, IL-6, and TNF-a in the supernatants were then determined by ELISA. B. MH.S cells were sorted for CD64 highest or lowest 1% expressing cells. The levels of IL-6 in IgG IC treated supernatant of CD64hi and CD64lo MH.S cells were then determined. C. CD64hi and CD64lo RAW264.7 cells were treated with or without LPS for 4 h. The levels of IL-1β in the supernatants were then determined by ELISA. Values are presented as mean ± SEM. n = 3-6, ****p < 0.0001 compared to control group. The experiments were repeated 3 times.
MiR-127 attenuates IgG IC-induced lung injury in vivo
We have shown that the IgG IC induced inflammation is inhibited by miR-127 in vitro. We next investigated whether miR-127 mediates regulation of inflammatory response in complement mediated acute lung injury in vivo. We administered miR-127 probes or control (CEMR67 or PBS) intranasally 2 hours before onset of the IgG IC, and then analyzed the extent of lung inflammation 4 h after onset of IgG IC model. Neither CEMR67 nor PBS had any effect. Flow cytometry revealed that more than half of macrophages had taken up Fam-labeled miR-127 (Fig. S4A). RT-PCR revealed an accumulation of miR-127 in the lungs of the miR-127 treatment group (Fig. S4B). miR-127 treated macrophages expressed less cell surface CD64 when compared with control oligonucleotide CEMR67 (or PBS) treated macrophages. (Fig. S4C). IgG IC induced a typical lung pathology (30, 33) characterized by lung hemorrhage, edema, fibrin deposition, and accumulation of inflammatory cells. However, treatment with miR-127 displayed a significant reduction in pathological changes (Fig. 5A). We collected bronchoalveolar lavage fluid (BALF) from mice of control, IgG IC model, and miR-127 treatment groups. Examination of the inflammatory response to IgG IC revealed a significant attenuation in total protein level (Fig. 5B) and Evans blue concentration (Fig. 5C). Furthermore, miR-127 also markedly reduced inflammatory cell infiltration (Fig. 5D-E) as well as neutrophil influx (Fig. 5F). These data suggest that miR-127 significantly attenuated IgG IC-induced lung injury.
Figure 5. miR-127 attenuated IgG IC induced acute lung injury in mice.
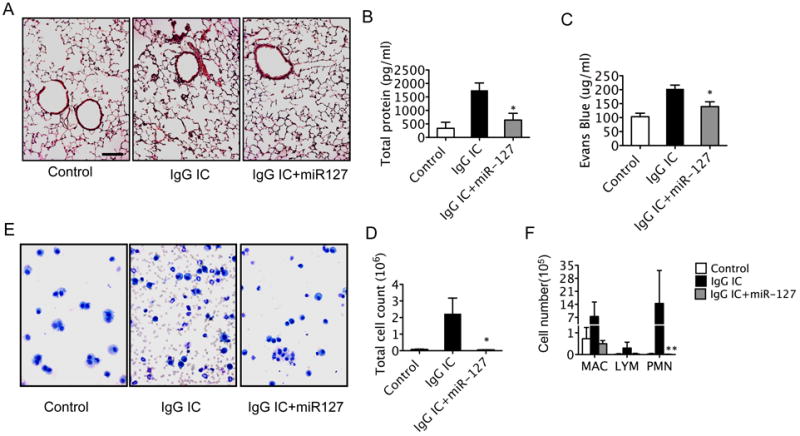
Mice received either PBS or miR-127 intranasally 2 h before onset of IgG IC model (10 μg/mouse anti-BSA in 40 μl PBS intranasal instillation and 5 mg/kg BSA together with 0.25% Evans blue in 200 μl PBS intravascular injection). At 4 h after IgG IC onset, mice were sacrificed and BALF was collected. Lungs tissue was fixed, embedded in paraffin, and sectioned for staining with hematoxylin-eosin. A. miR-127 decreases lung inflammation and alveolar hemorrhage on lung sections stained by hematoxylin and eosin. B. Total protein levels in BALF were performed by BCA assay. C. Evans blue concentrations in BALF were measured. D. Total cell counts in BALF were performed to determine inflammatory cell infiltration. E. Representative cytospin preparations stained with Protocol Hema3 stain set. F. Differential cell counts were performed on cytospin preparations of BAL (mac, macrophages; lym, lymphocytes; PMN, neutrophils). Values are presented as mean ± SEM. n = 3 - 6 in each group; *p < 0.05, **p < 0.01, compared to IgG IC group. The experiments were repeated 3 times.
