

Abstract
Off target binding or vector sequestration can significantly limit the efficiency of systemic virotherapy. We report here that systemically administered oncolytic measles virus (MV) was rapidly sequestered by the mononuclear phagocytic system (MPS) of the liver and spleen in measles receptor CD46-positive and CD46-negative mice. Since scavenger receptors on Kupffer cells are responsible for the elimination of blood-borne pathogens, we investigated here if MV uptake was mediated by scavenger receptors on Kupffer cells. Pretreatment of cells with poly(I), a scavenger receptor ligand, reduced MV expression by 99% in murine (J774A.1) macrophages and by 50% in human (THP-1) macrophages. Pre-dosing of mice with poly(I) reduced MPS sequestration of MV and increased circulating levels of MV by 4 to 15-folds at 2 minutes post virus administration. Circulating virus was still detectable 30 mins post infusion in mice predosed with poly(I) while no detectable MV was found at 5–10 min post infusion if mice did not receive poly(I). MPS blockade by poly(I) enhanced virus delivery to human ovarian SKOV3ip.1 and myeloma KAS6/1 xenografts in mice. Higher gene expression and improved control of tumor growth was noted early post therapy. Based on these results, incorporation of MPS blockade into MV treatment regimens is warranted.
Keywords: oncolytic measles virus, mononuclear phagocytic system, sequestration, poly(I), scavenger receptor, SKOV3ip.1, KAS6/1
Introduction
Tumor selective oncolytic viruses, which have multiple mechanisms of antitumor activity and target diverse pathways in the malignant cell, represent an exciting new class of targeted therapeutics for cancer therapy. Viruses can cause direct cell destruction and/or elicit innate and cellular antitumor immune responses that result in control of distant metastases.1–3 Based on promising phase II data, two oncolytic viruses, OncoVexGMCSF and Reolysin, are in phase III clinical testing, either as a single agent or in combination with standard chemotherapy in melanoma or head and neck cancer.4, 5
While oncolytic viruses hold much promise, there are several challenges associated with systemic virotherapy for the treatment of metastatic or surgically inaccessible tumors.6 Intravenously administrated viruses can quickly be inactivated by neutralizing antibodies in virus immune patients7, 8 Strategies that help viruses evade antibodies include coating viral particles with polyethylene glycol or polymers, hiding the virus in cell carriers or serotype switching.8–12 Viral particles can also be opsonized and sequestered by the reticuloendothelial system, resulting in shorter circulatory half-lives and reduced bioavailability to tumor cells.13–15 To minimize sequestration, strategies that deplete serum factors such as complement or Factor X (using cobra venom factor or warfarin), macrophages (using chldronate liposomes or adenoviral particles) or blockade of scavenger receptors (using polyinosinoic acid) have been attempted.16–19 Finally, the viral particles need to extravasate efficiently from the tumor blood vessels into the tumor parenchyma to initiate an infection. Indeed these are generic issues that apply to systemic administration of most viruses and various groups are developing innovative approaches to overcome these challenges.20–23
We and others have been developing the attenuated Edmonston B lineage of measles virus (MV) as a tumor selective oncolytic agent for cancer therapy.24 Two recombinant measles viruses modified to enable noninvasive monitoring, MV-CEA and MV-NIS, are currently undergoing clinical testing in patients with ovarian cancer, multiple myeloma or glioblastoma.25–30 Oncolytic MV infects cells by binding to one of two MV receptors, CD46 or SLAM (signaling lymphocyte activation molecule). The virus is highly fusogenic; infected cells fuse with their neighbors to form multinucleated syncytia, thereby facilitating viral spread through the infected culture.31 The virus has shown promising antitumor activity in numerous mouse xenograft models of human malignancies, including lymphoma, multiple myeloma, ovarian, colorectal, breast, prostate, liver cancer, and glioma.6, 29, 30, 32–37 Tumor selectivity of MV is in part due to high CD46 receptor density on the tumor cell surface, resulting in more extensive cytopathic effects compared to nontransformed cells.29, 38, 39 For example, CD138+ primary multiple myeloma cells express significantly higher levels of CD46 receptors per cell (49,130 molecules) than CD138 negative non-myeloma cells from the bone marrow (7,340 molecules).40 Potent cytopathic effects of extensive intercellular fusion were observed in measles-infected primary myeloma cells and not in the non-myeloma cells.40
Despite numerous studies evaluating the antitumor potency of MV, the in vivo fate of systemically administered oncolytic MV particles has not been reported before. In this study, we investigated the biodistribution of MV particles in two murine models; nontransgenic, CD46-negative mice and CD46-positive transgenic mice. CD46 negative athymic or SCID mice are used routinely in efficacy studies for evaluation of the antitumor activity of MV against human tumor xenografts. Since all nontransformed human cells except erythrocytes express CD46 at low levels, we have also used transgenic mice which express the human CD46 receptor with a human-like distribution and level of expression in this study. These CD46 transgenic mice have been extensively used in MV pathogenesis studies and in toxicology/pharmacology studies in support of Investigational New Drug (IND) applications to the Food and Drug Administration to test MV in phase I clinical trials. In the current study, we demonstrated that intravenously administered MV was rapidly sequestrated by macrophages in the liver and spleen of mice, and that sequestration was independent of CD46 expression, instead being mediated by virus binding to scavenger receptors on murine macrophages. Scavenger receptors form a large family of receptors (class A to H) that recognize negatively charged or opsonized materials and are responsible for the clearance of ligands in vivo, including DNA, damaged erythrocytes, and endotoxin.15, 43 Analysis of murine tissues has shown that most macrophage populations express scavenger receptor A (SR-A), which is found most abundantly on Kupffer cells, alveolar macrophages, and lamina propria macrophages in the gut.44 Ligands such as polyinosinic acid [poly(I)], a synthetic single-stranded RNA can bind to SR-A to competitively inhibit uptake of viruses, bacteria or oxidized erythrocytes by macrophages.18, 44, 45, 46 It appears that the base-quartet-stabilized four-stranded helix of poly(I) is a necessary structural determinant for binding to and inhibition of scavenger receptors. The negatively charged phosphates in polynucleotide quadruplexes may form a charged surface that is complementary to the positively charged surface of the collagenous ligand-binding domain of the scavenger receptor.47 Previous studies have shown that pre-administration of poly(I) prior to phage or adenovirus infusion decreased macrophage sequestration of the viral particles.18, 48, 49 In this study, pre-dosing mice with a poly(I) was performed to evaluate the impact of macrophage blockade on the circulatory half-life and antitumor activity of MV in two models of human cancer, multiple myeloma and ovarian cancer.
