

Abstract
The mechanism of the cell cycle regulatory peptidyl prolyl isomerase (PPIase), Pin1, was investigated using reduced-amide inhibitors designed to mimic the twisted-amide transition state. Inhibitors, R–pSer–Ψ[CH2N]–Pro–2-(indol-3-yl)-ethylamine, 1 (R = fluorenylmethoxycarbonyl, Fmoc), and 2 (R = Ac), of Pin1 were synthesized and bioassayed. Inhibitor 1 had an IC50 value of 6.3 μM, which is 4.5-fold better inhibition for Pin1 than our comparable ground state analogue, a cis-amide alkene isostere containing inhibitor. The change of Fmoc to Ac in 2 improved aqueous solubility for structural determination, and resulted in an IC50 value of 12 μM. The X-ray structure of the complex of 2 bound to Pin1 was determined to 1.76 Å resolution. The structure revealed that the reduced amide adopted a conformation similar to the proposed twisted-amide transition state of Pin1, with a trans-pyrrolidine conformation of the prolyl ring. A similar conformation of substrate would be destabilized relative to the planar amide conformation. Three additional reduced amides, with Thr replacing Ser, and l- or d-pipecolate (Pip) replacing Pro, were slightly weaker inhibitors of Pin1.
Pin1, a peptidyl-prolyl isomerase (PPIase) enzyme in the parvulin family, and the immunophilin PPIases, cyclophilin (CyP) and FK506 binding protein (FKBP), catalyze the cis-trans isomerization of Xaa-Pro amide bonds.(1-3) Pin1 has several demonstrated roles in regulating cell cycle progression.(4, 5) Pin1 regulates the cooperative transition of cells from G2 to M phase by interaction with a variety of cell cycle proteins, including Cdc25C phosphatase, which regulates the activity of cyclin-dependent kinase 1 (CDK1), the essential cell cycle regulatory kinase.(6-8) Pin1 specifically catalyzes the isomerization of phosphoSer/Thr–Pro amide bonds present in mitotic phosphoproteins.(6, 9) These activities make Pin1 a potential target for anti-cancer drugs.(10, 11)
PPIases accelerate prolyl cis-trans amide isomerization at a rate of ca. 106 s-1 faster than the thermal isomerization.(12) A twisted-amide mechanism has been proposed based on mechanistic studies of PPIases (Figure 1).(4, 12-18) It was observed that the ketone carbonyl of FKBP inhibitor FK506 was orthogonal to the amide plane in the bound conformation, so FK506 was proposed to act as a transition-state analogue of the twisted amide.(13) Secondary kinetic isotope effects, mutagenesis, spectroscopy, and theoretical calculations support the twisted amide mechanism for the CyP and FKBP PPIases.(12-18) Our goal is to understand the mechanism of Pin1 PPIase activity by designing specific inhibitors, and to use the best inhibitors to investigate cell cycle regulation by Pin1.
Figure 1.
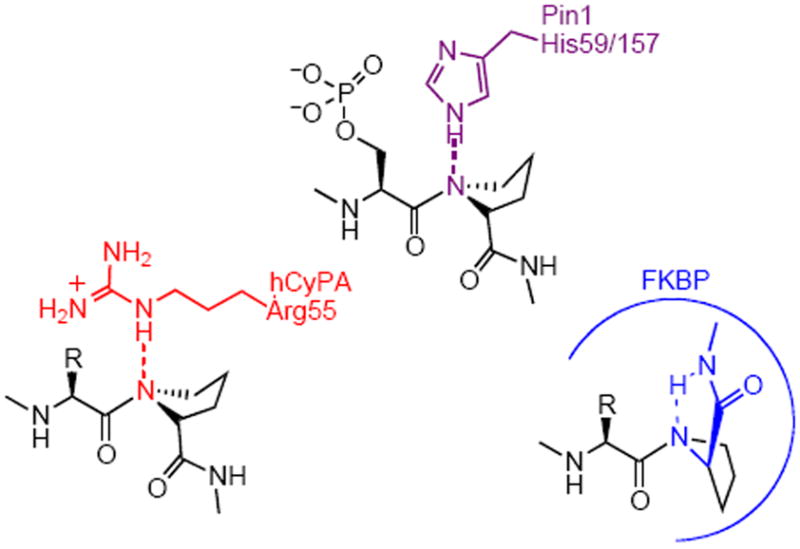
Proposed twisted-amide mechanisms for Pin1, hCyPA, and FKBP.(4, 13, 16-18)
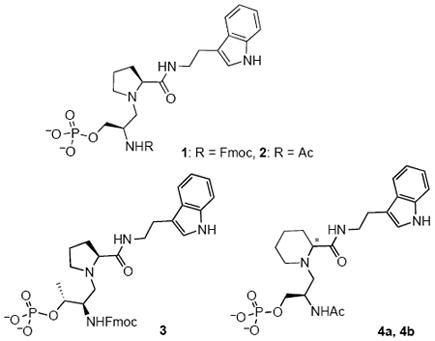
The reduced amide isostere concept was first reported in the development of potent inhibitors of human renin.(19) The broad use of reduced amide peptide isosteres in the design and synthesis of enzyme inhibitors has been reviewed.(20) To our knowledge, reduced amides have not yet been used as PPIase inhibitors. We now present pSer-Pro reduced amides 1, 2, 3, and 4 designed as transition-state analogue inhibitors of the PPIase activity of Pin1.