MiR-127 inhibits IgG IC-induced lung inflammation in vivo
Next we examined the role of miR-127 in the effect on cytokine release. Consistent with our in vitro observation, miR-127 significantly reduced cytokine release into the alveolar space. All major cytokines and chemokines (IL-6, TNF-α, MIP-1β, MIP-2, MCP-1, and KC) were reduced by miR-127 in IgG IC-induced lung injury (Fig. 6A).
Figure 6. miR-127 inhibited cytokine production and compliment component in IgG IC induced acute lung injury in vivo.
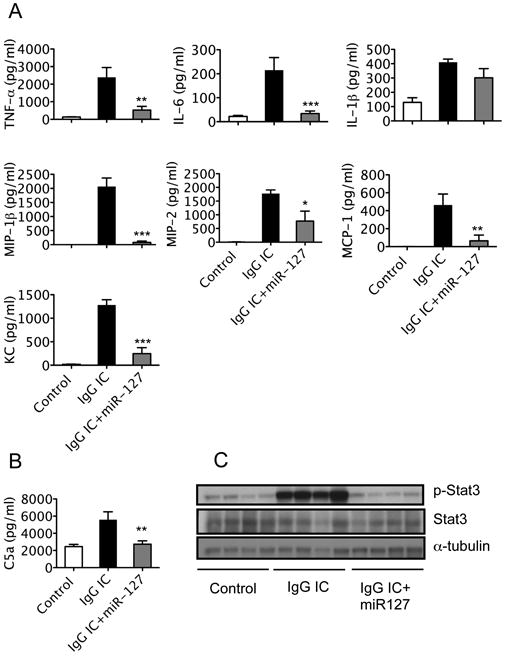
Mice received either PBS or miR-127 probes intranasally 2 h before onset of IgG IC model. At 4 h after IgG IC onset, mice were sacrificed and BALF or lung tissue was collected. A. Protein levels of IL-6, TNF-α, IL-1β, MIP-1β, MIP-2, MCP-1, and KC in BALF were measured with ELISA. B. Complement C5a protein in BALF was measured with ELISA. Values are presented as mean ± SEM. n = 3 - 6 in each group; *p < 0.05, **p < 0.01, ***p < 0.001 compared to IgG IC group. The experiments were repeated 3 times. C. Western blot was performed to determine STAT3 and phospho-STAT3 protein levels in lung tissues.
Complement activation is an important component of immune complex-mediated tissue injury (38). Complement 5 fragment a (C5a) has been demonstrated to be required for the full development of injury and neutrophil accumulation in an IgG IC model of lung injury (33, 39). Anti-C5a antibody reduced in lung vascular permeability and lung myeloperoxidase, and suppressed up-regulation of ICAM-1 and TNFα in the IgG immune complex model of lung injury (33). Anti-C5a antibody attenuated ARDS and some of the systemic manifestations of sepsis in nonhuman primates (40). Therefore, we wanted to determine whether complement C5a was affected. IgG immune complex markedly induced C5a in mouse BAL. miR-127 treatment significantly reduced C5a protein levels in the BAL (Fig. 6B).