Materials and Methods
Cells and viruses
The murine macrophage cell line J774A.1 (ATCC, TIB-67) and the human monocyte cell line THP-1 (ATCC, TIB-202) were obtained from American Type Culture Collection. J774A.1 was cultured in DMEM supplemented with 10% fetal bovine serum (FBS) and THP-1 was cultured in RPMI-1640 plus 10% FBS and 0.05 mM β-mercaptoethanol. Phorbol-12-myristate-13-acetate (PMA), 100 nM, was added to the culture media to differentiate THP-1 monocytes into macrophages. The human epithelial ovarian carcinoma, SKOV3ip.1 was maintained in α-MEM supplemented with 20% FBS and 2 nM L-glutamine. The human multiple myeloma cell line, KAS6/1, was cultured in 10% FBS RPMI-1640 medium supplemented with 1 ng/ml recombinant human interleukin-6 (R&D Systems, Minneapolis, MN).
Recombinant Edmonston strain MV expressing firefly luciferase (MV-Luc) and or the thyroidal sodium iodide symporter (MV-NIS) were propagated as previously described.26, 28 Viral titers were determined by TCID50 (median tissue culture infective dose) plaque-forming assay on Vero cells.50 Fluorescent DiI or DiO labeled MV particles were generated by adding Vybrant cell-labeling solution (Invitrogen, Carlsbad, CA) during virus propagation as previously described.51
In vitro infection and reporter-gene expression analysis
Murine macrophages (J774A.1), human monocytes (THP-1), and human macrophages (PMA treated THP-1) were seeded into 24-well culture plates at a density of 105 cells/well. After 24 h, cells were incubated for 1h at 37°C in the presence or absence of polyinosinic acid [poly(I)] or polyadenylic acid [poly(A)] (Sigma-Aldrich, St. Louis, MO). After 1h, MV-Luc was added (MOI = 0.5). Two hours after infection, the infection medium was replaced by standard growth medium and cells were incubated for 48h before assay for luciferase gene expression. For the luciferase assay, cells were lyzed with cell-culture lysis buffer (Promega Corporation, Madison, WI) and the lysates were analyzed with the luciferase assay system (Promega Corporation, Madison, WI) on a LumiCount luminometer (Packard). All data are expressed as relative light units (RLU).
Mice
All animal experiments were approved by and performed according to guidelines of the Institute Animal Care and Use Committee of the Mayo Foundation. Because murine tissues do not express measles virus receptors and hence cannot be infected by the virus, we have used two murine models in this study; the CD46 transgenic (CD46+) mouse, Ifnar-CD46Ge, which expresses the human CD46 receptor with the same tissue specificity52 and CD46 receptor negative (CD46−) immunocompromised athymic or SCID mice in which human tumor xenografts can be established. Ifnar-CD46Ge mice were bred in-house. Female athymic mice or ICR-SCID mice were purchased from Harlan (Indianapolis, IN) or Taconic (Germantown, NY), respectively.
The effect of poly(I) on MPS sequestration of MV were studied using athymic (CD46−) and Ifnar-CD46Ge (CD46+) mice. Mice were injected with PBS or PBS containing 0.2 mg poly(I) per mouse 5 min prior to injection of 106 TCID50 fluorescently labeled MV-Luc through either tail vein (athymic) or orbital plexus (Ifnar-CD46Ge). Since the CD46+ mice have a black tail which makes IV administration technically challenging, we have opted to administer the virus by the orbital plexus. Biodistribution studies on viral genomes have indicated comparable viral distribution by IV administration via the tail vein and RO (Peng, unpublished). Liver and spleen were harvested 3h after injection, snap frozen in OCT, and 5μm cryosections were photographed under fluorescence microscopy. Fluorescent MV-Luc positive cells were quantitated using Image J software (NIH). MV blood circulation times were determined by collecting an aliquot of blood via the retro-orbital plexus at 2, 5, 10, and 30 min after virus administration. Plasma was obtained and the amount of MV particles was determined by TCID50 assay on Vero cells.
For studying the effect of poly(I) on delivery of MV particles to the tumor site, athymic mice bearing subcutaneous SKOV3ip.1 tumors were given 0.2mg poly(I) 5 mins before 106 TCID50 MV-Luc was injected intravenously through the tail vein. For studying the effect of poly(I) on tumor growth, athymic mice bearing subcutaneous SKOV3ip.1 tumors and ICR-SCID mice bearing subcutaneous KAS6/1 tumors were given 0.2mg poly(I) i.v. 5 mins before i.v. administration of MV-NIS (107 TCID50 for SKOV3ip.1 tumors and 105 TCID50 for KS6/1 tumors).
Immunohistochemistry
Liver and spleen tissues from athymic mice and Ifnar-CD46Ge mice were frozen in optimal cutting temperature medium (OCT) and 5μm cryosections were cut onto slides. The tissues were fixed in acetone (−20°C) for 10 min and permeabilized using 0.01% Triton-X in PBS for 15 min. Blocking buffer containing 5% horse serum in PBS was applied for 20 min, and after which tissues were incubated with rat anti-murine CD68 antibody (Abcam Inc., Cambridge, MA) for 1h at room temperature. Slides were washed five times in PBS, and followed by incubation with Alexa 488 conjugated anti-rat antibody (Abcam Inc., Cambridge, MA) for 30 min.
Quantitative RT-PCR for measles nucleoprotein mRNA
Tissues from MV-NIS-injected mice were harvested 4h after virus injection and frozen in RNAlater solution (Ambion, Austin, TX). Total RNA was extracted from 10 to 20 mg of tissue, using RNeasy plus mini kits (Qiagen, Valencia, CA). Co-isolated DNA was removed by spinning through the gDNA Eliminator spin columns. A 61-base product of the MV-N gene was amplified using the iScript One-Step RT-PCR kit (Bio-Rad, Hercules, CA) as previously described.42
Statistical analysis
Statistical significance of experimental results was analyzed by unpaired student’s t test where indicated. A p value of <0.05 is considered statistically different.
