

SUMMARY
This review is focused on advances in understanding the biology of joint homeostasis and osteoarthritis (OA) pathogenesis mechanisms that have led to proof of concept studies on new therapeutic approaches. The three selected topics include angiogenesis in joint tissues, biomechanics and joint lubrication and mitochondrial dysfunction. This new information represents progress in the integration of mechanisms that control multiple aspects of OA pathophysiology.
Keywords: Angiogenesis, Autophagy, Lubricin, Mitochondria
Introduction
The OA disease process affects all joint tissues, leading to joint dysfunction and pain and systemic and organismal changes that reduce quality of life, initiate or aggravate co-morbidities and reduce life expectancy. Progress in understanding the biology of joint homeostasis and disease mechanisms is essential for the development of new approaches to prevent or treat OA. To cover the general topic ‘biology’ assigned to this review, areas of biology that are most relevant to OA were considered. These include developmental biology of joints and joint tissues, stem cell biology, cell and molecular biology of adult joint tissue homeostasis, mechanobiology, extracellular matrix biology, systems biology and the biology of aging. These areas of biology provide a knowledge base for research on the pathobiology of OA. OA biology can also be viewed on the basis of scale, ranging from molecular to cellular, tissue, organ and organism level processes that determine risk for onset and progression of the disease or are affected by the disease process.
As an in depth review of all areas of biology is not possible within this manuscript, three fields, angiogenesis, joint lubrication and mitochondrial homeostasis were selected for detailed review to illustrate how progress in biology has led to identification and preclinical validation of new targets for the treatment of OA.
Angiogenesis
The formation of new blood vessels has previously been recognized as essential for the development of synovial hyperplasia1 in arthritic joints where the synovial membrane becomes thickened, from increased proliferation of synovial fibroblasts, which also deposit new extracellular matrix. The formation of new blood vessels that supply oxygen and nutrients is not only necessary for the development of the hyperplastic synovium but also leads to tissue infiltration with blood-derived inflammatory cells and mediators. Synovial angiogenesis has been studied extensively in rheumatoid arthritis (RA). Many studies on angiogenesis compared RA, OA and normal synovium and the general conclusion is that angiogenesis correlates with the extent of synovial hyperplasia, which is most severe in RA, but also present in OA-affected joints2. The prominent role of synovial hyperplasia in RA has led to studies targeting mediators of angiogenesis such as vascular endothelial growth factor (VEGF) and this has been successful in RA animal models3.
The importance of angiogenesis in OA is further supported by recent observations that new blood vessels are not only formed in synovium but also in other tissues. Normal articular cartilage is avascular, and this has been related to the presence of angiogenesis inhibitors, such as thrombospondin, chondromodulin4, and collagen XVIII-derived endostatin5. OA articular cartilage is invaded at the osteochondral junction by blood vessels from the subchondral bone, which leads to increased blood vessel density in the non-calcified cartilage6. The osteochondral angiogenesis is also associated with subchondral bone marrow replacement by fibrovascular tissue expressing VEGF, and with increased nerve growth factor (NGF) expression within vascular channels that contain sensory nerve fibers6. In OA joints, increased angiogenesis is also observed in menisci along the fibrocartilage junction with increased penetration into the inner region7. These new blood vessels are also surrounded by sensory nerve fibers. The anterior cruciate ligaments from OA-affected human joints show increased numbers of blood vessels in the synovial sheath and within the ligament substance, where the new blood vessels are surrounded by cell aggregates8. Blood vessels and nerves also penetrate newly formed cartilage in osteophytes at the joint margins9.
OA joints contain increased levels of VEGF in articular cartilage and synovial fluid and several cytokines that stimulate VEGF production are present10, 11. The increased VEGF production in OA has also been linked to the shift in hypoxia inducible factors and the abnormal hypertrophic differentiation of OA chondrocytes12. Growth plate chondrocyte hypertrophy physiologically generates angiogenic factors to initiate vascular invasion of the hypertrophic zone13.