Signal transducer and activator of transcription 3 (STAT3) is a transcription factor transducing signals in response to cytokines and growth factors. STAT3 is activated in the lung during IgG IC-induced acute lung injury (30). Therefore, we wanted to determine if miR-127 treatment influences STAT3 activation. STAT3 and Phosphor-STAT3 (p-STAT3) protein levels were examined in total lung proteins. P-STAT3 protein levels were elevated in the IgG IC onset lungs, but diminished by miR127 treatment (Fig. 6C). Thus, miR-127 appears to function in attenuating IgG IC induced inflammatory response in vivo through the regulation of CD64.
Discussion
In this study, we found that miR-127 expression was regulated during inflammation both in vitro and in vivo. Functionally, miR-127 regulated cytokine release by macrophages in vitro and attenuated lung injury in an IgG IC-induced lung injury model in vivo. Mechanistically, miR-127 directly targeted 3′-UTR of CD64, resulting in the down-regulation of CD64.
Several studies have demonstrated that miRNAs regulate inflammation and the immune response. For example, a recent study showed that miR-21 was up-regulated in the lungs of mice with bleomycin-induced injury and also in the lungs of patients with idiopathic pulmonary fibrosis, and that miR-21 may regulate lung injury and fibrosis through targeting Smad7 (24). The down-regulation of let-7d was found in the patients with idiopathic pulmonary fibrosis and let-7d targets HMGA2 in epithelial cells (41). Let-7g was reported to be down-regulated in LPS-induced acute inflammation in circulating leukocytes (42). miR-126 promotes Th2 responses in the lung (25). miR-146a has been shown to play a role in innate immunity (43). However, the role of miR-127 in immune response and inflammation has not been investigated and the targets of miR-127 have not been identified.
There are limited reports suggesting that miR-127 may play a role in development (36) and tumor progression (37, 44, 45). We recently analyzed miRNA expression after bleomycin-induced lung injury with a miRNA array study (29) and identified that miR-127 expression was decreased at one day after bleomycin treatment. In this study, we showed that miR-127 expression is marginally down-regulated in IgG IC induced lung injury in vivo and decreased in macrophages when stimulated with inflammatory stimuli in vitro, suggesting miR-127 may play a role during inflammatory response. We focused our study on miR-127 regulation in IgG IC induced inflammation because putative mRNA targets predicted in silico were members of pathways implicated in FcγR mediated immune response in macrophages, including FcγRI, P13K cat class IA, VAV-1, PIP5KI, and Myosin II.
We provide several lines of evidence suggesting that FcγR1 is a genuine target of miR-127. First, using bioinformatics tools, targets of miR-127 were identified and CD64 was selected for experimental validation, given its known role in the inflammatory response (8). cDNA array analysis identified that CD64 was down-regulated in miR-127 over-expressing macrophages. We further showed that over-expression of miR-127 resulted in a decrease in mRNA, protein, and cell surface expression of CD64. Lastly, reduced luciferase activity was observed in a reporter system containing the full length of 3′ UTR of CD64, demonstrating that miR-127 directly targets CD64. However, we did not detect any effect of miR-127 inhibitor on IgG IC-induced cytokine release, cell surface CD64 expression, or CD64-luciferase activity in macrophages. It is possible that the endogenous miR-127 expression is low in lung tissue (36) as well as in macrophages, while CD64 expression is abundant on macrophages. Introduction of anti-miR-127 into macrophages may not be able to further reduce endogenous miR-127 levels, so that anti-miR-127 may not up-regulate CD64 expression in the cells. On the contrary, introduction of miR-127 mimics into cells can readily increase miR-127 levels in the cells and lead to significantly down-regulated CD64 expression.
By establishing stable or transient miR-127 over-expression in macrophage cell lines, we observed decreased TNF-α, IL-1β, or IL-6 release correlated with over-expression of miR-127 when stimulated with LPS, HA, PGN or IgG IC. We also present a functional role for miR127-CD64 axis in the signaling pathways induced in response to IgG IC. IgG IC or LPS treatment of sorted highest and lowest CD64 expression macrophages revealed a correlation of decreased cytokine releases and the loss of CD64.