Results
Systemically administered MV particles are sequestered by the mononuclear phagocytic system (MPS) of the liver and spleen
We have used two murine models in this study, CD46 negative athymic mice and Ifnar-CD46Ge mice genetically modified to express the human CD46 receptor with the same tissue specificity as in humans.52, 53 Use of both models allows us to evaluate the impact of CD46 expression on the biodistribution of MV particles. At 2, 5, 10 and 30 minutes post i.v. injection of MV in mice, blood samples were collected and the amount of infectious virus in the serum was analyzed by virus titration (TCID50) assay. There was a rapid decline in the amount of infectious MV from the circulation of both CD46 negative and CD46 positive mice (Figure 1). The half-life of circulating MV was about 1 min.
Figure 1.
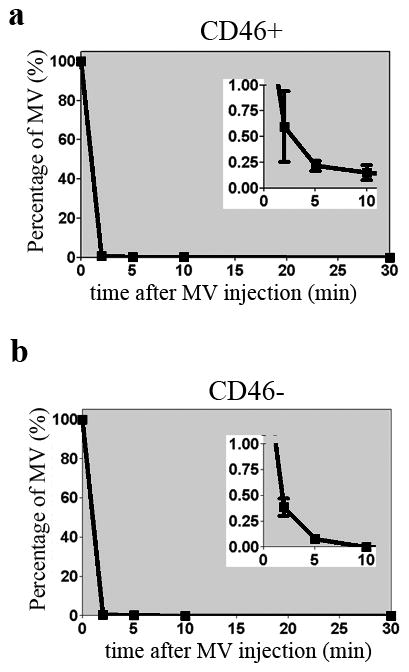
Systemically administered MV viruses are sequestered from circulation in both CD46 negative athymic mice and CD46 positive Ifnar-CD46Ge mice. Clearance of MV viruses from circulation post intravenous injection of 106 TCID50 MV-Luc into (a) CD46 positive mice or (b) CD46 negative mice. The decay graphs between 0–10 min are magnified and shown on the right upper corner as inserts.
To determine if disappearance of infectious virus was due to neutralization by murine blood, MV was added exogenously into whole blood harvested from athymic mice and Ifnar-CD46Ge mice or media (Opti-MEM) and incubated for 30 minutes at 37°C. Plasma was separated from whole blood on a Ficoll gradient and the amount of infectious virus was quantitated by TCID50 titration on Vero cells. As shown in Table 1, about 10% of input virus was found associated with lymphocytes from the CD46 positive mice. However, the majority (63–84%) of the input virus remained infectious in the plasma/platelets fraction even after 30 minutes of mixing at 37°C (Table 1), suggesting that the rapid disappearance of infectious MV from the circulation was not due to virus inactivation by mouse blood.
Table 1.
The amount (TCID50) of infectious MV recovered from the plasma, white blood cells (WBC) and red blood cell (RBC) fractions post incubation of MV with whole blood from CD46 negative or CD46 positive mice for 30 minutes at 37°C. The percentage of MV in the respective fractions is calculated against the input virus and shown in parentheses.
| Fraction | CD46− mice
|
CD46+ mice
|
||
|---|---|---|---|---|
| Exp. 1 | Exp. 2 | Exp. 1 | Exp. 2 | |
| Plasma | 1.3×106 (84%) | 1.3×106 (84%) | 9.5×105 (63%) | 9.5×105 (63%) |
| WBC | 2.6×104 (1.7%) | 2×104 (1.3%) | 1.5×105 (10%) | 1.1×105 (7.5%) |
| RBC | 1.2×104 (0.8%) | 4.3×103 (0.3%) | 7.4×104 (4.7%) | 5.3×104 (3.5%) |
To facilitate in vivo tracking of viral particles, MV was labeled fluorescently with either DiI or DiO lipophillic dyes and injected intravenously into mice. To ensure all circulating viruses have arrived at their destination, the mice were harvested at 3 hours post MV infusion. Major organs were examined under blue light to determine which tissues have taken up the DiO green fluorescently labeled MV. Both the liver and spleen were strongly positive for green fluorescent cells. Further analysis of the liver and spleen cryosections confirmed the presence of abundant green fluorescent cells in both CD46− athymic and CD46+ transgenic mice (Figure 2a). Green fluorescent cells were uniformly distributed in the liver parenchyma while the spleen showed a more distinct circular pattern around the white pulp.41 To determine if the cells were macrophages that had phagocytosed MV, co-localization studies was performed using immunohistochemical staining for CD68, a macrophage marker. Red fluorescent cells in the liver and spleen which had taken up the DiI labeled red fluorescent MV colocalized with CD68 positive cells, identifying macrophages as the cells responsible for uptake of Dil-MV (Figure 2b).
Figure 2.

Systemically administered MV particles are sequestered by macrophages in the liver and spleen of CD46 negative athymic and CD46 positive Ifnar-CD46Ge mice. (a) Fluorescence microscopy shows the sequestration of green fluorescent DiO-MV particles in the liver and spleen after intravenous delivery. Control = no virus given. 50X magnification. (b) Representative confocal microscopy images of liver and spleen cryosections showing colocalization of red fluorescent DiI-MV particles with CD68 positive (Alexa-488/green color) macrophages. 400X magnification. (c) Quantitative RT-PCR data showing the biodistribution of MV genomes in the major organs of mice post intravenous delivery of 1.5×107 TCID50 MV-NIS. Results are expressed as mean ± SD (n=3 mice). *indicate that the two groups are statistically different (p<0.05).
To more accurately measure the relative levels of MV uptake by the respective organs, quantitative RT-PCR for MV-nucleocapsid mRNA was performed. Very high amounts of viral particles were found in the liver (3×105 copies/μg total RNA) and spleen (1.5×105 copies/μg total RNA) of CD46+ mice (Figure 2c). In contrast to the athymic mice, a significantly higher amount of MV (6×105 copies/μg total RNA) was also found in the lungs of Ifnar-CD46Ge mice (p<0.05). Lower amounts of MV genomes were also detected in the kidneys and brains of Ifnar-CD46Ge mice, likely as a result of MV interaction with CD46 on these tissues.53 In CD46− athymic mice, viral particles were found mainly in the liver (3×105 copies/μg total RNA) and spleen (8×104 copies/μg total RNA). Taken together, these data indicate that a significant portion of systemically administered MV can be taken up by cells of the mononuclear phagocytic system of the liver and spleen independent of CD46 expression.