This new information that neovascularization is a feature of several tissues in OA joints and the association of angiogenesis with increased new sensory nerve fiber formation supports the hypothesis that these processes contribute to both structural change and pain in OA14. To test this hypothesis in the rat meniscectomy model of OA, PPI-2458, a specific inhibitor of endothelial cell proliferation was administered. PPI-2458 reduced synovial and osteochondral angiogenesis, synovial inflammation, cartilage damage and osteophyte formation. Importantly, this treatment also reduced pain behavior14. This study was the first to establish simultaneous benefit of an angiogenesis inhibitor on structure and pain modification in an OA model.
Several previous studies support angiogenesis inhibition as a promising approach for OA. Adenovirus-mediated thrombospondin-1 gene transfer in an OA model in rats significantly reduced microvessel density, inflammation, and suppressed OA progression15. The limitation of this study was that thrombospondin-1 has activities in addition to inhibition of angiogenesis that may contribute to the observed effects. In a cartilage repair model, the use of stem cells that overexpressed the VEGF antagonist sFlt-1 reduced angiogenesis and improved new cartilage tissue formation without osteophyte development16. Angiogenesis inhibition through chondromodulin prevented chondrocyte hypertrophy and stabilized the articular chondrocyte phenotype17.
Collectively, these new observations motivate further studies on the use of more specific and potent angiogenesis inhibitors in OA to better understand interactions between angiogenesis, cartilage and bone remodeling and pain mechanisms. The availability of antiangiogenic drugs that are approved for certain types of cancer and eye diseases18 facilitates clinical evaluation of this approach in OA.
The Mitochondria-Autophagy Connection
Much of the progress in understanding OA pathogenesis is related to mechanisms that are activated in established disease. More recent efforts are directed at defining early changes that predispose or lead to the onset of OA19. As the majority patients develop OA as a function of increasing age, it is essential to understand aging-related changes in cell function. The phenomenon of cell aging includes reduced cell proliferation and extracellular matrix synthesis and increased production of reactive oxygen species, inflammatory mediators and extracellular matrix degrading enzymes20. This altered pattern of gene and mediator expression has been designated as the senescence-associated phenotype21.
Two major mechanisms, telomere and mitochondrial dysfunction contribute to cellular aging and senescence. Shortening of telomeres in highly proliferative organs activates p53-dependent growth arrest, senescence and apoptosis22. Tissues with lower cell proliferation rates such as muscle or cartilage are equally affected by the aging process, largely through reactive oxygen species (ROS) induced genotoxic stress. A new model, which links telomere shortening, p53 activation and mitochondrial dysfunction was proposed in 2011. According to this model, the telomere-p53 axis suppresses PGC-1α or PGC-1β, which are the master regulators of mitochondrial biogenesis and metabolism and this leads to deficient ATP production and increased ROS generation, contributing to abnormal gene expression and cell death23, 24. Mitochondria also have an important role in pro-inflammatory signaling, as they initiate formation of inflammasomes and activation of other inflammatory pathways25.
A series of prior studies demonstrated mitochondrial dysfunction in various OA models and in human OA. In addition, mitochondrial DNA mutations are increased in OA chondrocytes26. During the past year, new insight was provided on genetic risk factors and mechanisms of mitochondrial dysfunction, and defects in the protection against and removal of abnormal mitochondria in OA.
Rego-Perez et. al. had previously reported that mitochondrial DNA haplogroups contribute to OA risk as disease prevalence in haplogroup H was significantly higher compared to haplogroup J. In their recent study, they showed that serum levels of MMP-13 were significantly higher in patients with OA, and carriers of haplogroup H27. These observations provide further support for a genetic risk factor that can manifest as mitochondrial dysfunction.
Scott et. al. analyzed expression of all three forms of superoxide dismutase, enzymes that protect against ROS-mediated cell damage. The expression of these 3 enzymes was downregulated in cartilage from human OA joints and guinea pigs with spontaneous OA. In particular, the mitochondrial SOD2 was reduced and the decline in SOD2 preceded OA-like lesions in the animal model28.
