

Abstract
Toll-like receptors (TLRs) are innate sentinels required for clearance of bacterial and fungal infections of the cornea, but their role in viral immunity is currently unknown. We report TLR signaling is expendable in HSV-1 containment as depicted by plaque assays of knockout mice (MyD88−/−, Trif−/− and MyD88−/− Trif−/− DKO) resembling wild type controls. To identify the key sentinel in viral recognition of the cornea, in vivo knockdown of the DNA sensor IFI-16/p204 in corneal epithelium was performed and resulted in a loss of IRF-3 nuclear translocation, interferon-α production, and viral containment. The sensor appears to have a similar function in other HSV clinically-relevant sites such as the vaginal mucosa in which a loss of p204/IFI-16 results in significantly more HSV-2 shedding. Thus, we have identified an IRF-3 dependent, IRF-7 and TLR - independent innate sensor responsible for HSV containment at the site of acute infection.
Introduction
Herpes simplex virus type-1 (HSV-1) is a widespread, neurotropic, double-stranded DNA virus affecting more than 60% of the world’s population.1 The virus is an important clinical pathogen due to its ability to induce significant morbidity in the central nervous system and cornea of both the immunocompetent and immunosuppressed host.1, 2 Following mucocutaneous contact, HSV-1 initiates infection by first invading host epithelium, replicating, and then gaining entry into sensory fibers where the virus is transported in a retrograde fashion to neuronal cell bodies housed in the trigeminal ganglia.3 In most cases, the virus will then persist as a latent infection for the life of the host periodically reactivating to send infectious virions in an anterograde manner down various branches of the trigeminal nerve to erupt as “cold sores” on or near the labium. In rare but medically significant cases, HSV-1 is transported through the ophthalmic division of the trigeminal nerve to the immunologically privileged cornea where the virus initiates a sequence of inflammatory events that can eventually lead to corneal blindness due to significant immune-mediated scaring.2
During the primary infection and subsequent HSV-1 reactivation in the cornea, in vitro studies suggest innate cell membrane and cell compartment sensors (i.e. toll-like receptors [TLRs]) are activated in response to specific viral invariant structures as displayed by responsiveness to TLR-3 agonists.4, 5 Once activated, TLRs in the cornea are thought to initiate signaling cascades through a myeloid differentiation primary response gene 88 (MyD88)- and/or TIR-domain-containing adapter-inducing interferon (IFN)-β (Trif) adaptor protein-dependent manner. The pathways then act to elicit NF-κB and IFN-regulatory factor 3 family activation to drive production of critical antiviral effector molecules such as double-stranded RNA dependent protein kinase (PKR), RNase L, and Mx proteins by way of type 1 IFN signaling.5–9 In human corneas, TLR mRNA expression is up-regulated during active herpetic stromal keratitis,10 and treatment of human corneal epithelial cell lines with polyinosinic-polycytidylic acid [poly (I:C)], a TLR-3 agonist, induces IFN-β production.4 Furthermore, glucocorticoid treatment is thought to diminish TLR-3 signaling and subsequently enhance ocular susceptibility to viral infection.11 Conversely, others have reported a role for TLR signaling in initiating immunopathology in the cornea.12 Taken together, these results suggest a significant role for TLR signaling in both the containment of HSV-1 as well as pathologic outcomes of the cornea. However, none of the aforementioned studies implicating TLRs as the innate sensor of HSV-1 in the cornea incorporated in vivo models capable of identifying the innate sentinel initiating IFN production and the sensor’s role in containing viral replication.
Thus, we set out to ascertain the in vivo role of TLR signaling during the innate immune response to HSV-1 in the cornea using mice deficient in TLR adaptor proteins (MyD88−/−, Trif−/−, or both [DKO]) to negate any TLR contribution to viral immunity. We hypothesized that type I IFN production required to prohibit viral replication was activated via signals emanating from TLRs and was the initial immune component necessary to control virus replication. Consequently, a loss of TLR signaling would result in a reduction in IFN production and an increase in HSV-1 susceptibility. Contrary to our hypothesis, we found signals initiated by TLR recognition of HSV-1 were expendable, while the recently described nuclear-localized macrophage DNA sensor, p204/IFN inducible protein 16 [IFI-16]13, 14 mediated viral surveillance and innate immunity of the corneal epithelium crucial in the initial control of acute HSV-1 infection.
Results
Loss of TLR signaling has no effect on viral containment
Previous work has shown a TLR-dependent signaling requirement in clearing bacterial and fungal infections of the cornea.15, 16 Likewise, in vitro literature suggests that a loss of TLR pattern recognition and subsequent loss of activation of downstream signaling cascades disrupt HSV-1 containment in the cornea.4, 11 In order to clarify the role of TLRs in the cornea in response to invading viruses, mice deficient in TLR adaptor proteins (MyD88−/− and Trif−/−) were ocularly infected with HSV-1 and compared to WT controls and mice with an ablation of type 1 IFN signaling [CD118−/−] rendering them highly susceptible to HSV-1 dissemination.17 Five days post infection (pi), MyD88−/− and Trif−/− mice possessed similar levels of virus in the cornea as WT controls but significantly less infectious virus than CD118−/− mice (Fig. 1A). To determine the earliest time point a difference could be appreciated between WT and CD118−/− mice, samples obtained at 24 hours (hr) pi revealed no distinguishable difference between the two (Fig. 1B). However, by 48 hrs pi, CD118−/− mice harbored substantially more HSV-1 in the cornea compared to WT controls (Fig. 1C). Similar to five days pi, MyD88−/− and Trif−/− viral levels mirrored those of WT controls and were considerably lower than those of CD118−/− mice at 48 hr pi (Fig. 1C). Higher inoculums of virus had similar outcomes (Supplementary Fig. 1). To negate any TLR pathway redundancy, MyD88−/− and Trif−/− mice were backcrossed to produce double knockouts (DKO). Similar to WT, MyD88−/−, and Trif−/− mice, MyD88/Trif DKO mice had equivalent levels of virus recovered in the cornea at 48 hr pi (Fig. 1C). Taken together, these results clearly demonstrated TLR recognition was not required for resistance to HSV-1 infection of the cornea.
Figure 1. The loss of TLR signaling has not affect on viral containment in the cornea.
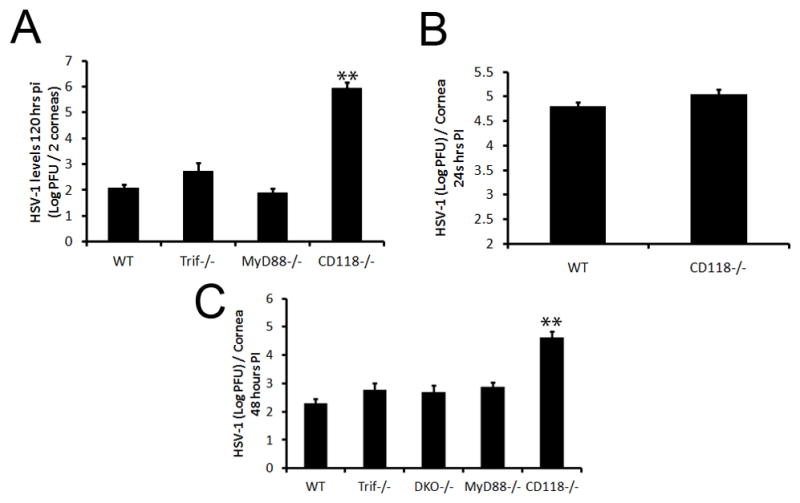
WT, Trif−/−, MyD88−/−, DKO and CD118−/− mice were infected with 1,000 PFU HSV-1/eye. At the indicated time pi, mice were euthanized and corneas harvested. Viral levels in the cornea were then determined by plaque assay at (A) 120 (B), 24, and (C), 48 hr pi. Bars represent 2–4 experiments of 2–4 corneas per group/experiment and are expressed as the mean log PFU/cornea ± SEM. **, p < 0.01; *, p < 0.05 when comparing all groups.