Scavenger receptors are involved in MV particles uptake in macrophages
The rapid clearance of MV from the circulation and sequestration into the liver and spleen is likely a result of virus binding to scavenger receptors on macrophages. To evaluate the role of scavenger receptors in MV uptake, macrophages were pre-treated with synthetic single-stranded RNA, poly(I) or polyadenylic acid [poly(A)]. Poly(I) is a broad-specificity scavenger receptor inhibitor while poly(A) inhibits a more limited subset of scavenger receptors.15 In particular, poly(I) is able to bind scavenger receptor class A while poly(A) does not.15 Human monocyte (THP-1), human macrophage (THP-1 treated with PMA), and murine macrophage (J774A.1) cell lines were pre-incubated with poly(I) or poly(A) before infection with MV encoding the firefly luciferase gene (MV-Luc). In the absence of poly(I) or poly(A), MV-Luc was able to infect and express in J774A.1 cells (~104 RLU) even though murine macrophages do not express the measles CD46 receptor (Figure 3), although the level of luciferase activity in J774.1 cells was 10 times lower than in CD46 positive human THP-1 macrophages (data not shown). Addition of poly(I) significantly (p <0.05) inhibited luciferase gene expression levels in all three cell types (Figure 3). In J774A.1 cells, the luciferase expression level was completely inhibited by poly(I) or poly(A), indicating that MV uptake by murine macrophages was predominantly due to scavenger receptors. Interestingly, MV gene expression in THP-1 human macrophages was inhibited 50% by poly(I), suggesting that scavenger receptors (or other receptors blocked by poly I) can contribute to a significant portion of viral entry into these cells. Poly(A) which binds to a smaller subset of scavenger receptor did not significantly inhibit MV-Luc uptake by human cells.
Figure 3.
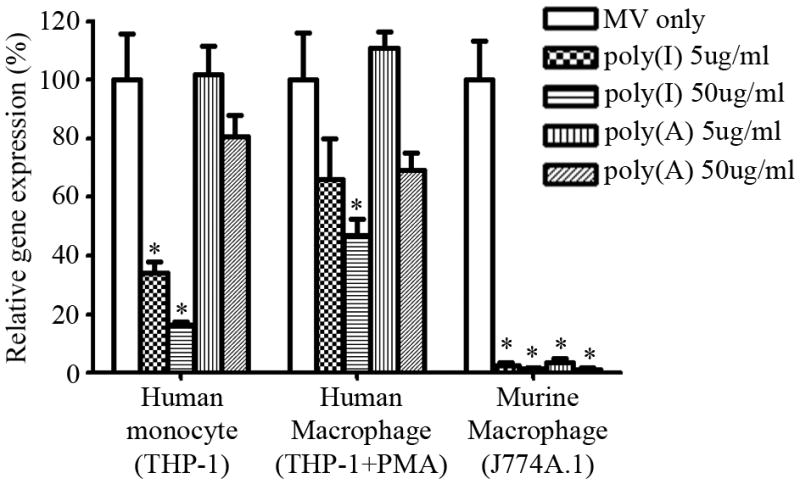
Poly(I) reduces MV-Luc infection of macrophages in vitro. Relative luciferase gene-expression levels in human monocyte (THP-1), human macrophage (THP-1 treated with PMA), and murine macrophage (J774A.1) cell lines in the absence (MV-Luc only) or presence of poly(A) or poly(I) and MV-Luc. Gene expression is presented as a percentage of that in cells infected with MV-Luc in the absence of poly(I). Results are expressed as the mean ± SD of three different experiments. * indicate statistically different (p<0.05) against MV only group.
Inhibition of the scavenger receptors reduced MV sequestration by macrophages in the liver and spleen
Based on the above findings, we next determined if inhibition of scavenger receptors would reduce MV sequestration in vivo. Mice were injected i.v. with poly(I) or saline 5 mins before i.v. administration of DiI-MV. Livers and spleens were harvested 3h later and numbers of cells that had taken up red fluorescent Dil-MV in the cryosections were quantitated. Compared to mice not given poly(I), there was significantly fewer numbers of red fluorescent cells in livers and spleens of mice pre-dosed with poly(I). Accumulation of Dil-MV particles was reduced by 65% (liver) and 75% (spleen) in athymic mice and by 35% in livers or spleens of Ifnar-CD46Ge mice (Figure 4a). Next, we evaluated whether blockade of scavenger receptors by poly(I) improved circulation half life of MV in mice. After i.v. administration of poly(I) and MV, blood samples were collected and analyzed for infectious virus by TCID50 assay. At the early time point of 2 minutes, pre-administration of poly(I) increased the maximum level of virus present in the blood by about 15-fold in athymic mice and by about 4-fold in Ifnar-CD46Ge mice (Figure 4b and 4c).
Figure 4.

Poly(I) reduces uptake of MV particles by the liver and spleen, enhancing bioavailability of MV in the blood of mice. CD46-negative and CD46-positive mice were injected intravenously with PBS or 0.2 mg poly(I) per mouse 5 min prior to administration of 106 TCID50 DiI-MV-Luc. (a) Numbers of red fluorescent cells (due to uptake of DiI-MV) in liver and spleen cryosection were counted (pixels) using the Image J software. Data is presented as a percentage of that in mice given MV only. Blood from (b) CD46-negative or (c) CD46-positive mice were collected at 2, 5, 10 and 30 min after MV injection and analyzed for infectious virus by TCID50 assay. Results are expressed as mean ± SD (n=3 mice). *indicate statistically different (p<0.05) against no poly(I).
Inhibition of the scavenger receptors enhanced measles virus delivery to the tumor and retards tumor growth
To determine if the reduced sequestration by MPS and prolonged circulation times impacted delivery of MV to the tumor site, MV-Luc was administered systemically into athymic mice bearing SKOV3ip.1 tumors after the mice had been pre-dosed with poly(I) or saline. Bioluminescent imaging data indicated that there was no detectable luciferase signal in the subcutaneous SKOV3ip.1 tumors in mice given MV-Luc alone (Figure 5a, b). In contrast, all three mice given poly(I) and MV-Luc showed robust luciferase signal in the SKOV3ip.1 tumors by day 5 and luciferase gene expression remained strong at day 10. These data suggest that pre-dosing with poly(I) enhanced MV delivery to the tumor site allowing the virus to initiate a sustained infection.
Figure 5.