The principal mechanism for removal of dysfunctional mitochondria is autophagy, or specifically, mitophagy29. Autophagy is mediated by formation of double-membrane vesicles, termed autophagosomes that sequester organelles, proteins, or portions of the cytoplasm. Autophagosomes then fuse with lysosomes, the sequestered contents are degraded by lysosomal enzymes and recycled as a source of energy30.
Autophagy is compromised in OA cartilage and this is in part related to reduced expression of autophagy regulators31. New observations by Terkeltaub et. al. indicate that AMPK, which is a key regulator of autophagy activation under conditions of low energy or other stress conditions is reduced in OA cartilage and in chondrocytes following treatment with IL-1β or TNFα. AMPK activators attenuate dephosphorylation of AMPKα and procatabolic responses in chondrocytes induced by these cytokines32.
The scenario suggested by these new observations is that mitochondrial DNA haplotype H, acquired mitochondrial DNA mutations and the telomere-p53-PGC1 pathway contribute to mitochondrial dysfunction, which is further aggravated by deficient anti-oxidant defenses and impaired autophagy (Figure 1). Addressing this pathway with anti-oxidants is a feasible and effective approach in preclinical models33. An alternative approach would be removal of dysfunctional mitochondria by enhancing autophagy. A target for enhancing autophagy is the enzyme mammalian target of rapamycin (mTOR), which is the key upstream regulator of autophagy. Activated mTOR prevents autophagy activation and its inhibition by rapamycin induces autophagy34. In genetically heterogeneous mice, rapamycin extended life span even when administration was initiated late in life, providing further support for the importance of autophagy in aging35, 36.
Fig. 1. Dysfunctional mitochondria in OA pathogenesis.
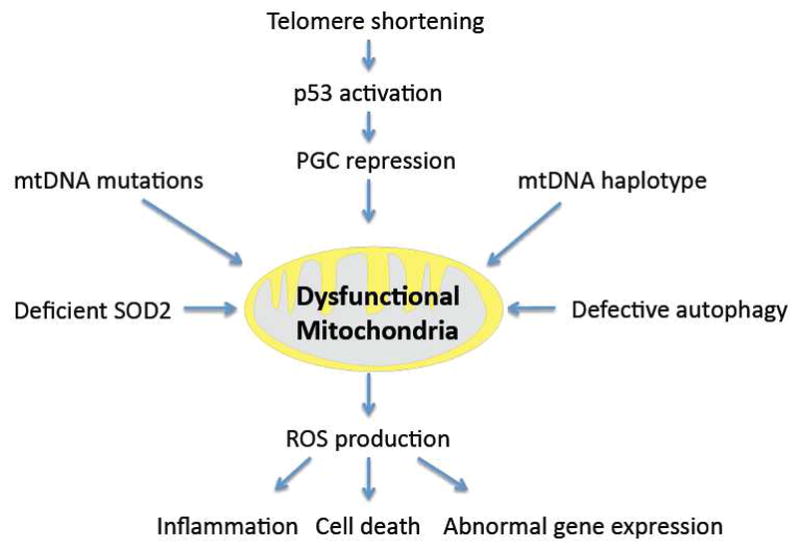
Multiple factors contribute to mitochondrial dysfunction, including inhibition of mitochondrial biogenesis through suppression of key mitochondrial transcription factors PGC. Risk for mitochondrial dysfunction is also increased in individuals with certain mitochondrial DNA haplotypes and through acquired mitochondrial DNA mutations. As a result of defective autophagy, dysfunctional mitochondria accumulate and produce increased amounts of reactive oxygen species, which promote inflammatory responses, abnormal gene expression and cell death. The impact of mitochondrial dysfunction is further aggravated by reduced levels of SOD2 and deficient anti-oxidant defenses.