Experimental Procedures
Synthesis
Pin1 inhibition assays
Pin1 inhibition assays were performed as published.(21) The Km value for the cis substrate of 183 ± 9 μM was previously reported.(21) Inhibitors were pre-equilibrated in the 1.0 mL quartz cuvette at 4 °C for 10 min. For each inhibitor concentration, the assay was performed in duplicate. Inhibitor 1 was assayed at final concentrations of 3.4, 4.5, 6.8, 9.0, 14, 18, 23, 36, 68 μM, with 10 μL of stock in DMSO:H2O (4:3). Inhibitor 2 was assayed at final concentrations of 1.5, 4.7, 17.5, 35.0, 116 μM, with 10 μL of stock in 1:1 DMSO:H2O. Inhibitor 3 was assayed at final concentrations of 1.2, 9.7, 20.0, 39, 78, 160, 320, 470 μM, with 20 μL of stock in 4:3 DMSO:H2O. Inhibitor 4a was assayed at final concentrations of 3.5, 7.0, 14, 28, 42, 83 μM, with 10 μL of stock in 1:1 DMSO:H2O. Inhibitor 4b was assayed at final concentrations of 39, 78, 120, 250, 500, 1000 μM, with 10 μL of stock in 1:1 DMSO: H2O. The plot of % Inhibition vs. log [I] (μM) produced a sigmoidal curve by fitting all the experimental data to a dose response curve using TableCurve (version 3 for win32) (Figures S1 to S5 in Supporting Information). The IC50 values were derived from the curves at 50% inhibition of enzyme activity (Table 1).
Table 1.
Protease-coupled Pin1 assay results for inhibitors.
 | ||
|---|---|---|
| Pin1 Inhibitor | IC50 (μM) | |
| 1 | Fmoc–pSer–Ψ[CH2N]-Pro–tryptamine | 6.3 ± 0.4 |
| 2 | Ac–pSer–Ψ[CH2N]-Pro–tryptamine | 12 ± 2 |
| 3 | Fmoc–pThr–Ψ[CH2N]-Pro–tryptamine | 30 ± 2 |
| 4a | Ac–pSer–Ψ[CH2N]-(R/S)Pipa–tryptamine | 16 ± 2 |
| 4b | Ac–pSer–Ψ[CH2N]-(R/S)Pipa–tryptamine | 189 ± 17 |
Stereochemistry at Pip α-carbon was not determined.
Protein Purification, Crystallization, and Data Collection
Pin1 R14A mutant was expressed and purified as described previously.(9, 22) Crystals grew from 1.9–2.0 M ammonium sulfate, 50 mM HEPES, pH 7.5, and 0.5 % PEG-400 (v/v) within 4 d using vapor diffusion in 2 μL of sitting drops containing 10 mg/mL R14A Pin1. Inhibitor 2 was dissolved in the cryogenic buffer: 40% PEG-400 (v/v), 50 mM HEPES, pH 7.5, at a stock concentration of 30 μM. R14A Pin1 crystals were transferred into 2 μL of solutions of reduced amide 2 at 30, 15, and 7 μM in cryogenic buffer. Two ca. 200 μm R14A Pin1 crystals that were soaked in the 30 and 15 μM inhibitor 2 solutions for ca. 60 h, were transferred to 100–200 μm crystal freezing loops, flash frozen in liquid nitrogen, and shipped to Argonne National Lab. Data were collected at wavelength of 1.0 Å at 100 K on beam-line SER-CAT of the Advanced Photon Source (APS). Diffraction data were processed with HKL2000 and the statistics are summarized (Table 2).(23)
Table 2.