Poly(I) enhanced MV delivery to the tumor site and improved antitumor activity of MV at early time points post therapy. Mice with subcutaneous SKOV3ip.1 xenografts were injected intravenously with saline or poly(I), followed by a single dose of MV-Luc. Mice were imaged at day 5 and day 10 post MV on a Xenogen 200 machine. (a) Bioluminescent images showing higher MV-Luc gene expression in tumors of mice predosed with poly(I). (b) The luciferase activities in the tumors were quantitated and graphed. The efficacy of single dose MV-NIS therapy with or without poly(I) pre-dosing was evaluated by comparing tumor growth over time in (c) SKOV3ip.1 ovarian (N=10 mice per group) and (d) KAS 6/1 myeloma (N=8 mice per group) xenografts. Pretreatment of mice with poly(I) was associated with a significant retardation in tumor growth. *indicate statistically different (p<0.05) from saline group at the respective time point.
The impact of poly(I) on the antitumor activity of MV-NIS, a recombinant measles virus currently undergoing clinical testing in patients with ovarian cancer and myeloma, was assessed in two established tumor xenograft models, subcutaneous SKOV3ip1 ovarian (Figure 5c) and KAS6/1 myeloma (Figure 5d) tumors. Mice were randomized into three treatment groups and received 1 dose of saline, saline/MV-NIS, or poly(I)/MV-NIS. As shown in figure 5c and 5d, there was no significant difference in the tumor volumes between mice given saline or a subtherapeutic dose of MV-NIS. In contrast, pre-dosing with poly(I) resulted in a significant inhibition of tumor growth compared to the saline controls in both tumor models (P<0.05). Since the mice were given only a single subtherapeutic dose of virus, the tumors continued to grow in the treated animals and the Kaplan Meier survival curves were not significantly enhanced compared to the saline control.
Discussion
Numerous studies have demonstrated the antitumor activity of MV in human tumor xenografts established in mice but there have been no published reports on the fate of intravenously administered MV particles in research animals. Here, we demonstrated that a 30 minute exposure of MV to murine blood at 37°C did not result in significant reduction in viral infectivity and the majority (63–84%) of the input virus remained infectious in the plasma/platelets fraction. Upon systemic administration in mice, MV particles have a short circulating half-life of about 1 min due to rapid uptake of particles by macrophages in the liver and spleen. Quantitative RT-PCR for viral mRNA confirmed that the amounts of virus trapped in the liver and spleen were comparable in both CD46-positive transgenic mice and CD46-negative athymic mice, demonstrating that sequestration of MV by the MPS was not due to CD46. Immunohistochemical staining of the liver and spleen cryosections from athymic and Ifnar-CD46Ge mice indicated that MV particles were taken up by macrophages. Blocking scavenger receptors with a competitive ligand poly(I) in vitro on macrophage cell lines or in vivo in mice significantly reduced MV uptake by macrophages.
In the liver, specialized macrophages (Kupffer cells) are predominantly distributed on the lumenal surface of hepatic sinusoids and play an important role in removing potentially harmful materials and particulate matter such as bacterial endotoxins, micro-organisms, immune-complexes and tumor cells from the blood. Several viruses such as vesicular stomatitis virus, simian immunodeficiency virus, and adenoviruses were also found to be efficiently cleared from the circulation by Kupffer cells.13, 14, 54 Recently, a number of studies demonstrated that adenoviruses can use scavenger receptor A as a direct route to enter Kupffer cells.15, 49, 55 Administration of poly(I) into mice prior to adenovirus injection significantly reduced the capacity of Kupffer cells to trap adenoviral particles, although the impact of poly(I) pre-dosing on antitumor activity of the adenoviral vector was not evaluated in that study. Here, we confirmed that MV is able to bind to scavenger receptors on macrophage cells and this route of entry was inhibited more than 99% by poly(I) and poly(A) on the murine macrophage J774.1 cell line. Interestingly, addition of poly(I) but not poly(A) also inhibited 50% of MV gene expression in CD46 receptor positive human THP-1 macrophage cell line, indicating the scavenger receptor (or other receptors blocked by polyI) can mediate uptake of MV by human macrophages.
Pre-dosing of athymic mice and Ifnar-CD46Ge mice with poly(I) prolonged the circulatory half-life of MV, resulting in a 15-fold and 4-fold increase, respectively, of MV levels in the blood early post virus infusion. Though modest, this increase in MV circulating half-life had an impact on virus delivery to tumors, enhancing virus delivery to and infection of tumor xenografts. Importantly, poly(I) pre-dosing resulted in superior tumor control by MV-NIS in two different models of human malignancy (ovarian and myeloma), at least at the early time points post delivery. Since the aim of the experiment was to evaluate the impact of poly(I) on MV delivery, tumor bearing mice were given only one dose of MV-NIS and the survival curves of mice treated with or without poly(I) was not significantly different at the end of the experiment. Nonetheless, this single delivery of MV at subtherapeutic levels clearly demonstrated the advantage of incorporating poly(I) in the treatment regimen to increase the bioavailability of the systemically applied virus.
The safety and feasibility of using poly(I) as a potential pre-dosing agent prior to virotherapy in cancer patients remains to be evaluated. It is important to point out that poly(I) is a single stranded RNA and is less effective at inducing IFN-alpha in leukocytes compared to double stranded small RNA polyI:C.56 Systemic administration of poly(I) at 0.2 mg per mouse induced no observable toxicity in livers of athymic mice although transient higher serum levels of IL-6, MCP-1 and TNF-α were detected.55 Alternative ligands of scavenger receptor A could be tested in lieu of poly(I) if clinical testing is desired, for example, fucoidan46, 57 or using an alternative strategy to transiently eliminate macrophages using clodronate loaded liposomes.58 While polyI:C has been given as adjuvant in vaccination protocols in human studies59 and fucoidan has been given orally to humans60, 61, intravenous administration of poly(I), fucoidan or clodronate liposomes in humans has not been reported. Intravenous use of these agents will require testing in toxicology studies to evaluate feasibility for clinical use. Instead of blocking or saturating the mononuclear phagocytic system using ligands, one could employ other strategies such as shielding the viral vectors with polymers such as polyethylene glycol or HPMA copolymers to minimize vector recognition and uptake by the MPS.62, 63 Tumor homing cells have also been used as carriers to delivery oncolytic viruses to tumor xenografts64 and also protect the viruses from inactivation by serum factors such as complement and antiviral antibodies.10 The relative effectiveness of these different strategies at increasing the bioavailability of viruses and their applicability in clinical practice remains to be compared in future studies.
Acknowledgments
We gratefully acknowledge funding support from the NIH/NCI (CA118488, CA129193, CA100634, CA129966, CA125614), Mayo Foundation and the Mayo Clinic Comprehensive Cancer Center (CA15083).