Next we determined the role of miR-127 in an IgG IC induced acute lung injury model in vivo (8). miR-127 attenuated the inflammatory response and lung permeability in IgG IC induced acute lung injury, as indicated by a change in lung morphology, decreased inflammatory cell infiltration, cytokine releases, total protein level, Evans blue concentration, and complement activating protein C5a. C5a has been demonstrated to be required for the full development of injury and neutrophil accumulation in an IgG IC model of lung injury (39). It has shown that C5a directly activated chemokine production by neutrophils and macrophages. In the presence of IgG IC, C5a and C5b-C9 membrane attack complex cause a synergistic intrapulmonary generation of MIP-1β, MIP-2 and MCP-1 (46). Also, we performed Western blot to analyze the total STAT3 and phosphor-STAT3 levels in whole lung tissue. As previous reports indicate, STAT3 is activated in the lung during IgG IC induced acute lung injury, and its activity is chiefly regulated at the post-translational level (30). We found that miR127 could prevent IgG IC induced phosphorylation of STAT3.
In our study, we found that over expression of miR-127 leads to down-regulation of CD64 expression. CD64 is one member of the IgG receptor family. Binding of IgG to FcγRs triggers a wide variety of cellular functions including phagocytosis, antibody-dependent cellular cytotoxicity, and release of inflammatory mediators, as well as clearance of immune complexes and regulation of antibody production (7). FcγRs have been indicated to be important immune-regulatory receptors involved in various disease states of allergy, autoimmunity, and inflammation, especially IgG-mediated human disease (7). By using a variety of FcγR−/− mice, Ioan-Facsinay and colleagues were able to show that in the absence of FcγRI, IgG2a-IC-induced cellular processes of phagocytosis, cytokine release, cellular cytotoxicity, and antigen presentation are impaired (9). Furthermore, FcγRI contributes significantly to enhanced PMN infiltration in IC-induced peritonitis (47). Mice lacking FcγRI showed reduced inflammatory responses in an immune complex-induced reverse passive Arthus model (48). Given the importance of FcγRI in IgG IC-mediated inflammatory responses, our findings determined that miR-127 acts as an anti-inflammatory factor by targeting FcγRI both in vitro and in vivo. Interestingly, the microarray analysis of different gene expression revealed an increase of FcγRII subunits in MH.S-miR127 cells when compared with control cells. As indicated before, FcγRII is a negative regulator of immune complex signaling. It is reported that FcγRII prevents inappropriate activation of effector responses. Mice with FcγRII deficiency were hyperresponsive to sub-threshold levels of cytotoxic antibodies (49) and immune complexes (50). This suggests to us that miR-127 may result in increased expression of FcRI and decreased expression FcRII, which collaboratively inhibit the inflammatory response. Hence, our data show the potential of miR-127 as a target for the treatment of inflammatory diseases such as ALI and ARDS.
As we focused on the role of miR-127 in immune complex-induced lung inflammation, our data suggest CD64 is the target of miR-127. We have also showed that miR-127 may have a role in the inflammatory response induced by LPS, PGN, and hyaluronan fragments. It is possible that the effect of miR-127 on cytokine production stimulated by TLR ligands is through CD64, since it has been suggested that CD64 contributes to protection from bacterial infection (9) and there are crosstalk pathways between TLRs and FcγRs (51). However, it is more plausible that miR-127 targets a set of genes, of which CD64 plays a key role in immune complex-induced inflammation. In addition to CD64, there are at least 24 other genes putatively targeted by miR-127 involved in immune response, based on in silico analysis. By GeneGo pathway analysis (www.genego.com), these genes are involved in Phosphatidylinositol (3,4,5)-triphosphate (PIP3) signaling, nuclear factor of activated T-cells (NFAT) signaling, CD28 signaling, and triggering receptor expressed on myeloid cells 1 (TREM1) signaling. By targeting a set of genes including CD64, miR-127 may therefore play a role in regulation of the inflammatory response to a variety of IgG receptor as well as TLR stimulation.