Pin1–2 complex X-ray data collection and refinement statistics.
| Crystal Data | Pin1–2 (A3) |
|---|---|
| Space Group | P3121 |
| Unit Cell | |
| a (Å) | 68.6 |
| b (Å) | 68.8 |
| c (Å) | 79.8 |
| α=β (°) | 90.0 |
| γ (°) | 120 |
|
| |
| Data collection | |
|
| |
| X-ray source | APSa |
| Resolution (Å) | 1.76 (48-1.76)b |
| Rsym (%) | 4.8 (47.2) |
| Completeness (%) | 93.9 (82.6) %b |
|
| |
| Refinement statistics | |
|
| |
| R-work (%) | 22.1 |
| R-free (%) | 25.7 |
| RMS Bond lengths (Å) | 0.018 |
| RMS Bond angles (°) | 1.751 |
| No. of ligand or cofactor molecules | 1 RZDc |
| 1 PE4c | |
| No. of water molecules | 92 |
| Average B overall (Å2) | 31.2 |
Advanced Photon Source, SER-CAT beamline, Argonne National Laboratories.
The values in parentheses are for the highest resolution shell.
RZD is compound 2. PE4 is PEG-400.
Structure Solution and Refinement
The crystal structure of Pin1 R14A mutant in complex with inhibitor 2 was determined by molecular replacement using the Pin1 R14A structure with high-affinity peptidomimetic inhibitor d-peptide as a search model (PDB ID: 2ITK) using the program AmoRe(24) available in the CCP4 software package (Table 2).(25) Molecular replacement solutions were refined with REFMAC, reserving 5% of the measured and reduced structure factor amplitudes as an unbiased test set for cross validation (Rfree).(26) The location of the inhibitor was clear in Fo-Fc maps even after the first round of refinement with only apo Pin1 R14A as model. The inhibitor model was built into the electron density using Coot.(27) SigmaA-weighted electron density maps (2Fo-Fc and Fo-Fc) were calculated after each cycle of refinement and carefully inspected to guide model rebuilding using Coot. The final models were evaluated by PROCHECK.(28)
Results
Design of Inhibitors
The tertiary amine of a reduced amide, lacking the amide carbonyl, would adopt a twisted-amide-like conformation, and could act as a stable hydrogen-bond acceptor, analogous to the transition state. The Fmoc group for 1 was chosen because it could be analogous to an aromatic residue on the N-terminus, and it was used as the N-terminal protecting group during synthesis. In compound 3, pThr was substituted for pSer. Substitution of the Pro in compound 2 with racemic (R/S)-Pip gave a pair of diastereomers, 4a and 4b.
Synthesis and Assay of Inhibitors
The reduced amide inhibitors were synthesized in 6 steps for Fmoc derivatives 1 and 3, and 9 steps for acetyl derivatives 2 and 4, with reduction of the amide to the tertiary amine with borane·THF in the key step (Supporting Information).(29, 30) The α-chymotrypsin protease-coupled assay with succinyl–Ala–Glu–cis-Pro–Phe–pNA as substrate was used to measure inhibition of Pin1,(21) and the IC50 values of 1, 2, 3, and 4a, and 4b for Pin1 were determined (Table 1).