References
- 1.Chiocca EA. The host response to cancer virotherapy. Current opinion in molecular therapeutics. 2008;10(1):38–45. [PubMed] [Google Scholar]
- 2.Liu TC, Galanis E, Kirn D. Clinical trial results with oncolytic virotherapy: a century of promise, a decade of progress. Nature clinical practice. 2007;4(2):101–17. doi: 10.1038/ncponc0736. [DOI] [PubMed] [Google Scholar]
- 3.Melcher A, Parato K, Rooney CM, Bell JC. Thunder and lightning: immunotherapy and oncolytic viruses collide. Mol Ther. 2011;19(6):1008–16. doi: 10.1038/mt.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park BH, Hwang T, Liu TC, Sze DY, Kim JS, Kwon HC, et al. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. The lancet oncology. 2008;9(6):533–42. doi: 10.1016/S1470-2045(08)70107-4. [DOI] [PubMed] [Google Scholar]
- 5.Senzer NN, Kaufman HL, Amatruda T, Nemunaitis M, Reid T, Daniels G, et al. Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J Clin Oncol. 2009;27(34):5763–71. doi: 10.1200/JCO.2009.24.3675. [DOI] [PubMed] [Google Scholar]
- 6.Russell SJ, Peng KW. Measles virus for cancer therapy. Current topics in microbiology and immunology. 2009;330:213–41. doi: 10.1007/978-3-540-70617-5_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu C, Russell SJ, Peng KW. Systemic therapy of disseminated myeloma in passively immunized mice using measles virus-infected cell carriers. Mol Ther. 2010;18(6):1155–64. doi: 10.1038/mt.2010.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hemminki O, Bauerschmitz G, Hemmi S, Lavilla-Alonso S, Diaconu I, Guse K, et al. Oncolytic adenovirus based on serotype 3. Cancer Gene Ther. 2011;18(4):288–96. doi: 10.1038/cgt.2010.79. [DOI] [PubMed] [Google Scholar]
- 9.Doronin K, Shashkova EV, May SM, Hofherr SE, Barry MA. Chemical modification with high molecular weight polyethylene glycol reduces transduction of hepatocytes and increases efficacy of intravenously delivered oncolytic adenovirus. Hum Gene Ther. 2009;20(9):975–88. doi: 10.1089/hum.2009.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mader EK, Maeyama Y, Lin Y, Butler GW, Russell HM, Galanis E, et al. Mesenchymal stem cell carriers protect oncolytic measles viruses from antibody neutralization in an orthotopic ovarian cancer therapy model. Clin Cancer Res. 2009;15(23):7246–55. doi: 10.1158/1078-0432.CCR-09-1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Power AT, Wang J, Falls TJ, Paterson JM, Parato KA, Lichty BD, et al. Carrier cell-based delivery of an oncolytic virus circumvents antiviral immunity. Mol Ther. 2007;15(1):123–30. doi: 10.1038/sj.mt.6300039. [DOI] [PubMed] [Google Scholar]
- 12.Miest TS, Yaiw KC, Frenzke M, Lampe J, Hudacek AW, Springfeld C, et al. Molecular therapy: the journal of the American Society of Gene Therapy. 2011. Envelope-chimeric Entry-targeted Measles Virus Escapes Neutralization and Achieves Oncolysis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alemany R, Suzuki K, Curiel DT. Blood clearance rates of adenovirus type 5 in mice. The Journal of general virology. 2000;81(Pt 11):2605–9. doi: 10.1099/0022-1317-81-11-2605. [DOI] [PubMed] [Google Scholar]
- 14.Brunner KT, Hurez D, Mc CR, Benacerraf B. Blood clearance of P32-labeled vesicular stomatitis and Newcastle disease viruses by the reticuloendothelial system in mice. J Immunol. 1960;85:99–105. [PubMed] [Google Scholar]
- 15.Xu Z, Tian J, Smith JS, Byrnes AP. Clearance of adenovirus by Kupffer cells is mediated by scavenger receptors, natural antibodies, and complement. Journal of virology. 2008;82(23):11705–13. doi: 10.1128/JVI.01320-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ikeda K, Ichikawa T, Wakimoto H, Silver JS, Deisboeck TS, Finkelstein D, et al. Oncolytic virus therapy of multiple tumors in the brain requires suppression of innate and elicited antiviral responses. Nature medicine. 1999;5(8):881–7. doi: 10.1038/11320. [DOI] [PubMed] [Google Scholar]
- 17.Shashkova EV, Doronin K, Senac JS, Barry MA. Macrophage depletion combined with anticoagulant therapy increases therapeutic window of systemic treatment with oncolytic adenovirus. Cancer research. 2008;68(14):5896–904. doi: 10.1158/0008-5472.CAN-08-0488. [DOI] [PubMed] [Google Scholar]
- 18.Haisma HJ, Kamps JA, Kamps GK, Plantinga JA, Rots MG, Bellu AR. Polyinosinic acid enhances delivery of adenovirus vectors in vivo by preventing sequestration in liver macrophages. The Journal of general virology. 2008;89(Pt 5):1097–105. doi: 10.1099/vir.0.83495-0. [DOI] [PubMed] [Google Scholar]
- 19.Meijer C, Wiezer MJ, Diehl AM, Schouten HJ, Schouten HJ, Meijer S, et al. Kupffer cell depletion by CI2MDP-liposomes alters hepatic cytokine expression and delays liver regeneration after partial hepatectomy. Liver. 2000;20(1):66–77. doi: 10.1034/j.1600-0676.2000.020001066.x. [DOI] [PubMed] [Google Scholar]
- 20.Baker AH, McVey JH, Waddington SN, Di Paolo NC, Shayakhmetov DM. The influence of blood on in vivo adenovirus bio-distribution and transduction. Molecular therapy: the journal of the American Society of Gene Therapy. 2007;15(8):1410–6. doi: 10.1038/sj.mt.6300206. [DOI] [PubMed] [Google Scholar]
- 21.Kueberuwa G, Cawood R, Seymour LW. Blood compatibility of enveloped viruses. Current opinion in molecular therapeutics. 2010;12(4):412–20. [PubMed] [Google Scholar]