In summary, we describe for the first time that a miRNA was involved in IgG IC induced lung injury. Specifically, we identify that miR-127 targeted CD64. miR-127 administration inhibited the inflammatory response of IgG immune-complex induced lung injury. We believe that these findings have important implications in the regulation of the adaptive immune response in acute lung injury. Further investigation into the miR-127-CD64 axis would pave the way to developing a novel therapeutic approach to the treatment of infectious and non-infectious lung inflammation.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health grants R01-HL 060539 and R01-HL 077291 (to P.W.N). T.X. was supported in part by a Ph.D. student fellowship from the China Scholarship Council.
References
- 1.Matthay MA, Zemans RL. The acute respiratory distress syndrome: pathogenesis and treatment. Annu Rev Pathol. 2011;6:147–163. doi: 10.1146/annurev-pathol-011110-130158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wheeler AP, Bernard GR. Acute lung injury and the acute respiratory distress syndrome: a clinical review. Lancet. 2007;369:1553–1564. doi: 10.1016/S0140-6736(07)60604-7. [DOI] [PubMed] [Google Scholar]
- 3.Crosby LM, Waters CM. Epithelial repair mechanisms in the lung. Am J Physiol Lung Cell Mol Physiol. 2010;298:L715–731. doi: 10.1152/ajplung.00361.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Opitz B, van Laak V, Eitel J, Suttorp N. Innate immune recognition in infectious and noninfectious diseases of the lung. Am J Respir Crit Care Med. 2010;181:1294–1309. doi: 10.1164/rccm.200909-1427SO. [DOI] [PubMed] [Google Scholar]
- 5.Jiang D, Liang J, Noble PW. Regulation of non-infectious lung injury, inflammation, and repair by the extracellular matrix glycosaminoglycan hyaluronan. Anat Rec (Hoboken) 2010;293:982–985. doi: 10.1002/ar.21102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA, Homer RJ, Goldstein DR, Bucala R, Lee PJ, Medzhitov R, Noble PW. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med. 2005;11:1173–1179. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 7.Gessner JE, Heiken H, Tamm A, Schmidt RE. The IgG Fc receptor family. Ann Hematol. 1998;76:231–248. doi: 10.1007/s002770050396. [DOI] [PubMed] [Google Scholar]
- 8.Gao H, Neff T, Ward PA. Regulation of lung inflammation in the model of IgG immune-complex injury. Annu Rev Pathol. 2006;1:215–242. doi: 10.1146/annurev.pathol.1.110304.100155. [DOI] [PubMed] [Google Scholar]
- 9.Ioan-Facsinay A, de Kimpe SJ, Hellwig SM, van Lent PL, Hofhuis FM, van Ojik HH, Sedlik C, da Silveira SA, Gerber J, de Jong YF, Roozendaal R, Aarden LA, van den Berg WB, Saito T, Mosser D, Amigorena S, Izui S, van Ommen GJ, van Vugt M, van de Winkel JG, Verbeek JS. FcgammaRI (CD64) contributes substantially to severity of arthritis, hypersensitivity responses, and protection from bacterial infection. Immunity. 2002;16:391–402. doi: 10.1016/s1074-7613(02)00294-7. [DOI] [PubMed] [Google Scholar]
- 10.Eagen JW, Memoli VA, Roberts JL, Matthew GR, Schwartz MM, Lewis EJ. Pulmonary hemorrhage in systemic lupus erythematosus. Medicine (Baltimore) 1978;57:545–560. doi: 10.1097/00005792-197811000-00005. [DOI] [PubMed] [Google Scholar]
- 11.Okroj M, Heinegard D, Holmdahl R, Blom AM. Rheumatoid arthritis and the complement system. Ann Med. 2007;39:517–530. doi: 10.1080/07853890701477546. [DOI] [PubMed] [Google Scholar]