In articular cartilage, autophagy is not only deficient in aging31 but also in response to mechanical injury37. In an in vitro model, rapamycin treatment of cartilage explants that were exposed to high levels of mechanical load reduced cell death and extracellular matrix damage37. Rapamycin also suppressed biochemically induced cell death and nitric oxide production in response to proinflammatory cytokines. The first study to address potential therapeutic benefits of rapamycin in vivo used a knee instability-induced OA model in mice. Rapamycin treatment reduced the severity of OA and this was associated with preservation of cartilage cellularity38. These findings are the first to suggest that rapamycin is a promising new approach towards chondroprotection. Additional studies are required to directly link the rapamycin effects to removal of dysfunctional mitochondria and to analyze its efficacy in models of aging-related OA.
Chondroprotection through Lubrication
Articular cartilage functions as a surface that allows joint movement under conditions of extremely low friction. Hyaluronic acid (HA) and lubricin are two major joint lubricants and effective “boundary lubricants” that exhibit low friction and protect surfaces from wear39. Detailed mechanisms how HA is anchored and immobilized at the cartilage surface had not been established. Lubricin adsorbs to various types of substrates and forms complexes with itself and HA. Lubricin is thus expected to form cross-links with the surface-immobilized, partially entangled HA layer forming a cross-linked HA-lubricin complex. However, as the physical bond between HA and lubricin is relatively weak, it would be expected to dissociate under normal friction and shear forces40. The study by Greene et. al. examined mechanisms related to low friction and wear protection. Their results support a new concept of an adaptive mechanically controlled lubrication mechanism. According to this model, compression causes diffusion of nominally free HA out of the cartilage but then becomes physically trapped at the interface by the increasingly constricted collagen pore network. The mechanically trapped HA-lubricin complex now acts as an effective chemically bound lubricant, reducing the friction force slightly, but more importantly, eliminating wear damage to the cartilage surfaces. This concept illustrates that the lubrication system as a whole requires both HA and lubricin to function optimally.
Besides their role in lubrication, both lubricin and HA have additional functions in the joint. Lubricin regulates synovial cell adhesion and proliferation. Lubricin mutations are linked to the camptodactyly-arthropathy-coxa vara-pericarditis syndrome, which features synovial hyperplasia and failure of joint function41. Mice that are deficient in lubricin not only show degradation of the cartilage superficial zone but also hyperplasia of intimal cells in the synovium. Purified or recombinant lubricin inhibits the synoviocyte proliferation in vitro42. Interestingly, a splice form of lubricin is a growth factor for endothelial cells43. Future studies will need to define in more detail the lubricin splice forms, their receptors and activities on cells involved in OA. A large number of studies already reported on cellular effects of HA, which include anti-inflammatory, cytoprotective and anabolic activities that are mediated by cell surface receptors CD44 and RHAMM44, 45.
Both, HA and lubricin are deficient in arthritic joints39. Lubricin may be degraded and synthesis appears to be reduced, due to loss of joint lining cells, which are the main producers or due to inhibition of synthesis by inflammatory cytokines.
Based on the protective functions of HA and lubricin, their deficiency in arthritis joints and their synergistic activities, recent studies examined these molecules individually and in combination in animal models of joint injury and OA. Flannery et. al. reported that intraarticular injection of recombinant lubricin protected against cartilage degradation in a rat model of OA induced meniscectomy and medial collateral ligament transection46. Jay et. al. used the anterior cruciate ligament transection model in rats to compare three forms of lubricin, recombinant human lubricin, synovial fluid lubricin and lubricin produced by cultured synoviocytes47. Injected lubricin bound to the cartilage surface. All three forms of lubricin reduced the severity of cartilage damage but only the effect of lubricin from synoviocytes was statistically significant. Lubricin also protected against glycosaminoglycan depletion, collagen degradation and loss of cells in the cartilage superficial zone47. Teeple et. al. used the same OA model in rats to test lubricin purified from human synovial fluid and also compared it to and combined it with HA. These results confirmed the protective effect of lubricin but HA did not have protective effects and the therapeutic benefit of lubricin was enhanced by the addition of exogenous HA48. Previous studies on HA effects in OA models showed variable results, ranging from protective to deleterious effects48.