- 22.Nakashima H, Kaur B, Chiocca EA. Directing systemic oncolytic viral delivery to tumors via carrier cells. Cytokine Growth Factor Rev. 2010;21(2–3):119–26. doi: 10.1016/j.cytogfr.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tseng JC, Granot T, DiGiacomo V, Levin B, Meruelo D. Enhanced specific delivery and targeting of oncolytic Sindbis viral vectors by modulating vascular leakiness in tumor. Cancer Gene Ther. 2010;17(4):244–55. doi: 10.1038/cgt.2009.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lech PJ, Russell SJ. Use of attenuated paramyxoviruses for cancer therapy. Expert review of vaccines. 2010;9(11):1275–302. doi: 10.1586/erv.10.124. [DOI] [PubMed] [Google Scholar]
- 25.Dingli D, Bergert ER, Bajzer Z, O’Connor MK, Russell SJ, Morris JC. Dynamic iodide trapping by tumor cells expressing the thyroidal sodium iodide symporter. Biochemical and biophysical research communications. 2004;325(1):157–66. doi: 10.1016/j.bbrc.2004.09.219. [DOI] [PubMed] [Google Scholar]
- 26.Dingli D, Peng KW, Harvey ME, Greipp PR, O’Connor MK, Cattaneo R, et al. Image-guided radiovirotherapy for multiple myeloma using a recombinant measles virus expressing the thyroidal sodium iodide symporter. Blood. 2004;103(5):1641–6. doi: 10.1182/blood-2003-07-2233. [DOI] [PubMed] [Google Scholar]
- 27.Galanis E, Hartmann LC, Cliby WA, Long HJ, Peethambaram PP, Barrette BA, et al. Phase I trial of intraperitoneal administration of an oncolytic measles virus strain engineered to express carcinoembryonic antigen for recurrent ovarian cancer. Cancer research. 2010;70(3):875–82. doi: 10.1158/0008-5472.CAN-09-2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peng KW, Facteau S, Wegman T, O’Kane D, Russell SJ. Non-invasive in vivo monitoring of trackable viruses expressing soluble marker peptides. Nature medicine. 2002;8(5):527–31. doi: 10.1038/nm0502-527. [DOI] [PubMed] [Google Scholar]
- 29.Peng KW, TenEyck CJ, Galanis E, Kalli KR, Hartmann LC, Russell SJ. Intraperitoneal therapy of ovarian cancer using an engineered measles virus. Cancer research. 2002;62 (16):4656–62. [PubMed] [Google Scholar]
- 30.Phuong LK, Allen C, Peng KW, Giannini C, Greiner S, TenEyck CJ, et al. Use of a vaccine strain of measles virus genetically engineered to produce carcinoembryonic antigen as a novel therapeutic agent against glioblastoma multiforme. Cancer research. 2003;63(10):2462–9. [PubMed] [Google Scholar]
- 31.Lamb RA, Paterson RG, Jardetzky TS. Paramyxovirus membrane fusion: lessons from the F and HN atomic structures. Virology. 2006;344(1):30–7. doi: 10.1016/j.virol.2005.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blechacz B, Splinter PL, Greiner S, Myers R, Peng KW, Federspiel MJ, et al. Engineered measles virus as a novel oncolytic viral therapy system for hepatocellular carcinoma. Hepatology (Baltimore, Md) 2006;44(6):1465–77. doi: 10.1002/hep.21437. [DOI] [PubMed] [Google Scholar]
- 33.Grote D, Russell SJ, Cornu TI, Cattaneo R, Vile R, Poland GA, et al. Live attenuated measles virus induces regression of human lymphoma xenografts in immunodeficient mice. Blood. 2001;97(12):3746–54. doi: 10.1182/blood.v97.12.3746. [DOI] [PubMed] [Google Scholar]
- 34.Hoffmann D, Bangen JM, Bayer W, Wildner O. Synergy between expression of fusogenic membrane proteins, chemotherapy and facultative virotherapy in colorectal cancer. Gene therapy. 2006;13(21):1534–44. doi: 10.1038/sj.gt.3302806. [DOI] [PubMed] [Google Scholar]
- 35.McDonald CJ, Erlichman C, Ingle JN, Rosales GA, Allen C, Greiner SM, et al. A measles virus vaccine strain derivative as a novel oncolytic agent against breast cancer. Breast cancer research and treatment. 2006;99(2):177–84. doi: 10.1007/s10549-006-9200-5. [DOI] [PubMed] [Google Scholar]
- 36.Msaouel P, Iankov ID, Allen C, Morris JC, von Messling V, Cattaneo R, et al. Engineered measles virus as a novel oncolytic therapy against prostate cancer. The Prostate. 2009;69(1):82–91. doi: 10.1002/pros.20857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peng KW, Ahmann GJ, Pham L, Greipp PR, Cattaneo R, Russell SJ. Systemic therapy of myeloma xenografts by an attenuated measles virus. Blood. 2001;98(7):2002–7. doi: 10.1182/blood.v98.7.2002. [DOI] [PubMed] [Google Scholar]
- 38.Anderson BD, Nakamura T, Russell SJ, Peng KW. High CD46 receptor density determines preferential killing of tumor cells by oncolytic measles virus. Cancer research. 2004;64(14):4919–26. doi: 10.1158/0008-5472.CAN-04-0884. [DOI] [PubMed] [Google Scholar]
- 39.Ong HT, Hasegawa K, Dietz AB, Russell SJ, Peng KW. Evaluation of T cells as carriers for systemic measles virotherapy in the presence of antiviral antibodies. Gene therapy. 2007;14(4):324–33. doi: 10.1038/sj.gt.3302880. [DOI] [PubMed] [Google Scholar]
- 40.Ong HT, Timm MM, Greipp PR, Witzig TE, Dispenzieri A, Russell SJ, et al. Oncolytic measles virus targets high CD46 expression on multiple myeloma cells. Experimental hematology. 2006;34(6):713–20. doi: 10.1016/j.exphem.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 41.Peng KW, Frenzke M, Myers R, Soeffker D, Harvey M, Greiner S, et al. Biodistribution of oncolytic measles virus after intraperitoneal administration into Ifnar-CD46Ge transgenic mice. Hum Gene Ther. 2003;14(16):1565–77. doi: 10.1089/104303403322495070. [DOI] [PubMed] [Google Scholar]
- 42.Myers RM, Greiner SM, Harvey ME, Griesmann G, Kuffel MJ, Buhrow SA, et al. Preclinical pharmacology and toxicology of intravenous MV-NIS, an oncolytic measles virus administered with or without cyclophosphamide. Clinical pharmacology and therapeutics. 2007;82(6):700–10. doi: 10.1038/sj.clpt.6100409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murphy JE, Tedbury PR, Homer-Vanniasinkam S, Walker JH, Ponnambalam S. Biochemistry and cell biology of mammalian scavenger receptors. Atherosclerosis. 2005;182(1):1–15. doi: 10.1016/j.atherosclerosis.2005.03.036. [DOI] [PubMed] [Google Scholar]