X-ray Crystallography
The aqueous solubility of inhibitor 1 was very poor; it was difficult to incorporate the inhibitor into the crystals in soaking experiments. Inhibitor 2 was synthesized with an acetyl on the N-terminus to improve the solubility. In previous studies, we discovered that crystals of wild type Pin1, though they diffract readily, are very sensitive towards ligand soaking (Y. Zhang and J.P. Noel, unpublished results). The Pin1 R14A mutant, designed as an entropy reduction mutation, promotes crystal packing as space group P3121, rather than P4321 as in wild type protein.(31) R14A crystals show higher stability upon ligand incorporation.(31) The mutation of Arg14 to Ala does not interfere with the enzyme isomerization activity or ligand binding.(32)
The high-resolution crystal structure of Pin1 in complex with inhibitor 2 was determined and refined to 1.76 Å resolution (Table 2, Figure 2). Inhibitor 2 bound to the Pin1 PPIase catalytic domain (Figure 2). One or the other epimers at nitrogen of reduced amide 2 should have lower energy upon binding in the enzyme active site. Both the (SN)- and (RN)-Pro epimers of inhibitor 2 were modeled with the data, but only the (SN)-Pro model fit to the electon density map, demonstrating that the (SN)-Pro had the correct bound stereochemistry (Figure 2). The (RN)-epimer of 2 was flexibly superimposed on the (SN)-epimer structure at the tryptamine-N, tryptamine-Cα, Pro-Cα, pseudo-Ser-Cα, and phosphate-P atoms using Sybyl 8.1.1 (Figure 2).(33) The RMS deviation for all atoms was 0.63 Å. The phosphate was bound by Lys63, Arg68, and Arg69 (Figure 3), and the prolyl ring was bound in a pocket by His59, Leu122, Met130, and Phe124 (Figure 3). Distances from Pin1 potential side-chain hydrogen-bond donors to the prolyl nitrogen were measured (Figure 4).
Figure 2.
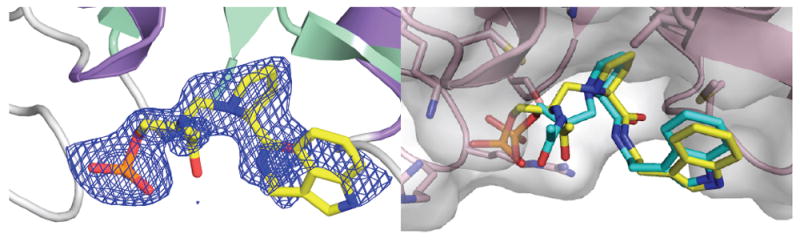
X-ray crystal structure of reduced amide 2 (yellow) in complex with Pin1 at 1.76 Å. Left: The structure of reduced amide (SN)-2 (yellow) bound to Pin1 is shown as SIGMAA-weighted 2Fo - Fc electron-density map (blue) contoured at 1σ. Right: Prolyl nitrogen (RN)-epimer of 2 (turquoise) flexibly superimposed on the (SN)-epimer structure (yellow). Only the (SN)-epimer fit the electron density. (Created with MacPyMOL 2006.(41))
Figure 3.
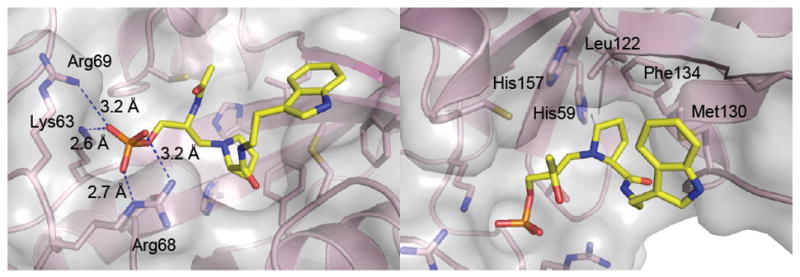
Anchor points for inhibitor 2 in the Pin1 active site. Left: Phosphate binding pocket. Salt bridge distances from Lys63, Arg68, and Arg69 to the phosphate oxygens of 2 are shown. Right: The proline-ring binding pocket is composed of hydrophobic residues: His157, His59, Leu122, Phe134, and Met130. Met130 also has a hydrophobic interaction with the tryptamine indole ring. (Created with MacPyMOL 2006.(41))
Figure 4.