- 12.Borza DB, Neilson EG, Hudson BG. Pathogenesis of Goodpasture syndrome: a molecular perspective. Seminars in nephrology. 2003;23:522–531. doi: 10.1053/s0270-9295(03)00131-1. [DOI] [PubMed] [Google Scholar]
- 13.McCluskey RT. Immunopathogenetic mechanisms in renal disease. Am J Kidney Dis. 1987;10:172–180. doi: 10.1016/s0272-6386(87)80171-3. [DOI] [PubMed] [Google Scholar]
- 14.Krupa A, Fudala R, Stankowska D, Loyd T, Allen TC, Matthay MA, Gryczynski Z, Gryczynski I, Mettikolla YV, Kurdowska AK. Anti-chemokine autoantibody:chemokine immune complexes activate endothelial cells via IgG receptors. Am J Respir Cell Mol Biol. 2009;41:155–169. doi: 10.1165/rcmb.2008-0183OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dreisin RB, Schwarz MI, Theofilopoulos AN, Stanford RE. Circulating immune complexes in the idiopathic interstitial pneumonias. N Engl J Med. 1978;298:353–357. doi: 10.1056/NEJM197802162980701. [DOI] [PubMed] [Google Scholar]
- 16.Soda K, Ando M, Sakata T, Sugimoto M, Nakashima H, Araki S. C1q and C3 in bronchoalveolar lavage fluid from patients with summer-type hypersensitivity pneumonitis. Chest. 1988;93:76–80. doi: 10.1378/chest.93.1.76. [DOI] [PubMed] [Google Scholar]
- 17.Kurdowska A, Miller EJ, Noble JM, Baughman RP, Matthay MA, Brelsford WG, Cohen AB. Anti-IL-8 autoantibodies in alveolar fluid from patients with the adult respiratory distress syndrome. J Immunol. 1996;157:2699–2706. [PubMed] [Google Scholar]
- 18.Krupa A, Walencka MJ, Shrivastava V, Loyd T, Fudala R, Frevert CW, Martin TR, Kurdowska AK. Anti-KC autoantibody:KC complexes cause severe lung inflammation in mice via IgG receptors. Am J Respir Cell Mol Biol. 2007;37:532–543. doi: 10.1165/rcmb.2006-0395OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Valencia-Sanchez MA, Liu J, Hannon GJ, Parker R. Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev. 2006;20:515–524. doi: 10.1101/gad.1399806. [DOI] [PubMed] [Google Scholar]
- 20.Lindsay MA. microRNAs and the immune response. Trends Immunol. 2008;29:343–351. doi: 10.1016/j.it.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 21.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006;103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nahid MA, Satoh M, Chan EK. Mechanistic Role of MicroRNA-146a in Endotoxin-Induced Differential Cross-Regulation of TLR Signaling. J Immunol. 2010 doi: 10.4049/jimmunol.1002311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tili E, Michaille JJ, Adair B, Alder H, Limagne E, Taccioli C, Ferracin M, Delmas D, Latruffe N, Croce CM. Resveratrol decreases the levels of miR-155 by upregulating miR-663, a microRNA targeting JunB and JunD. Carcinogenesis. 2010;31:1561–1566. doi: 10.1093/carcin/bgq143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu G, Friggeri A, Yang Y, Park YJ, Tsuruta Y, Abraham E. miR-147, a microRNA that is induced upon Toll-like receptor stimulation, regulates murine macrophage inflammatory responses. Proc Natl Acad Sci U S A. 2009;106:15819–15824. doi: 10.1073/pnas.0901216106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mattes J, Collison A, Plank M, Phipps S, Foster PS. Antagonism of microRNA-126 suppresses the effector function of TH2 cells and the development of allergic airways disease. Proc Natl Acad Sci U S A. 2009;106:18704–18709. doi: 10.1073/pnas.0905063106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neilson JR, Zheng GX, Burge CB, Sharp PA. Dynamic regulation of miRNA expression in ordered stages of cellular development. Genes Dev. 2007;21:578–589. doi: 10.1101/gad.1522907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li QJ, Chau J, Ebert PJ, Sylvester G, Min H, Liu G, Braich R, Manoharan M, Soutschek J, Skare P, Klein LO, Davis MM, Chen CZ. miR-181a is an intrinsic modulator of T cell sensitivity and selection. Cell. 2007;129:147–161. doi: 10.1016/j.cell.