IFN production despite a loss of TLR signaling
Since MyD88−/− and Trif−/− mice did not succumb to HSV-1 dissemination as seen in CD118−/− mice (Supplementary Fig. 1, data not shown), the results would suggest other innate sensors were central to the activation of signaling pathways that result in type I IFN production and resistance to infection. We previously reported that IFN-α is secreted by infected resident cells and cells juxtaposed to infected cells of the corneal epithelium following HSV-1 infection, while IFN-β expression was restricted to cells residing in the limbus of the cornea.18 To investigate whether IFN-α production was altered or diminished in TLR adaptor-deficient mice, corneal whole mounts were evaluated by confocal microscopy. IFN-α production in Trif−/− and CD118−/− mice was similar to WT controls and higher than areas devoid of HSV lesions in all mice (Fig. 2A). IFN-α levels were reduced in the cornea of infected MyD88−/− mice but still detectable relative to isotype controls and sites lacking HSV-1 (Fig. 2A). This reiterated our previously published work in which infected cells (yellow arrows) and cells abutting infected cells (white arrows) were the primary source of IFN-α (Fig. 2A). To further explore the consequences of a reduction in IFN-α expression in MyD88−/− mice, we evaluated downstream IFN-inducible pathways including IFN stimulatory gene-54 (ISG54) and oligosynthetase 1a (OAS1a) expression in the cornea following infection. In response to HSV-1 infection, OAS1a expression was similar between Trif−/− and WT mice at 24 hours pi (Fig. 2B). By comparison, OAS1a expression was elevated in MyD88−/− mouse corneas compared to WT or Trif−/− mouse samples at 24 hours pi although the levels did not reach significance. By 48 hours pi, OAS1a expression was no different between the three groups. ISG54 expression was not significantly different comparing WT to MyD88−/− and Trif−/− mice at 24 and 48 hours pi (Fig. 2B). As the downstream effector molecule of OAS activation, RNase L, contributes to viral surveillance in the cornea following HSV-1 infection,6 the reduction in IFN-α expression observed in the cornea of MyD88−/− mice is not to a point that impacts on IFN-inducible, anti-viral pathways investigated. This data suggested an innate sensor other than TLRs as the principle sentinel in the cornea in response to HSV-1.
Figure 2. Unaltered IFN response despite a loss of TLR signaling.

(A) WT, Trif−/−, MyD88−/−, and CD118−/− mice were infected with 1,000 PFU HSV-1/eye. Forty-four to forty-eight hr pi, IFN production (green) was examined by confocal imaging relative to HSV-1 lesions (red) in the cornea (n = 3–6 corneas/group). Images are representative of 2 independent experiments acquired with a 400x objective with a digital zoom of 10x. White arrows depict uninfected IFN-α expressing cells; yellow arrows depict HSV-infected IFN-α expressing cells; white bars, 50 μm. (B) Mice were infected with HSV-1 and 24 or 48 hrs pi, corneas were evaluated for mRNA expression of ISG54 and OAS1a by RT-PCR. Bars are normalized to uninfected controls and represent 2 independent experiments of 2–4 corneas per group plotted as relative expression ± SEM.
p204/IFI-16 recognition of HSV-1 in epithelium facilitates viral surveillance
While the loss of TLR signaling had minimal to no effect on viral containment and IFN-α production, the sensor that drives IFN-α production in the cornea during the acute HSV-1 infection remained elusive. To identify potential candidate sentinels in the cornea initiating IFN production, five innate sensors/adaptor proteins (DNA-dependent activator of IFN-regulatory factors [DAI], mitochondrial antiviral signaling protein [MAVS], absent in melanoma 2 [AIM2], retinoic acid inducible gene I [RIG-I], and IFN-inducible protein-16 [IFI-16/p204]) were evaluated by RT-PCR for up-regulation following HSV-1 infection. DAI was the most highly induced sensor with a greater than 40-fold increase following infection (Supplementary Fig. 2). However, initial in vivo siRNA knockdown experiments of DAI19 had minimal to no effect on HSV-1 containment (Fig. 3E, Supplementary Fig. 2). The DNA sensor of macrophages, p204 [IFI16, the human homologue of mouse p204], next became the primary focus due to prior literary precedent depicting a role for the protein in initiating IFN production through an IRF-3 dependent mechanism.13, 14, 20, 21 To first establish the presence or absence of the mainly nuclear sensor, confocal microscopy and subsequent 3-D reconstructions of the cornea were used to localize p204 to corneal epithelium with nuclear, perinuclear, and cytoplasmic staining (Fig. 3A). In order to evaluate the role of p204 in innate recognition of HSV-1 in the cornea, initial analysis of the success of in vivo siRNA knockdown of the targeted protein was performed. The results showed a consistent reduction in global p204 protein expression greater than 50% of control transfected and infected mice (Fig. 3B, Fig. 4A). Furthermore, cytoplasmic IRF-3 levels (Fig. 3C) and IRF-3 nuclear translocation was severely diminished in WT mice treated with siRNA to p204 with corresponding HSV-1 titers significantly elevated compared to those of WT mice transfected with non-specific siRNA at 48 hours pi (Fig. 3D–E). If normalized, the viral load difference between mice treated with nonspecific siRNA and siRNA to p204 were comparable to the differences seen between WT and CD118−/− mice (Fig. 1C). Furthermore, STING−/− mice22 contained significantly more virus in the cornea than WT controls yet had similar levels of virus as CD118−/− mice (Fig. 3F). To confirm these results, antibody treatment, which has been shown to inhibit intracellular proteins,23 was used and cytosolic p204 inhibition resulted in a rise in viral load (Fig. 4B–C). p204 is known to translocate from the nucleus to the cytoplasm following viral infection14 and the importance of this trafficking was further underscored by cytoplasmic antibody neutralization. To further substantiate a p204-dependent pathway finding in response to DNA viruses, in vivo (cornea) siRNA knockdown of STING (Supplementary Fig. 2), the adaptor protein of p204,14 was evaluated for infectious content. The results show targeting STING expression resulted in a significant reduction in viral containment compared to controls as well as those of DAI knockdowns (Fig. 3E). IFN production was then evaluated in transfected Trif−/− mice to negate siRNA activation of TLR-3 signaling and subsequent IFN production.24 Forty-eight hours pi, IFN production was impaired in mice treated with siRNA to p204 compared to mice transfected with nonspecific siRNA (Fig. 3G). Taken together, these results suggested p204 was the initiator of HSV-1 immunity in the cornea of mice. To test the role of p204 in response to RNA viral infection, WT and CD118−/− mice were infected with vesicular stomatitis virus (VSV). Forty-eight hours pi, CD118−/− mice had significantly more VSV transcript expression than WT highlighting IFN production in RNA defense (Fig. 5A). However, the loss of p204 did not lead to an increased susceptibility to the VSV (Fig. 5B). To ascertain whether the human equivalent of p204, IFI-16, resides in human corneal epithelium as well, uninfected telomerase-immortalized human corneal epithelial (THCE) cells were subjected to confocal microscopy and Western blot analysis. The results clearly detected IFI-16 expression principally in the nucleus of the THCE cells (Fig. 6A) substantiated by the detection of a protein consistent with the apparent molecular weight of IFI-16 of 90 kD (Fig. 6B). To firmly establish a direct interaction between IFI-16 and foreign DNA, IFI-16 pulldown of transfected biotinylated DNA in THCE cells was performed clearly showing binding of IFI-16 to foreign, GC-rich DNA suggesting a role of the protein in cell immune surveillance of GC-rich viruses such as HSV-1 (Fig. 6D). To substantiate the role of IFI-16, knockdown of IFI-16 in THCE cells (Fig. 6E) resulted in a loss of viral containment not seen when RIG-I and TLR-9 knockdown was performed with similar efficiency (Fig. 6C, data not shown). A loss of IFI-16 also resulted in a loss of nuclear translocation of IRF-3 (Fig. 6F–H) but not nuclear translocation of IRF-7 in which nuclear protein levels are nearly indistinguishable [IFI-16 knockdown 98% of control] (Fig. 6I). It should also be noted the mere infection of THCE cells with HSV-1 resulted in a loss of IFI-16 expression suggesting a viral targeting of necessary host immune proteins (Fig. 6E). To confirm that a loss of IFI-16 signaling reduced IFN signals, IFN-α driven-CXCL10 expression25 was evaluated and a loss of IFI-16 resulted in a loss of CXCL10 mRNA expression (Fig. 6J). Therefore, the aforementioned results suggest a similar role of IFI-16 in humans as p204 in mice relative to HSV-1 surveillance within the cornea. However, the critical role of p204/IFI-16 is not restricted to corneal epithelium, but the sensor likely initiates responses in the epithelium at other sites including the vaginal mucosa; another site commonly infected by HSV viruses. Specifically, a reduction of p204 expression in WT mouse vaginal epithelium by siRNA (Fig. 7C) results in a significant elevation in virus recovered from the vaginal lumen 48 hr pi compared to siRNA control-treated or siRNA targeting DAI expression (Fig. 7A–B). To evaluate whether the sensor is also expressed in epithelium of the skin, confocal microscopy was used to identify the presence of IFI-16 in human primary keratinocytes (Fig. 7D). While untested, this suggested that the sensor may play a role in HSV recognition of the skin.