These studies shed new light on the role of joint lubricants as potential OA therapies. The potential synergy between HA and lubricin suggested by molecular models has yet to be demonstrated in animal models. A specific clinical indication that appears to represent a promising opportunity for lubricants as therapeutics in posttraumatic arthritis, which is associated with an acute deficiency of lubricants49.
Conclusions
These selected examples illustrate how progress in specific areas of biology can lead to identification of OA therapeutic targets, which can be readily validated in animal models. The findings from the past year add to an extensive list of OA drug candidates that were effective in animal models. A similar review in 2010 highlighted other signaling pathways in aging and the role of abnormal hypertrophic differentiation50. A major challenge in this field is how to select among these pathways and molecules the most promising candidates for testing in clinical trials. The three examples discussed above represent distinct aspects of OA pathogenesis. Similarly diverse were the pathways that have been targeted in completed clinical trials, including inhibitors of matrix degrading enzymes, bone resorption or IL-1. Clinical trails with these drugs either failed to show conclusive efficacy or were associated with adverse events51, 52. These clinical results raise important questions in OA biology. Are drug targets valid and can methods be developed, for example based on systems biology approaches, to select or discover more promising drug targets? Do currently used animal models provide a meaningful representation of human OA? Are clinical trials design parameters in regard to patient risk factor profile and stage in the disease process appropriate? Can stages in the disease process and corresponding stage-specific interventions be identified? Should OA prevention and treatment start much earlier in the disease process than in previous studies? Answering these questions remains a challenge in OA biology and a prerequisite for the first successful clinical trial on a disease-modifying OA drug.
Acknowledgments
Role of the funding source
The funding bodies had no role in the design of the study, data collection, analysis and interpretation of the data, the writing of the manuscript or in the decision to submit the manuscript.
I apologize to all colleagues whose work could not be cited due to space limitations. My research is supported by grants NIH grants AR058954, AR056026 and AG007996.
Footnotes
Author contributions
Martin Lotz drafted the review article and approved the final version to be published.
Conflict of interest
The author declares that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Szekanecz Z, Besenyei T, Paragh G, Koch AE. New insights in synovial angiogenesis. Joint Bone Spine. 2010;77:13–9. doi: 10.1016/j.jbspin.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marrelli A, Cipriani P, Liakouli V, Carubbi F, Perricone C, Perricone R, et al. Angiogenesis in rheumatoid arthritis: A disease specific process or a common response to chronic inflammation? Autoimmun Rev. 2011;10:595–8. doi: 10.1016/j.autrev.2011.04.020. [DOI] [PubMed] [Google Scholar]
- 3.Thairu N, Kiriakidis S, Dawson P, Paleolog E. Angiogenesis as a therapeutic target in arthritis in 2011: learning the lessons of the colorectal cancer experience. Angiogenesis. 2011;14:223–34. doi: 10.1007/s10456-011-9208-2. [DOI] [PubMed] [Google Scholar]
- 4.Shukunami C, Hiraki Y. Chondromodulin-I and tenomodulin: the negative control of angiogenesis in connective tissue. Curr Pharm Des. 2007;13:2101–12. doi: 10.2174/138161207781039751. [DOI] [PubMed] [Google Scholar]