- 44.Peiser L, Gordon S. The function of scavenger receptors expressed by macrophages and their role in the regulation of inflammation. Microbes Infect. 2001;3(2):149–59. doi: 10.1016/s1286-4579(00)01362-9. [DOI] [PubMed] [Google Scholar]
- 45.Thelen T, Hao Y, Medeiros AI, Curtis JL, Serezani CH, Kobzik L, et al. The class A scavenger receptor, macrophage receptor with collagenous structure, is the major phagocytic receptor for Clostridium sordellii expressed by human decidual macrophages. J Immunol. 2010;185(7):4328–35. doi: 10.4049/jimmunol.1000989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Terpstra V, van Berkel TJ. Scavenger receptors on liver Kupffer cells mediate the in vivo uptake of oxidatively damaged red blood cells in mice. Blood. 2000;95(6):2157–63. [PubMed] [Google Scholar]
- 47.Pearson AM, Rich A, Krieger M. Polynucleotide binding to macrophage scavenger receptors depends on the formation of base-quartet-stabilized four-stranded helices. J Biol Chem. 1993;268(5):3546–54. [PubMed] [Google Scholar]
- 48.Finger AN, Bisoffi M, Wetterwald A, Gautschi E, Hohenfeld U, Klima I, et al. Scavenger receptor block as strategy for the identification of bone marrow homing phages by panning in vivo random peptide phage displayed libraries. J Immunol Methods. 2002;264(1–2):173–86. doi: 10.1016/s0022-1759(02)00089-3. [DOI] [PubMed] [Google Scholar]
- 49.Haisma HJ, Bellu AR. Pharmacological interventions for improving adenovirus usage in gene therapy. Molecular pharmaceutics. 2011;8(1):50–5. doi: 10.1021/mp100310h. [DOI] [PubMed] [Google Scholar]
- 50.Hadac EM, Peng KW, Nakamura T, Russell SJ. Reengineering paramyxovirus tropism. Virology. 2004;329(2):217–25. doi: 10.1016/j.virol.2004.08.036. [DOI] [PubMed] [Google Scholar]
- 51.Ong HT, Trejo TR, Pham LD, Oberg AL, Russell SJ, Peng KW. Intravascularly administered RGD-displaying measles viruses bind to and infect neovessel endothelial cells in vivo. Mol Ther. 2009;17(6):1012–21. doi: 10.1038/mt.2009.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mrkic B, Pavlovic J, Rulicke T, Volpe P, Buchholz CJ, Hourcade D, et al. Measles virus spread and pathogenesis in genetically modified mice. Journal of virology. 1998;72(9):7420–7. doi: 10.1128/jvi.72.9.7420-7427.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kemper C, Leung M, Stephensen CB, Pinkert CA, Liszewski MK, Cattaneo R, et al. Membrane cofactor protein (MCP; CD46) expression in transgenic mice. Clinical and experimental immunology. 2001;124(2):180–9. doi: 10.1046/j.1365-2249.2001.01458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang L, Dailey PJ, Gettie A, Blanchard J, Ho DD. The liver is a major organ for clearing simian immunodeficiency virus in rhesus monkeys. Journal of virology. 2002;76(10):5271–3. doi: 10.1128/JVI.76.10.5271-5273.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koski A, Rajecki M, Guse K, Kanerva A, Ristimaki A, Pesonen S, et al. Systemic adenoviral gene delivery to orthotopic murine breast tumors with ablation of coagulation factors, thrombocytes and Kupffer cells. The journal of gene medicine. 2009;11(11):966–77. doi: 10.1002/jgm.1373. [DOI] [PubMed] [Google Scholar]
- 56.Zare F, Magnusson M, Mollers LN, Jin T, Tarkowski A, Bokarewa M. Single-stranded polyinosinic acid oligonucleotides trigger leukocyte production of proteins belonging to fibrinolytic and coagulation cascades. J Leukoc Biol. 2008;84(3):741–7. doi: 10.1189/jlb.0506345. [DOI] [PubMed] [Google Scholar]
- 57.Nakamura T, Suzuki H, Wada Y, Kodama T, Doi T. Fucoidan induces nitric oxide production via p38 mitogen-activated protein kinase and NF-kappaB-dependent signaling pathways through macrophage scavenger receptors. Biochemical and biophysical research communications. 2006;343(1):286–94. doi: 10.1016/j.bbrc.2006.02.146. [DOI] [PubMed] [Google Scholar]
- 58.Van Rooijen N, Sanders A. Kupffer cell depletion by liposome-delivered drugs: comparative activity of intracellular clodronate, propamidine, and ethylenediaminetetraacetic acid. Hepatology (Baltimore, Md) 1996;23(5):1239–43. doi: 10.1053/jhep.1996.v23.pm0008621159. [DOI] [PubMed] [Google Scholar]
- 59.Okada H, Kalinski P, Ueda R, Hoji A, Kohanbash G, Donegan TE, et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with {alpha}-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J Clin Oncol. 2011;29(3):330–6. doi: 10.1200/JCO.2010.30.7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Irhimeh MR, Fitton JH, Lowenthal RM. Pilot clinical study to evaluate the anticoagulant activity of fucoidan. Blood Coagul Fibrinolysis. 2009;20(7):607–10. doi: 10.1097/MBC.0b013e32833135fe. [DOI] [PubMed] [Google Scholar]
- 61.Li B, Lu F, Wei X, Zhao R. Fucoidan: structure and bioactivity. Molecules (Basel, Switzerland) 2008;13(8):1671–95. doi: 10.3390/molecules13081671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fisher KD, Seymour LW. HPMA copolymers for masking and retargeting of therapeutic viruses. Advanced drug delivery reviews. 2010;62(2):240–5. doi: 10.1016/j.addr.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 63.Kreppel F, Kochanek S. Modification of adenovirus gene transfer vectors with synthetic polymers: a scientific review and technical guide. Mol Ther. 2008;16(1):16–29. doi: 10.1038/sj.mt.6300321. [DOI] [PubMed] [Google Scholar]
- 64.Power AT, Bell JC. Taming the Trojan horse: optimizing dynamic carrier cell/oncolytic virus systems for cancer biotherapy. Gene therapy. 2008;15(10):772–9. doi: 10.1038/gt.2008.40. [DOI] [PubMed] [Google Scholar]