Figure 3. p204/IFI-16 expressed in the corneal epithelium drives resistance to HSV-1.

(A) Uninfected WT mice were evaluated for p204 expression (red) by confocal microscopy and the sensor was found in the nuclear (blue), perinuclear, and cytoplasmic regions of corneal epithelium using a 400x objective with a digital zoom of 10x. The lower panels are 3-D reconstructions of the cornea. S, stroma; E, epithelium; white bars, 50 μm; UI, uninfected. (B) In vivo siRNA knockdown of p204 was then performed in infected WT mice and consistently greater than 50% of levels of infected mice transfected with non-specific siRNA. Following siRNA knockdown of p204 and HSV-1 infection, IRF-3 cytoplasmic levels were diminished in siRNA transfected groups (C); however, nuclear translocation of IRF-3 (D) was severely diminished 48 hours pi in the nuclear extract of WT corneas treated exclusively with si-p204 as compared to those of control siRNA-treated or uninfected mice. n, nuclear. (E) A loss of p204 or STING following siRNA knockdown resulted in significantly more infectious virus in the cornea compared to WT mice treated with non-specific siRNA or siRNA to DAI (n = 4–8 corneas) as determined by plaque assay. *, p<.05 comparing STING or p204 to DAI or control, values are representative of 2–3 independent experiments. (F) WT, STING−/− and CD118−/− mice were infected with HSV-1 (1,000 pfu/cornea) and viral load determined by plaque assay 48 hrs pi. Results represent 2 experiments of 2–4 corneas/group/experiment and are expressed as the mean log PFU/cornea ± SEM. **, p<.01 compared to WT. (G) Trif−/− mice were transfected with non-specific or anti-p204 siRNA and subsequently infected with HSV-1 (1,000 pfu/cornea). Forty-eight hr pi IFN production (green) was analyzed by confocal microscopy in relation to herpetic lesions (red) in the cornea. White bars, 50 μm.
Figure 4. Cytoplasmic p204 neutralization results in an elevation in HSV-1 in the cornea.
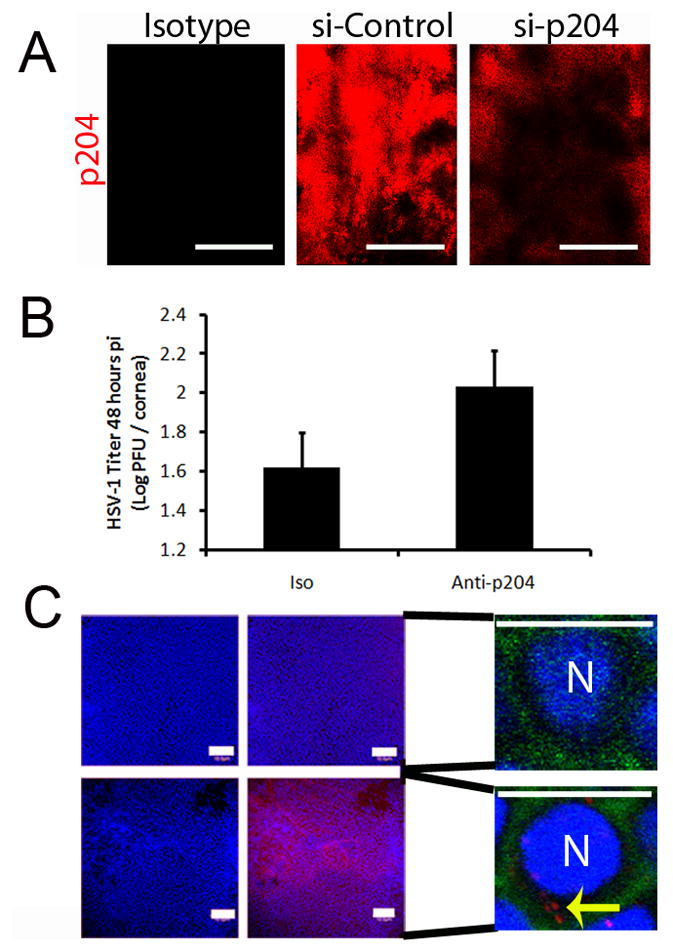
(A) To assess siRNA knockdown of p204, WT mice were transfected with either nonspecific siRNA or to p204. Twenty-four hr post-transfection, knockdown was evaluated by confocal microscopy. White bar, 100 μm. (B) WT mice were infected with HSV-1 (1,000 pfu/cornea) and subsequently given a subconjunctival injection of isotype or anti-mouse p204 polyclonal antibody (10 μg) to neutralize cytosolic protein. Forty-eight hr pi corneas were harvested and evaluated for viral titer by plaque assay. Bars represent 2 experiments of 4 corneas per group and are expressed as the mean log PFU/cornea ± SEM. (C, left panels) To confirm the ability of the p204 antibody (red) to access cellular cytoplasm, WT mice were infected with HSV-1 and given a subconjunctival p204 injection (lower panel) or PBS (upper panel). The corneas were then fixed and stained with a DyLight549-conjugated anti-rabbit secondary and DAPI (blue, nuclei). Images represent two independent experiments of 2–4 corneas per group and were imaged at a magnification of 200x. White bars, 100 μm. (C, right panels) To establish cellular location of the injected antibody, infected corneas were given a subconjunctival injection of p204 antibody or PBS and were then stained with phalloidin (green, cell perimeters), p204 (red), and DAPI (blue) and imaged at 600x with a digital zoom of 10. White bar, 10 μm; N, nuclei, yellow arrows, cytoplasmic location of p204 antibody.
Figure 5. p204 drives innate resistance to HSV but not VSV in the cornea.