- 5.Feng Y, Wu YP, Zhu XD, Zhang YH, Ma QJ. Endostatin promotes the anabolic program of rabbit chondrocyte. Cell Res. 2005;15:201–6. doi: 10.1038/sj.cr.7290287. [DOI] [PubMed] [Google Scholar]
- 6.Walsh DA, McWilliams DF, Turley MJ, Dixon MR, Franses RE, Mapp PI, et al. Angiogenesis and nerve growth factor at the osteochondral junction in rheumatoid arthritis and osteoarthritis. Rheumatology. 2010;49:1852–61. doi: 10.1093/rheumatology/keq188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ashraf S, Wibberley H, Mapp PI, Hill R, Wilson D, Walsh DA. Increased vascular penetration and nerve growth in the meniscus: a potential source of pain in osteoarthritis. Ann Rheum Dis. 2011;70:523–9. doi: 10.1136/ard.2010.137844. [DOI] [PubMed] [Google Scholar]
- 8.Hasegawa A, Otsuki S, Pauli C, Miyaki S, Patil S, Steklov N, et al. Anterior cruciate ligament changes in human joint in aging and osteoarthritis. Arthritis Rheum. 2011 doi: 10.1002/art.33417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suri S, Gill SE, Massena de Camin S, Wilson D, McWilliams DF, Walsh DA. Neurovascular invasion at the osteochondral junction and in osteophytes in osteoarthritis. Ann Rheum Dis. 2007;66:1423–8. doi: 10.1136/ard.2006.063354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wei T, Kulkarni NH, Zeng QQ, Helvering LM, Lin X, Lawrence F, et al. Analysis of early changes in the articular cartilage transcriptisome in the rat meniscal tear model of osteoarthritis: pathway comparisons with the rat anterior cruciate transection model and with human osteoarthritic cartilage. Osteoarthritis Cartilage. 2010;18:992–1000. doi: 10.1016/j.joca.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 11.Pesesse L, Sanchez C, Henrotin Y. Osteochondral plate angiogenesis: a new treatment target in osteoarthritis. Joint Bone Spine. 2011;78:144–9. doi: 10.1016/j.jbspin.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 12.Saito T, Kawaguchi H. HIF-2alpha as a possible therapeutic target of osteoarthritis. Osteoarthritis Cartilage. 2010;18:1552–6. doi: 10.1016/j.joca.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 13.Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423:332–6. doi: 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- 14.Ashraf S, Mapp PI, Walsh DA. Contributions of angiogenesis to inflammation, joint damage and pain in a rat model of osteoarthritis. Arthritis Rheum. 2011 doi: 10.1002/art.30422. [DOI] [PubMed] [Google Scholar]
- 15.Hsieh JL, Shen PC, Shiau AL, Jou IM, Lee CH, Wang CR, et al. Intraarticular gene transfer of thrombospondin-1 suppresses the disease progression of experimental osteoarthritis. J Orthop Res. 2010;28:1300–6. doi: 10.1002/jor.21134. [DOI] [PubMed] [Google Scholar]
- 16.Matsumoto T, Cooper GM, Gharaibeh B, Meszaros LB, Li G, Usas A, et al. Cartilage repair in a rat model of osteoarthritis through intraarticular transplantation of muscle-derived stem cells expressing bone morphogenetic protein 4 and soluble Flt-1. Arthritis Rheum. 2009;60:1390–405. doi: 10.1002/art.24443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klinger P, Surmann-Schmitt C, Brem M, Swoboda B, Distler JH, Carl HD, et al. Chondromodulin 1 stabilizes the chondrocyte phenotype and inhibits endochondral ossification of porcine cartilage repair tissue. Arthritis Rheum. 2011;63:2721–31. doi: 10.1002/art.30335. [DOI] [PubMed] [Google Scholar]
- 18.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lotz MK, Carames B. Autophagy and cartilage homeostasis mechanisms in joint health, aging and OA. Nat Rev Rheumatol. 2011;7:579–87. doi: 10.1038/nrrheum.2011.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loeser RF. Aging and osteoarthritis: the role of chondrocyte senescence and aging changes in the cartilage matrix. Osteoarthritis Cartilage. 2009;17:971–9. doi: 10.1016/j.joca.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Campisi J, Andersen JK, Kapahi P, Melov S. Cellular senescence: A link between cancer and age-related degenerative disease? Semin Cancer Biol. 2011 doi: 10.1016/j.semcancer.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kirkwood TB. Understanding the odd science of aging. Cell. 2005;120:437–47. doi: 10.1016/j.cell.2005.01.027. [DOI] [PubMed] [Google Scholar]