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 28.Thai TH, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, Murphy A, Frendewey D, Valenzuela D, Kutok JL, Schmidt-Supprian M, Rajewsky N, Yancopoulos G, Rao A, Rajewsky K. Regulation of the germinal center response by microRNA-155. Science. 2007;316:604–608. doi: 10.1126/science.1141229. [DOI] [PubMed] [Google Scholar]
- 29.Xie T, Liang J, Guo R, Liu N, Noble PW, Jiang D. Comprehensive microRNA analysis in bleomycin-induced pulmonary fibrosis identifies multiple sites of molecular regulation. Physiol Genomics. 2011 doi: 10.1152/physiolgenomics.00222.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao H, Guo RF, Speyer CL, Reuben J, Neff TA, Hoesel LM, Riedemann NC, McClintock SD, Sarma JV, Van Rooijen N, Zetoune FS, Ward PA. Stat3 activation in acute lung injury. J Immunol. 2004;172:7703–7712. doi: 10.4049/jimmunol.172.12.7703. [DOI] [PubMed] [Google Scholar]
- 31.Ventura A, Meissner A, Dillon CP, McManus M, Sharp PA, Van Parijs L, Jaenisch R, Jacks T. Cre-lox-regulated conditional RNA interference from transgenes. Proc Natl Acad Sci U S A. 2004;101:10380–10385. doi: 10.1073/pnas.0403954101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watson A, Latchman D. Gene Delivery into Neuronal Cells by Calcium Phosphate-Mediated Transfection. Methods. 1996;10:289–291. doi: 10.1006/meth.1996.0105. [DOI] [PubMed] [Google Scholar]
- 33.Mulligan MS, Schmid E, Beck-Schimmer B, Till GO, Friedl HP, Brauer RB, Hugli TE, Miyasaka M, Warner RL, Johnson KJ, Ward PA. Requirement and role of C5a in acute lung inflammatory injury in rats. J Clin Invest. 1996;98:503–512. doi: 10.1172/JCI118818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang D, Liang J, Campanella GS, Guo R, Yu S, Xie T, Liu N, Jung Y, Homer R, Meltzer EB, Li Y, Tager AM, Goetinck PF, Luster AD, Noble PW. Inhibition of pulmonary fibrosis in mice by CXCL10 requires glycosaminoglycan binding and syndecan-4. J Clin Invest. 2010;120:2049–2057. doi: 10.1172/JCI38644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang D, Liang J, Hodge J, Lu B, Zhu Z, Yu S, Fan J, Gao Y, Yin Z, Homer R, Gerard C, Noble PW. Regulation of pulmonary fibrosis by chemokine receptor CXCR3. J Clin Invest. 2004;114:291–299. doi: 10.1172/JCI16861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bhaskaran M, Wang Y, Zhang H, Weng T, Baviskar P, Guo Y, Gou D, Liu L. MicroRNA-127 modulates fetal lung development. Physiol Genomics. 2009;37:268–278. doi: 10.1152/physiolgenomics.90268.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tryndyak VP, Ross SA, Beland FA, Pogribny IP. Down-regulation of the microRNAs miR-34a, miR-127, and miR-200b in rat liver during hepatocarcinogenesis induced by a methyl-deficient diet. Mol Carcinog. 2009;48:479–487. doi: 10.1002/mc.20484. [DOI] [PubMed] [Google Scholar]
- 38.Hammerschmidt DE, Weaver LJ, Hudson LD, Craddock PR, Jacob HS. Association of complement activation and elevated plasma-C5a with adult respiratory distress syndrome. Pathophysiological relevance and possible prognostic value. Lancet. 1980;1:947–949. doi: 10.1016/s0140-6736(80)91403-8. [DOI] [PubMed] [Google Scholar]