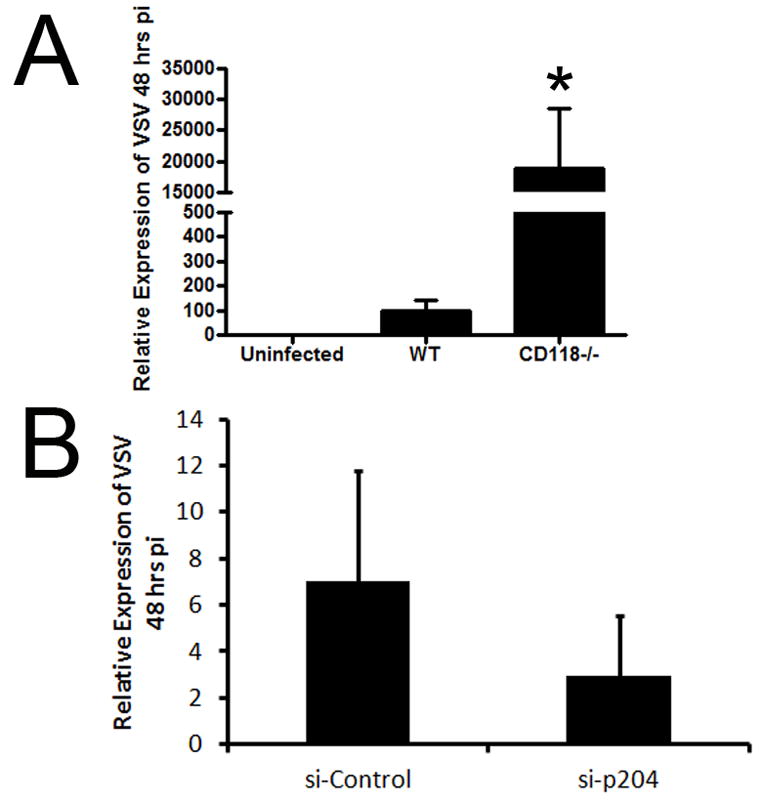
(A) WT and CD118−/− corneal sensitivity to VSV was compared 48 hours pi following infection with 1,000 PFU/eye VSV by RT-PCR. *, p < 0.05 comparing WT to CD118−/−. (B) WT mice were transfected with either nonspecific siRNA or targeted to p204. Twenty four hr post transfection, corneas were scarified and infected with VSV. Corneas were isolated 48 hr pi and VSV levels were then evaluated by RT-PCR. Expression was normalized to β-actin levels. Bars represent 2 independent experiments of 2–4 corneas per group.
Figure 6. Human IFI-16 drives innate resistance in the cornea to HSV-1.
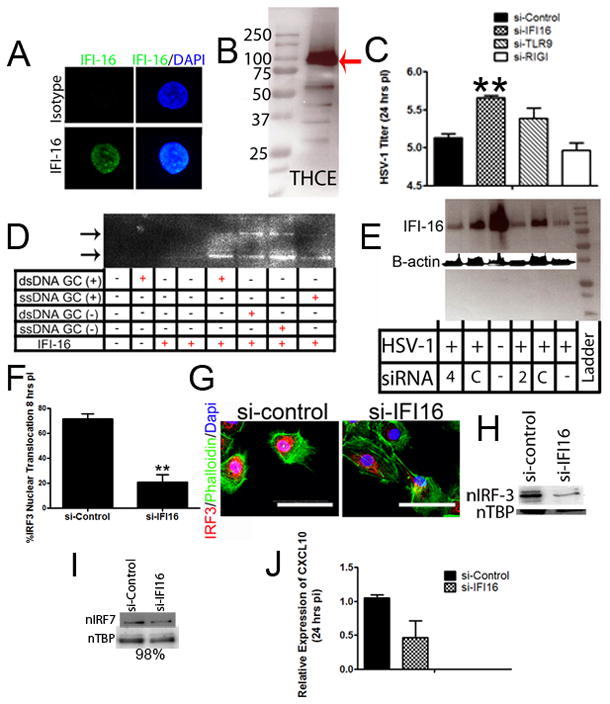
(A) The human homologue, IFI-16 (green), was localized to the nucleus (blue) of uninfected THCE cells by confocal imaging using a 400x objective and digital zoom of 10x. White bars, 200 μm. (B) Imaging was confirmed by Western blot of uninfected THCE cells indicating the presence of the approximately 90 kD protein as highlighted by the red arrow. (C) THCE cells were transfected with 2 pmols of control siRNA or siRNA to IFI-16, TLR9, or RIG-I and infected 4 hr later. Twenty-four hr pi, infectious content was evaluated by plaque assay (n = 4–6/group, 2 independent experiments). **, p < 0.01 comparing siRNA IFI-16 to siRNA control-treated groups. (D) To confirm a direct interaction between the DNA sensor (IFI-16) and HSV-1, ChIP pulldown of a biotinylated 50mer of DNA (single or double stranded, GC or non-GC rich) with IFI-16 antibody was performed in THCE cells and then subsequently identified by western blot analysis. (E) IFI-16 knockdown was performed in THCE cells with varying concentrations of control and anti-IFI-16 siRNA (2 or 4 pmols). Knockdown was confirmed by Western blot. (F–H) Following IFI-16 knockdown, IRF-3 nuclear translocation (G) was severely diminished 8 hr pi as seen by confocal microscopy (quantified in F) and western blot analysis (H). n, nuclear; white bar, 50 μm. Images and blots are representative of 2–4 independent experiments. (I) However, IRF-7 nuclear translocation was no different (levels 98% of control) comparing IFI-16 knockdown to control transfected THCE cells. Image represents 2 independent experiments. (J) THCE cells were transfected and infected. Twenty-four hours pi, CXCL10 expression was evaluated by RT-PCR. Results are representative of 2 independent experiments of 2–3 samples/group and expressed as the relative expression ± SEM.
Figure 7. p204/IFI-16 drives epithelial resistance to HSV.

To evaluate the role of p204/IFI-16 in other epithelial tissues, WT mice were transfected and infected with 2,000 PFU/vagina. Viral shedding into the vaginal lumen (A) was evaluated by plaque assay 48 hr pi. Results represent 2 independent experiments of 2 mice per group and are expressed as the mean log PFU/tissue ± SEM. *, p < 0.05 when comparing si-DAI to si-p204; black line represents levels seen in control-transfected mice. (B–C) RT-PCR or Western blot analysis was used to confirm knockdown efficiency of DAI and p204 in the vaginal tissue respectively. (D) To identify the presence of IFI-16 (green) in human skin, adult primary keratinocytes were stained and imaged at 200x with a digital zoom of 2.5. Images represent 2 independent experiments of 1–2 samples per group. White bars, 10 μm. KD, knockdown.
Discussion
In the study described herein, we have established to the best of our knowledge the first in vivo, TLR-independent innate immune response linked to containment of a clinically important viral pathogen in the cornea and genital epithelium. Despite a loss of TLR adaptor proteins and subsequent downstream TLR signaling, MyD88−/− and 3Trif−/− mice showed competent IFN production in the cornea that resulted in viral surveillance equivalent to that of WT animals. In mice that are unresponsive to type I IFN (CD118−/−), HSV-1 replicates in an uninhibited fashion and rapidly disseminates throughout the host underscoring the critical role of IFNs in viral immunity.17, 26
In tissues and cells found outside the cornea, HSV-1 surveillance is facilitated by TLRs, cytosolic RNA and DNA sensors, mitochondrial associated proteins and nuclear-localized sensors that act to drive IFN production necessary in viral containment.14, 19, 27 The presence of several of these sentinels in a single cell type highlights the considerable redundancy found in most cells. However, in certain tissues like the brain, TLR3 seems to mediate a major portion of HSV-1 surveillance. Patients with TLR3 deficiencies are more susceptible to herpes encephalitis than healthy controls yet not other tested pathogens28–30 emphasizing the importance of TLR3 in the central nervous system in response to HSV-1. In the cornea, p204 facilitates the innate immune response to HSV-1 evident by increased susceptibility to virus replication when protein expression is compromised. In fact, if viral titers of siRNA-treated mice are normalized to unaltered WT levels 48 hours pi, viral levels in mice treated with siRNA to p204 closely resemble those found in highly susceptible CD118−/− mice (5.4 to 4.99), while non-specific siRNA treatment results in levels identical to WT (2.4 to 2.4). This suggests IFI-16/p204 is the principle initiator of IFN production in the cornea. Thus, while a new sensor, DDX41, has recently been shown to signal through STING-dependent manner similar to that of p204,31 the loss of p204 seems to facilitate the major if not sole contribution to innate immunity of the cornea as seen by a loss of viral containment similar to that of both CD118−/− and STING−/− mice. These results were confirmed by antibody inhibition of p204 resulting in a rise in infectious HSV-1 highlighting the importance of the cytoplasmic fraction of p204. p204 is a protein localized to epithelium and some hematopoietic lineages and has been shown to inhibit ribosomal RNA transcription, to play a role in cell growth arrest, and most recently, to drive IFN production in response to intracellular HSV-1 DNA.13, 14, 32, 33 Consequently, it seems plausible that a loss of this single protein, p204, and its downstream signaling events in the corneal epithelium, and epithelium in general, could explain the inability of CD118−/− mice to mount an effective innate immune response. Specifically, p204 binding of HSV DNA motifs recruits STING that acts to drive IFN production through an IRF-3 dependent, IRF-7 independent manner.14, 34 IFNs then initiate JAK/STAT signaling to induce antiviral protein translation.35 A disruption in this cascade results in uncontrolled viral replication and spread into the central nervous system ultimately resulting in encephalitis and death of the host.