- 23.Sahin E, Colla S, Liesa M, Moslehi J, Muller FL, Guo M, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470:359–65. doi: 10.1038/nature09787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sahin E, Depinho RA. Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature. 2010;464:520–8. doi: 10.1038/nature08982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tschopp J. Mitochondria: Sovereign of inflammation? Eur J Immunol. 2011;41:1196–202. doi: 10.1002/eji.201141436. [DOI] [PubMed] [Google Scholar]
- 26.Kim J, Xu M, Xo R, Mates A, Wilson GL, Pearsall AWt, et al. Mitochondrial DNA damage is involved in apoptosis caused by pro-inflammatory cytokines in human OA chondrocytes. Osteoarthritis Cartilage. 2010;18:424–32. doi: 10.1016/j.joca.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 27.Rego-Perez I, Fernandez-Moreno M, Deberg M, Pertega S, Fernandez-Lopez C, Oreiro N, et al. Mitochondrial DNA haplogroups and serum levels of proteolytic enzymes in patients with osteoarthritis. Ann Rheum Dis. 2011;70:646–52. doi: 10.1136/ard.2010.133637. [DOI] [PubMed] [Google Scholar]
- 28.Scott JL, Gabrielides C, Davidson RK, Swingler TE, Clark IM, Wallis GA, et al. Superoxide dismutase downregulation in osteoarthritis progression and end-stage disease. Ann Rheum Dis. 2010;69:1502–10. doi: 10.1136/ard.2009.119966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science. 2011;333:1109–12. doi: 10.1126/science.1201940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mizushima N. Physiological functions of autophagy. Curr Top Microbiol Immunol. 2009;335:71–84. doi: 10.1007/978-3-642-00302-8_3. [DOI] [PubMed] [Google Scholar]
- 31.Carames B, Taniguchi N, Otsuki S, Blanco FJ, Lotz M. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum. 2010;62:791–801. doi: 10.1002/art.27305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Terkeltaub R, Yang B, Lotz M, Liu-Bryan R. Chondrocyte AMP-activated protein kinase activity suppresses matrix degradation responses to proinflammatory cytokines interleukin-1beta and tumor necrosis factor alpha. Arthritis Rheum. 2011;63:1928–37. doi: 10.1002/art.30333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakagawa S, Arai Y, Mazda O, Kishida T, Takahashi KA, Sakao K, et al. N-acetylcysteine prevents nitric oxide-induced chondrocyte apoptosis and cartilage degeneration in an experimental model of osteoarthritis. J Orthop Res. 2010;28:156–63. doi: 10.1002/jor.20976. [DOI] [PubMed] [Google Scholar]
- 34.Stanfel MN, Shamieh LS, Kaeberlein M, Kennedy BK. The TOR pathway comes of age. Biochim Biophys Acta. 2009;1790:1067–74. doi: 10.1016/j.bbagen.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–5. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller RA, Harrison DE, Astle CM, Baur JA, Boyd AR, de Cabo R, et al. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci. 2011;66:191–201. doi: 10.1093/gerona/glq178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carames B, Taniguchi N, Seino D, Blanco FJ, D’Lima D, Lotz M. Mechanical injury suppresses autophagy regulators and its pharmacological activation results in chondroprotection. Arthritis Rheum. 2011 doi: 10.1002/art.33444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carames B, Hasegawa A, Taniguchi N, Miyaki S, Blanco FJ, Lotz M. Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann Rheum Dis. 2011 doi: 10.1136/annrheumdis-2011-200557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hui AY, McCarty WJ, Masuda K, Firestein GS, Sah RL. A systems biology approach to synovial joint lubrication in health, injury, and disease. Wiley Interdiscip Rev Syst Biol Med. 2011 doi: 10.1002/wsbm.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Greene GW, Banquy X, Lee DW, Lowrey DD, Yu J, Israelachvili JN. Adaptive mechanically controlled lubrication mechanism found in articular joints. Proc Natl Acad Sci U S A. 2011;108:5255–9. doi: 10.1073/pnas.1101002108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marcelino J, Carpten JD, Suwairi WM, Gutierrez OM, Schwartz S, Robbins C, et al. CACP, encoding a secreted proteoglycan, is mutated in camptodactyly-arthropathy-coxa vara-pericarditis syndrome. Nat Genet. 1999;23:319–22. doi: 10.1038/15496. [DOI] [PubMed] [Google Scholar]
- 42.Rhee DK, Marcelino J, Baker M, Gong Y, Smits P, Lefebvre V, et al. The secreted glycoprotein lubricin protects cartilage surfaces and inhibits synovial cell overgrowth. J Clin Invest. 2005;115:622–31. doi: 10.1172/JCI200522263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu YJ, Lu SH, Xu B, Yang RC, Ren Q, Liu B, et al. Hemangiopoietin, a novel human growth factor for the primitive cells of both hematopoietic and endothelial cell lineages. Blood. 2004;103:4449–56. doi: 10.1182/blood-2003-06-1825. [DOI] [PubMed] [Google Scholar]
- 44.Knudson CB, Knudson W. Hyaluronan and CD44: modulators of chondrocyte metabolism. Clin Orthop Relat Res. 2004:S152–62. [PubMed] [Google Scholar]
- 45.Bastow ER, Byers S, Golub SB, Clarkin CE, Pitsillides AA, Fosang AJ. Hyaluronan synthesis and degradation in cartilage and bone. Cell Mol Life Sci. 2008;65:395–413. doi: 10.1007/s00018-007-7360-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Flannery CR, Zollner R, Corcoran C, Jones AR, Root A, Rivera-Bermudez MA, et al. Prevention of cartilage degeneration in a rat model of osteoarthritis by intraarticular treatment with recombinant lubricin. Arthritis Rheum. 2009;60:840–7. doi: 10.1002/art.24304. [DOI] [PubMed] [Google Scholar]
- 47.Jay GD, Fleming BC, Watkins BA, McHugh KA, Anderson SC, Zhang LX, et al. Prevention of cartilage degeneration and restoration of chondroprotection by lubricin tribosupplementation in the rat following anterior cruciate ligament transection. Arthritis Rheum. 2010;62:2382–91. doi: 10.1002/art.27550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Teeple E, Elsaid KA, Jay GD, Zhang L, Badger GJ, Akelman M, et al. Effects of supplemental intra-articular lubricin and hyaluronic acid on the progression of posttraumatic arthritis in the anterior cruciate ligament-deficient rat knee. Am J Sports Med. 2011;39:164–72. doi: 10.1177/0363546510378088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Elsaid KA, Machan JT, Waller K, Fleming BC, Jay GD. The impact of anterior cruciate ligament injury on lubricin metabolism and the effect of inhibiting tumor necrosis factor alpha on chondroprotection in an animal model. Arthritis Rheum. 2009;60:2997–3006. doi: 10.1002/art.24800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van den Berg WB. Osteoarthritis year 2010 in review: pathomechanisms. Osteoarthritis Cartilage. 2011;19:338–41. doi: 10.1016/j.joca.2011.01.022. [DOI] [PubMed] [Google Scholar]
- 51.Matthews GL, Hunter DJ. Emerging drugs for osteoarthritis. Expert Opin Emerg Drugs. 2011;16:479–91. doi: 10.1517/14728214.2011.576670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Le Graverand-Gastineau MP. Disease modifying osteoarthritis drugs: facing development challenges and choosing molecular targets. Curr Drug Targets. 2010;11:528–35. doi: 10.2174/138945010791011893. [DOI] [PubMed] [Google Scholar]