- 39.Guo RF, Ward PA. Role of C5a in inflammatory responses. Annu Rev Immunol. 2005;23:821–852. doi: 10.1146/annurev.immunol.23.021704.115835. [DOI] [PubMed] [Google Scholar]
- 40.Stevens JH, O'Hanley P, Shapiro JM, Mihm FG, Satoh PS, Collins JA, Raffin TA. Effects of anti-C5a antibodies on the adult respiratory distress syndrome in septic primates. J Clin Invest. 1986;77:1812–1816. doi: 10.1172/JCI112506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pandit KV, Corcoran D, Yousef H, Yarlagadda M, Tzouvelekis A, Gibson KF, Konishi K, Yousem SA, Singh M, Handley D, Richards T, Selman M, Watkins SC, Pardo A, Ben-Yehudah A, Bouros D, Eickelberg O, Ray P, Benos PV, Kaminski N. Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2010;182:220–229. doi: 10.1164/rccm.200911-1698OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schmidt WM, Spiel AO, Jilma B, Wolzt M, Muller M. In vivo profile of the human leukocyte microRNA response to endotoxemia. Biochem Biophys Res Commun. 2009;380:437–441. doi: 10.1016/j.bbrc.2008.12.190. [DOI] [PubMed] [Google Scholar]
- 43.Williams AE, Perry MM, Moschos SA, Larner-Svensson HM, Lindsay MA. Role of miRNA-146a in the regulation of the innate immune response and cancer. Biochem Soc Trans. 2008;36:1211–1215. doi: 10.1042/BST0361211. [DOI] [PubMed] [Google Scholar]
- 44.Yan LX, Huang XF, Shao Q, Huang MY, Deng L, Wu QL, Zeng YX, Shao JY. MicroRNA miR-21 overexpression in human breast cancer is associated with advanced clinical stage, lymph node metastasis and patient poor prognosis. RNA. 2008;14:2348–2360. doi: 10.1261/rna.1034808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dixon-McIver A, East P, Mein CA, Cazier JB, Molloy G, Chaplin T, Andrew Lister T, Young BD, Debernardi S. Distinctive patterns of microRNA expression associated with karyotype in acute myeloid leukaemia. PLoS One. 2008;3:e2141. doi: 10.1371/journal.pone.0002141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun L, Guo RF, Gao H, Sarma JV, Zetoune FS, Ward PA. Attenuation of IgG immune complex-induced acute lung injury by silencing C5aR in lung epithelial cells. FASEB J. 2009;23:3808–3818. doi: 10.1096/fj.09-133694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heller T, Gessner JE, Schmidt RE, Klos A, Bautsch W, Kohl J. Cutting edge: Fc receptor type I for IgG on macrophages and complement mediate the inflammatory response in immune complex peritonitis. J Immunol. 1999;162:5657–5661. [PubMed] [Google Scholar]
- 48.Barnes N, Gavin AL, Tan PS, Mottram P, Koentgen F, Hogarth PM. FcgammaRI-deficient mice show multiple alterations to inflammatory and immune responses. Immunity. 2002;16:379–389. doi: 10.1016/s1074-7613(02)00287-x. [DOI] [PubMed] [Google Scholar]
- 49.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 2000;6:443–446. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 50.Clynes R, Maizes JS, Guinamard R, Ono M, Takai T, Ravetch JV. Modulation of immune complex-induced inflammation in vivo by the coordinate expression of activation and inhibitory Fc receptors. The Journal of experimental medicine. 1999;189:179–185. doi: 10.1084/jem.189.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rittirsch D, Flierl MA, Day DE, Nadeau BA, Zetoune FS, Sarma JV, Werner CM, Wanner GA, Simmen HP, Huber-Lang MS, Ward PA. Cross-talk between TLR4 and FcgammaReceptorIII (CD16) pathways. PLoS pathogens. 2009;5:e1000464. doi: 10.1371/journal.ppat.1000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.