Although innate redundancy appears widespread, it is not as extensive in some cells as others, possibly determined by embryological origins. TLRs have gained significant interest since their role in innate immunity was first established.36 However, they represent only one of several known viral sensors and may be expendable in certain tissues in response to specific pathogens as we report in the cornea in reference to HSV-1 infection. It may not be the major sensor, but p204 plays a significant role in other epidermal tissues such as the stratified squamous epithelium of skin and vaginal mucosa where HSV replicates and elicits orolabial and genital lesions. This is highlighted by the observation in vaginal tissue that CD118−/− mice are extremely susceptible to HSV-237 and in mice lacking the p204 adaptor protein, STING, infection with HSV-1 results in more significant pathology and loss of IFN production.34
In relation to patients suffering from herpetic stromal keratitis (HSK), treatment with TLR antagonists may be of some clinical benefit. While untested in our model, in vitro literature suggests TLRs are functional4, 10, 11 and in vivo can initiate destructive inflammatory conditions.12, 38, 39 We hypothesize that inhibiting TLRs in the cornea could lead to better clinical outcomes in those suffering from herpes infections of the cornea due to a reduction in inflammation.12 In the current study, we show that despite a loss of TLR signaling, initial control of HSV-1 replication remained sufficient. Thus, patients treated with TLR antagonists would likely have similar viral loads as untreated patients yet less corneal inflammation if innate control of HSV-1 in humans mimics that of mice as it appears from our in vitro data. To further support our hypothesis, glucocorticoids are used clinically to effectively reduce inflammation in HSK and have been suggested to diminish TLR3 signaling in vitro.11 A reduction in TLR3 signaling in the cornea may be one mechanism of action of broad-spectrum steroid treatments in reducing inflammation in HSK. While TLR inactivation may be controversial, the need for more specific medications to alleviate corneal pathology due to HSK will continue to rise as do the number of immunocompromised patients that suffer from a higher incidence of herpes infections relative to the general public.40
In conclusion, we propose that p204 activates IFN-α production through a STING-dependent manner in response to HSV-1 within resident cells of the cornea and likely other epithelial tissues. IFN-α drives the expression of antiviral pathways including PKR and RNase L which work in concert to orchestrate an antiviral state in the cornea41 and reduce the lytic replication of the virus proximal to the site of entry.
Materials and Methods
Mice and Virus
C57BL/6J wild type (WT) and Trif deficient (Ticam−/−, Trif−/−; C57BL/6J background) mice were purchased from Jackson Laboratory and housed in the Dean McGee Eye Institute’s and Presbyterian Health Foundation’s animal facilities alongside STING deficient (STING−/−)22, MyD88−/−42 and CD118−/− mice.43 Trif−/− and MyD88−/− mice were mated to generate double-knockouts. Animal treatment was consistent with the National Institutes of Health Guidelines on the Care and Use of Laboratory Animals, while all experimental procedures were approved by the University of Oklahoma Health Sciences Center and Dean A. McGee Eye Institutes’ Institutional Animal and Care Use Committees. HSV-1 McKrae and a clinical isolate of HSV-2 were passed as previously described.17, 37 Vesicular stomatitis virus (Indiana strain) was originally obtained from Dr. Robert Fleischmann (Univ. Minnesota).
In vivo HSV-1, -2, and VSV infection
Six to ten week old male and female mice were anesthetized by an intraperitoneal (i.p.) injection of xylazine (6.6 mg/kg) and ketamine (100 mg/kg). Corneas were scarified with a 25 gauge 1½″ needle and tear film blotted. HSV-1 McKrae or vesicular stomatitis virus were resuspended (1,000 PFU/eye) in 3 μLs of sterile 1X PBS and topically applied onto each cornea. Female mice were vaginally infected with 2,000 PFU/vagina resuspended in 20 μl of 1X PBS 5 days following Depo-Provera® (Pfizer) treatment.
Plaque Assays
The corneas and 10 μl vaginal washes of infected mice were harvested at designated times, resuspended in RPMI 1640 media containing 10% FBS and antibiotic/antimycotic reagents (Invitrogen) (referred to as normal media), and subjected to homogenization with a tissue miser for approximately 20–30 seconds as previously described.17 Supernatant was then clarified with a 10,000 x g spin for 1.5 minutes and then serially diluted onto a confluent lawn of HSV-1 susceptible green monkey kidney (Vero) cells. Plaques were allowed to develop for 24–36 hours and then counted with the aid of an inverted Zeiss microscope.
ChIP Assay
Briefly, THCE cells were transfected (as seen elsewhere) with 4 pmols of biotinylated (on 5′ end) single-stranded or double-stranded GC rich or ΔGC DNA 50mers. Twenty-four hours post-transfection, cells were fixed with paraformaldehyde, subjected to glycine, and washed three times. The cells were then lysed in SDS lysis buffer with protease inhibitors. Samples were then incubated with IFI-16 antibody (Santa Cruz) for 2 hours at 4°C except those samples used as controls. The samples were then incubated with Protein A magnetic beads for 1 hour at 4°C and then passed over a magnetic column. Samples were rinsed with several different washes per manufacturer’s instructions (Miltenyi Biotec). Following elution, samples were run on an SDS gel and the biotin was probed for using Strepavidin conjugated to horseradish peroxidase. Sequences for DNA 50mers are as follows:
| DNA 50mers | |
|---|---|
| GC Rich | 5′-ATGGCTCCGCCCCGCCGGGACCCCGCCCCCGGCCCGCCCCGGGCT-3′ |
| ΔGC | 5′-ATGGCTCTGTGTTGCCGGGACCTGTGTTCCGGCCTGTGTTGGGCT-3′ |
Confocal Imaging
WT, Trif−/−, MyD88−/−, and CD118−/− corneas were harvested at the indicated time pi. The corneas were fixed for 30 minutes with 4% paraformaldehyde at 4°C and subsequently washed three times with 1% Triton X-100 (BioRad) in PBS (PBS-Triton). Samples were then blocked overnight with either donkey serum (Jackson Immunology) or Fc block (BD Pharmingen) at 4°C and washed with PBS-Triton. Following the final wash, purified rabbit polyclonal antibody to p20413 or HSV-1 (Dakocytomation) was incubated overnight at 4°C. The corneas were washed again and secondary antibodies conjugated to DyLite549 fluorochrome (Jackson Immunology) and Fitc-conjugated isotype, anti-IFN-α, or anti-IFN-β (PBL Biomedical) antibodies were incubated overnight at 4°C. Following three washes, DAPI (Vector Labs) staining of nuclei was then performed overnight. Corneas were then mounted and imaged using an Olympus IX81-FV500 epiflourescent/confocal laser-scanning microscope. Images were analyzed with Fluoview software (Olympus). 3-D reconstruction was performed by successive z-stack compilation using Fluoview software. Telomerase-immortalized human corneal epithelial (THCE) cells (a gift from Dr. Jerry Shay, UT Southwestern)44 and adult primary keratinocytes (PromoCell) were fixed and imaged in the same fashion except anti-IFI-16 (human p204 homolog) was purchased from Santa Cruz (clone 1G7) and human serum (Jackson Immunology) was used as the isotype control. Anti-IRF-3 polyclonal antibody recognizes both phosphorylated and unphosphorylated protein (Abcam). Phalloidin dye (Invitrogen) labels F-actin. To quantify IRF-3 nuclear translocation, images were captured, and all cells were counted in the visual field for IRF-3 translocation (co-localization with DAPI stain in the nucleus). The number of cells counted was then compared to the total number of cells in each visual field to calculate percent nuclear translocation.
In vitro transfection and infection
THCE cells were grown in 6-well plates to 50–70% confluency in keratinocyte serum free medium containing bovine pituitary extract and epithelial growth factor (KSFM) at which point transfected with 2 or 4 pmols of either nonspecific or siRNA specific to IFI-16, TLR-9, or RIG-I (Invitrogen) in KFSM. Sequences are included below. Following a four-hour incubation, KFSM was removed and replaced with fresh KFSM. Twenty-four hours post-transfection, cells were infected with an MOI of 0.001 of HSV-1 (strain McKrae). KFSM was then replaced after 1 hour at 37°C with fresh KFSM, and cells and supernatant harvested 24 hours pi. Cells were then used to confirm knock down by Western blot of IFI-16 and viral titer to evaluate the role of IFI-16 in human corneal epithelium in response to HSV-1 infection.
| siRNA | |
|---|---|
| si-Control | 5′-UUCUCCGAACGUGUCACGUTT-3′ |
| si-hIFI16 | 5′-GGUGCUGAACGCAACAGAAUCAUUU-3′ |
| si-p204 | 5′-CGGAGAGGAAUAAAUUCAUT-3′ |
| si-STING | 5′-AACUGUUUCCGUCUGUGGGUUCUUG-3′ |
| si-DAI | 5′-ACAGUCCAGACAGUCCACAUCAAAU-3′ |
| si-hTLR9 | 5′-CUGGAAGAGCUAAACCUGA-3′ |
| si-hRIGI | 5′-CAAGAAGAGUACCACUUAA-3′ |
p204 antibody inhibition
WT mice were infected with 1,000 PFU/eye. Thirty minutes pi, the mice were given a subconjunctival injection of 10 μg of rabbit isotype or anti-p204 antibody (rabbit polyclonal). Forty-eight hours pi, the corneas were harvested and evaluated for viral content by plaque assay. For antibody detection, mice were subjected to the treatment described above, and corneas were harvested, fixed, and blocked as previously described. The corneas were then imaged by confocal microscopy.
In vivo transfection
Five microliters of lipofectamine (Invitrogen) was mixed with 2 μl of sterile DMEM lacking serum and antibiotics, while 1 μl of control (Negative Control siRNA), anti-p204 siRNA, anti-DAI, or anti-STING (1 pmol; Stealth RNAi siRNA, Invitrogen) was mixed with 2 μl of DMEM. The solutions were then mixed after a 5 minute incubation and topically applied to anesthetized mice whose corneas had been scarified with a 25 gauge 1 ½″ needle. Twenty-four hours after transfection, mice were infected with HSV-1 and evaluated for knockdown (consistently greater than 50% of control transfected and infected WT mice) by Western blot using a Kodak Imager or RT-PCR, viral titer, or IFN production. Vaginal mucosa was transfected in much the same way but volume doubled. Briefly, 5 days after treatment with Depo-Provera®,37 female mouse vaginal tracts were swabbed and 2 μl of nonspecific or specific si-RNA (si-DAI or si-p204) was mixed with 10 μl of lipofectamine resuspended in 8 μl serum free DMEM. This was then applied intravaginally. Twenty-four hr post-transfection, the mice were infected with 2,000 pfu/vagina HSV-2 (clinical isolate, Charity Hospital, New Orleans, LA).
IRF-3/7 Nuclear Translocation
Corneas were harvested 48 hrs pi from infected and uninfected mice and sonicated in cell lysis buffer with protease inhibitor (Calbiochem) resuspended in T-PER (Thermo Scientific) for 20 seconds. The samples were then rotated for 1 hour at 4°C and subsequently centrifuged at 10,000 x g for 15 minutes. The supernatant was removed for analysis, while cell lysis buffer (Santa Cruz) was added to the pellet. Sample buffer (BioRad) was then added and the mixture was boiled for 5 minutes. The samples were then analyzed for protein content and β-actin and/or TATA-binding protein were used as the loading controls. Nuclear translocation of IRF-3 and -7 was evaluated in THCE cells 8 hrs pi as described for mouse corneas.
RT-PCR
Total RNA was isolated from WT (unaltered or transfected), MyD88−/−, Trif−/−, and CD118−/− corneas and vaginal tissue at the indicated time pi using Trizol per manufacturer’s guidelines (Invitrogen). cDNA was synthesized using avian myeloblastosis reverse transcriptase (Promega) with oligo (dT) primers(Promega) and quantitative real-time PCR was performed with an iCycler (Bio-Rad) as previously described.45 Primer sequences can be found below or as previously described.46–48
| β-actinF | 5′-CTTCTACAATGAGCTGCGTGTG-3′ |
| β-actinR | 5′-TTGAAGGTCTCAAACATGATCTGG-3′ |
| STINGF | 5′-CCTAGCCTCGCACGAACTTG-3′ |
| STINGR | 5′-CGCACAGCCTTCCAGTAGC-3′ |
| DAIF | 5′-GGAAGATCTACCACTCACGTC-3′ |
| DAIR | 5′-CCTTGTTGGCAGATCATGTTG-3′ |
| AIM2F | 5′-TGGAACAATTGTGAATGGGCT-3′ |
| AIM2R | 5′-CACCAACACCTCCATTGT-3′ |
| RIGIF | 5′-CCACAAACTTGGAGAGTCACG-3′ |
| RIGIR | 5′-GAGTTTACAGATATGTTGCATG-3′ |
| MAVSF | 5′-CAAGAGCAAGAACCAGAACTG-3′ |
| MAVSR | 5′-GTGGAAGGGGCTGGAAGG-3′ |
| p204F | 5′-TCAGTTTCAGTAGCCACGGTAGCA-3′ |
| p204R | 5′-TGGTCCCAAACAAGTGATGGTGC-3′ |
| IFI16F | 5′-CCGTTCATGACCACGAGCATAGG-3′ |
| IFI16R | 5′TCAGTCTTGGTTTCAACGTGGT-3′ |
| VSVF | 5′-AATGACGATGAGACGATGCAATC-3′ |
| VSVR | 5′-CAAGTCACGCGTGACCATCT-3′ |
| Oas1aF | 5′-CTTTGATGTCCTGGGTCATGT-3′ |
| Oas1aR | 5′-GCTCCGTGAAGCAGGTAGAG-3′ |
| ISG54F | 5′-TCTGATTCTGAGGCCTTGCA-3′ |
| ISG54R | 5′-TGCTGACCTCCTCCATTCTC-3′ |
Western Blots
Corneas and vaginal tissue were isolated from infected and uninfected mice. Samples were then resuspended in Radio-Immunoprecipitation buffer containing protease inhibitor (Santa Cruz) and subjected to a brief homogenization with a tissue miser. Samples were then allowed to solubilize for 20 minutes at 4°C and centrifuged for 15–20 minutes. Supernatant was evaluated for protein content (BioRad). Following electrophoresis at 99V, protein samples were transferred to PVDF membranes soaked in methanol for 1 hour at 99V. Membranes were washed in Tris buffer (pH 7.6) and primary antibody [IRF-3 and IRF-7, Abcam] (1:1000 dilution) incubated overnight at 4°C. After 3 washes in Tris buffer (pH 7.6), samples were treated with secondary antibody (1:5000 dilution) for 1 hour at room temperature and washed again 3 times. Signal intensity was then evaluated (Pierce) on a Kodak imager.
Statistical analysis
Statistical analysis was conducted using a GBSTAT one-way analysis of variance followed by an ad hoc Tukey’s t test in experiments with more than two groups, while a Student’s t test or two-way analysis of variance (THCE IRF-3 translocation only) was used to evaluate significance between only two variables. Significance was defined as a p value less than 0.05 throughout the paper.
Supplementary Material
(A) To first confirm a loss of MyD88 adaptor protein did not modify ocular susceptibility to HSV, WT, MyD88−/− and CD118−/− mice were infected with 10,000 PFU/eye HSV-1. Forty-eight hours pi, corneas were harvested and evaluated for viral content by plaque assay. Bars represent 2 experiments of 2–4 corneas per group and are expressed as the mean log PFU/cornea ± SEM. **, p < 0.01 when comparing all groups. (B) WT, MyD88−/−, CD118−/− mice were infected with 1,000 PFU HSV-1/eye. Five days pi, mice were euthanized and trigeminal ganglia (TG) harvested. Results are presented as the mean log PFU/TG ± SEM for 3 experiments of 2–4 TGs per group. **, p < 0.01 when comparing all groups.
(A) WT mice were infected with HSV-1 (1,000 pfu/cornea) and corneas were harvested 0 or 24 hr pi. Relative expression of DAI, AIM2, RIG-I, MAVS, STING, and p204 were evaluated by RT-PCR and normalized to β-actin levels and uninfected controls. Bars represent 2 independent experiments of 2–4 corneas per group. To assess knockdown efficiency of DAI and STING, RT-PCR of transfected corneas was compared as depicted in (B), representing 2 independent experiments of 2–4 corneas/experiment. Values are expressed as the percent of control transfected mice.
Acknowledgments
We would like to thank Sara Moore, Linh Sramek, and Gabby Nguyen for technical support and Drs. Helen Rosenberg, Shizuro Akira, and Russell E. Vance for the CD118−/−, MyD88−/− and STING−/− mice respectively. They would also like to thank Todd Wuest for reagents. Principal support for the study was from NIH AI053108 to DJJC. CDC was supported in part by NIAID training grant AI007633. Additional support includes an OUHSC Presbyterian Health Foundation Presidential Professorship award to DJJC, NIH/NCRR P20 RR017703, and an unrestricted grant from Research to Prevent Blindness.
Footnotes
Disclosure
The authors have no conflict of interest to declare.
References
- 1.Conrady CD, Drevets DA, Carr DJ. Herpes simplex type I (HSV-1) infection of the nervous system: is an immune response a good thing? J Neuroimmunol. 2010;220(1–2):1–9. doi: 10.1016/j.jneuroim.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duan R, de Vries RD, Osterhaus AD, Remeijer L, Verjans GM. Acyclovir-resistant corneal HSV-1 isolates from patients with herpetic keratitis. J Infect Dis. 2008;198(5):659–663. doi: 10.1086/590668. [DOI] [PubMed] [Google Scholar]
- 3.Khanna KM, Lepisto AJ, Hendricks RL. Immunity to latent viral infection: many skirmishes but few fatalities. Trends Immunol. 2004;25(5):230–234. doi: 10.1016/j.it.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 4.Kumar A, Zhang J, Yu FS. Toll-like receptor 3 agonist poly(I:C)-induced antiviral response in human corneal epithelial cells. Immunology. 2006;117(1):11–21. doi: 10.1111/j.1365-2567.2005.02258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 6.Austin BA, James C, Silverman RH, Carr DJ. Critical role for the oligoadenylate synthetase/RNase L pathway in response to IFN-beta during acute ocular herpes simplex virus type 1 infection. J Immunol. 2005;175(2):1100–1106. doi: 10.4049/jimmunol.175.2.1100. [DOI] [PubMed] [Google Scholar]
- 7.Minkovitz JB, Pepose JS. Topical interferon alpha-2a treatment of herpes simplex keratitis resistant to multiple antiviral medications in an immunosuppressed patient. Cornea. 1995;14(3):326–330. doi: 10.1097/00003226-199505000-00017. [DOI] [PubMed] [Google Scholar]
- 8.Staeheli P, Haller O, Boll W, Lindenmann J, Weissmann C. Mx protein: constitutive expression in 3T3 cells transformed with cloned Mx cDNA confers selective resistance to influenza virus. Cell. 1986;44(1):147–158. doi: 10.1016/0092-8674(86)90493-9. [DOI] [PubMed] [Google Scholar]
- 9.Leib DA, Machalek MA, Williams BR, Silverman RH, Virgin HW. Specific phenotypic restoration of an attenuated virus by knockout of a host resistance gene. Proc Natl Acad Sci U S A. 2000;97(11):6097–6101. doi: 10.1073/pnas.100415697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jin X, Qin Q, Chen W, Qu J. Expression of toll-like receptors in the healthy and herpes simplex virus-infected cornea. Cornea. 2007;26(7):847–852. doi: 10.1097/ICO.0b013e318093de1f. [DOI] [PubMed] [Google Scholar]
- 11.Hara Y, Shiraishi A, Kobayashi T, Kadota Y, Shirakata Y, Hashimoto K, et al. Alteration of TLR3 pathways by glucocorticoids may be responsible for immunosusceptibility of human corneal epithelial cells to viral infections. Mol Vis. 2009;15:937–948. [PMC free article] [PubMed] [Google Scholar]
- 12.Sarangi PP, Kim B, Kurt-Jones E, Rouse BT. Innate recognition network driving herpes simplex virus-induced corneal immunopathology: role of the toll pathway in early inflammatory events in stromal keratitis. J Virol. 2007;81(20):11128–11138. doi: 10.1128/JVI.01008-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu CJ, Wang H, Lengyel P. The interferon-inducible nucleolar p204 protein binds the ribosomal RNA-specific UBF1 transcription factor and inhibits ribosomal RNA transcription. EMBO J. 1999;18(10):2845–2854. doi: 10.1093/emboj/18.10.2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol. 2010;11(11):997–1004. doi: 10.1038/ni.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hise AG, Daehnel K, Gillette-Ferguson I, Cho E, McGarry HF, Taylor MJ, et al. Innate immune responses to endosymbiotic Wolbachia bacteria in Brugia malayi and Onchocerca volvulus are dependent on TLR2, TLR6, MyD88, and Mal, but not TLR4, TRIF, or TRAM. J Immunol. 2007;178(2):1068–1076. doi: 10.4049/jimmunol.178.2.1068. [DOI] [PubMed] [Google Scholar]
- 16.Tarabishy AB, Aldabagh B, Sun Y, Imamura Y, Mukherjee PK, Lass JH, et al. MyD88 regulation of Fusarium keratitis is dependent on TLR4 and IL-1R1 but not TLR2. J Immunol. 2008;181(1):593–600. doi: 10.4049/jimmunol.181.1.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Conrady CD, Thapa M, Wuest T, Carr DJ. Loss of mandibular lymph node integrity is associated with an increase in sensitivity to HSV-1 infection in CD118-deficient mice. J Immunol. 2009;182(6):3678–3687. doi: 10.4049/jimmunol.0803878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Conrady CD, Jones H, Zheng M, Carr DJJ. A functional type I interferon pathway drives resistance to cornea herpes simplex virus type 1 infection by recruitment of leukocytes. J Biomed Res. 2011;25(2):111–129. doi: 10.1016/S1674-8301(11)60014-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448(7152):501–505. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- 20.Jones JW, Kayagaki N, Broz P, Henry T, Newton K, O’Rourke K, et al. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc Natl Acad Sci U S A. 2010;107(21):9771–9776. doi: 10.1073/pnas.1003738107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiu YH, Macmillan JB, Chen ZJ. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell. 2009;138(3):576–591. doi: 10.1016/j.cell.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sauer JD, Sotelo-Troha K, von Moltke J, Monroe KM, Rae CS, Brubaker SW, et al. The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect Immun. 2011;79(2):688–694. doi: 10.1128/IAI.00999-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edelson BT, Unanue ER. Intracellular antibody neutralizes Listeria growth. Immunity. 2001;14(5):503–512. doi: 10.1016/s1074-7613(01)00139-x. [DOI] [PubMed] [Google Scholar]
- 24.Cho WG, Albuquerque RJ, Kleinman ME, Tarallo V, Greco A, Nozaki M, et al. Small interfering RNA-induced TLR3 activation inhibits blood and lymphatic vessel growth. Proc Natl Acad Sci U S A. 2009;106(17):7137–7142. doi: 10.1073/pnas.0812317106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Padovan E, Spagnoli GC, Ferrantini M, Heberer M. IFN-alpha2a induces IP-10/CXCL10 and MIG/CXCL9 production in monocyte-derived dendritic cells and enhances their capacity to attract and stimulate CD8+ effector T cells. J Leukoc Biol. 2002;71(4):669–676. [PubMed] [Google Scholar]
- 26.Leib DA, Harrison TE, Laslo KM, Machalek MA, Moorman NJ, Virgin HW. Interferons regulate the phenotype of wild-type and mutant herpes simplex viruses in vivo. J Exp Med. 1999;189(4):663–672. doi: 10.1084/jem.189.4.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paludan SR, Bowie AG, Horan KA, Fitzgerald KA. Recognition of herpesviruses by the innate immune system. Nat Rev Immunol. 2011;11(2):143–154. doi: 10.1038/nri2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science. 2007;317(5844):1522–1527. doi: 10.1126/science.1139522. [DOI] [PubMed] [Google Scholar]
- 29.Guo Y, Audry M, Ciancanelli M, Alsina L, Azevedo J, Herman M, et al. Herpes simplex virus encephalitis in a patient with complete TLR3 deficiency: TLR3 is otherwise redundant in protective immunity. J Exp Med. 2011 doi: 10.1084/jem.20101568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Casrouge A, Zhang SY, Eidenschenk C, Jouanguy E, Puel A, Yang K, et al. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science. 2006;314(5797):308–312. doi: 10.1126/science.1128346. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Z, Yuan B, Bao M, Lu N, Kim T, Liu YJ. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol. 2011;12(10):959–965. doi: 10.1038/ni.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wei W, Clarke CJ, Somers GR, Cresswell KS, Loveland KA, Trapani JA, et al. Expression of IFI 16 in epithelial cells and lymphoid tissues. Histochem Cell Biol. 2003;119(1):45–54. doi: 10.1007/s00418-002-0485-0. [DOI] [PubMed] [Google Scholar]
- 33.Xin H, Curry J, Johnstone RW, Nickoloff BJ, Choubey D. Role of IFI 16, a member of the interferon-inducible p200-protein family, in prostate epithelial cellular senescence. Oncogene. 2003;22(31):4831–4840. doi: 10.1038/sj.onc.1206754. [DOI] [PubMed] [Google Scholar]
- 34.Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461(7265):788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Darnell JE, Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264(5164):1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 36.Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388(6640):394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 37.Conrady CD, Halford WP, Carr DJ. Loss of the type I interferon pathway increases vulnerability of mice to genital herpes simplex virus 2 infection. J Virol. 2011;85(4):1625–1633. doi: 10.1128/JVI.01715-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sabroe I, Parker LC, Dower SK, Whyte MK. The role of TLR activation in inflammation. J Pathol. 2008;214(2):126–135. doi: 10.1002/path.2264. [DOI] [PubMed] [Google Scholar]
- 39.Kurt-Jones EA, Chan M, Zhou S, Wang J, Reed G, Bronson R, et al. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc Natl Acad Sci U S A. 2004;101(5):1315–1320. doi: 10.1073/pnas.0308057100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oshry T, Lifshitz T. Intravenous acyclovir treatment for extensive herpetic keratitis in a liver transplant patient. Int Ophthalmol. 1997;21(5):265–268. doi: 10.1023/a:1006057204609. [DOI] [PubMed] [Google Scholar]
- 41.Al-Khatib K, Williams BR, Silverman RH, Halford W, Carr DJ. Distinctive roles for 2′,5′-oligoadenylate synthetases and double-stranded RNA-dependent protein kinase R in the in vivo antiviral effect of an adenoviral vector expressing murine IFN-beta. J Immunol. 2004;172(9):5638–5647. doi: 10.4049/jimmunol.172.9.5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, et al. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998;9(1):143–150. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- 43.Garvey TL, Dyer KD, Ellis JA, Bonville CA, Foster B, Prussin C, et al. Inflammatory responses to pneumovirus infection in IFN-alpha beta R gene-deleted mice. J Immunol. 2005;175(7):4735–4744. doi: 10.4049/jimmunol.175.7.4735. [DOI] [PubMed] [Google Scholar]
- 44.Robertson DM, Li L, Fisher S, Pearce VP, Shay JW, Wright WE, et al. Characterization of growth and differentiation in a telomerase-immortalized human corneal epithelial cell line. Invest Ophthalmol Vis Sci. 2005;46(2):470–478. doi: 10.1167/iovs.04-0528. [DOI] [PubMed] [Google Scholar]
- 45.Wuest TR, Carr DJ. VEGF-A expression by HSV-1-infected cells drives corneal lymphangiogenesis. J Exp Med. 2010;207(1):101–115. doi: 10.1084/jem.20091385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takeshita F, Suzuki K, Sasaki S, Ishii N, Klinman DM, Ishii KJ. Transcriptional regulation of the human TLR9 gene. J Immunol. 2004;173(4):2552–2561. doi: 10.4049/jimmunol.173.4.2552. [DOI] [PubMed] [Google Scholar]
- 47.Spurrell JC, Wiehler S, Zaheer RS, Sanders SP, Proud D. Human airway epithelial cells produce IP-10 (CXCL10) in vitro and in vivo upon rhinovirus infection. Am J Physiol Lung Cell Mol Physiol. 2005;289(1):L85–95. doi: 10.1152/ajplung.00397.2004. [DOI] [PubMed] [Google Scholar]
- 48.Hoene V, Peiser M, Wanner R. Human monocyte-derived dendritic cells express TLR9 and react directly to the CpG-A oligonucleotide D19. J Leukoc Biol. 2006;80(6):1328–1336. doi: 10.1189/jlb.0106011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) To first confirm a loss of MyD88 adaptor protein did not modify ocular susceptibility to HSV, WT, MyD88−/− and CD118−/− mice were infected with 10,000 PFU/eye HSV-1. Forty-eight hours pi, corneas were harvested and evaluated for viral content by plaque assay. Bars represent 2 experiments of 2–4 corneas per group and are expressed as the mean log PFU/cornea ± SEM. **, p < 0.01 when comparing all groups. (B) WT, MyD88−/−, CD118−/− mice were infected with 1,000 PFU HSV-1/eye. Five days pi, mice were euthanized and trigeminal ganglia (TG) harvested. Results are presented as the mean log PFU/TG ± SEM for 3 experiments of 2–4 TGs per group. **, p < 0.01 when comparing all groups.
(A) WT mice were infected with HSV-1 (1,000 pfu/cornea) and corneas were harvested 0 or 24 hr pi. Relative expression of DAI, AIM2, RIG-I, MAVS, STING, and p204 were evaluated by RT-PCR and normalized to β-actin levels and uninfected controls. Bars represent 2 independent experiments of 2–4 corneas per group. To assess knockdown efficiency of DAI and STING, RT-PCR of transfected corneas was compared as depicted in (B), representing 2 independent experiments of 2–4 corneas/experiment. Values are expressed as the percent of control transfected mice.