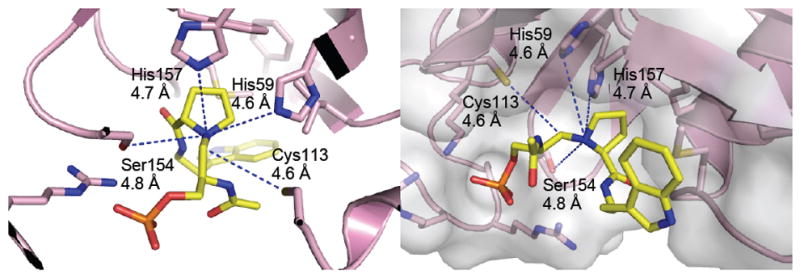
Two views of the Pin1–2 complex showing the distances from the active site Pin1 residues: His59, His157, and Ser154 to the prolyl nitrogen. None of the residues are within hydrogen-bonding distance of the prolyl nitrogen. The distance from Cys113 to the inhibitor CH2 group that substitutes for the substrate carbonyl is also shown.(Created with MacPyMOL 2006.(41))
Discussion
Design of Reduced Amide Inhibitors of Pin1
The concept of a transition-state analogue is very effective as a basis for designing potent enzyme inhibitors.(34) In related work, ketone inhibitors designed as electrophilic acceptors of the Pin1 active-site nucleophile Cys113 were ineffective inhibitors of Pin1, suggesting that Pin1 does not operate through a nucleophilic addition mechanism.(35) In the proposed twisted amide mechanism for PPIases, the pyrrolidine nitrogen is no longer in conjugation with the amide carbonyl; it is sp3 hybridized (pyramidal), and the lone pair is orthogonal to the carbonyl π-bond (Figure 1). In the transition state, the sp3 nitrogen could act as a transient hydrogen bond acceptor from donors in the active site of Pin1.(18)
Aromatic residues are preferred on both the C- and N-termini of Pin1 substrates, so initially Fmoc was chosen as the N-terminal group of 1, and the aromatic tryptamine at the C-terminus of the pSer–Pro core.(6, 22) Cis and trans alkene ground-state analogues, Ac–Phe–Phe–pSer–Ψ[CH=C]-Pro–Arg–NH2, were found to be good inhibitors of Pin1.(21, 36) The crystal structures of these two inhibitors, as well as d/l-pThr-Pip peptide inhibitors, in complex with Pin1 showed that the electron density for the N-terminal side chains was missing, indicating that the N-terminal residues were disordered and possibly contribute little to the binding affinity.(22) The reduced amide 2, with Ac instead of Fmoc at the N-terminus, was designed as a more water soluble analogue of 1. Three additional structurally modified reduced amides were designed as Pin1 inhibitors. Compound 3 with pThr instead of pSer was included since pThr–Pro is also a specified substrate and inhibitor motif of Pin1.(6, 22, 37) Inhibitory peptide-Pin1 complex structures suggested there is room for the bulkier Pip ring in the Pro binding pocket,(22) and Pip-containing peptides were 100-fold more potent inhibitors than their Pro analogues.(37) So we substituted Pro with Pip in diastereomers 4a and 4b.
Mechanistic Implications of the Pin1-2 Structure
To better understand the catalytic and inhibitory mechanisms of Pin1, the structure of the Pin1–inhibitor 2 complex was solved (Table 1). The bound inhibitor had the prolyl ring orthogonal to the methylene that replaced the carbonyl, a conformation that is expected of a twisted-amide transition state (Figure 2). The flexibility of the reduced amide backbone allowed it to mimic the ω angle of one possible transition state. There were two anchor points for binding inhibitor 2 (Figure 3). The phosphate moiety was bound in the basic site formed by the residues of Lys63 and Arg69 of Pin1, similar to peptidic inhibitors,(22) but with strong involvement of Arg68 as in the original Pin1 structure with SO42– bound (Figure 3).(9) Strong binding of the phosphate group may facilitate binding of the transition-state conformation. Similarly, the prolyl ring was cradled in a hydrophobic pocket formed by the side chains of His59, Leu122, Met130, and Phe134 (Figure 3). These two anchor points permit binding of either cis or trans pSer-Pro substrates, while allowing the peptide backbone significant flexibility for catalysis.(9, 22, 38) The tight-binding anchor points could serve to destabilize the substrate by stretching it into a trans-pyrrolidine conformation.
Notably, (SN)-2 adopted a trans-pyrrolidine conformation in the catalytic site, similar to our models of related ketone substrate-analogue inhibitors bound to Pin1.(35) The distance between the reduced amide CH2 carbon and the prolyl carbonyl carbon was 3.7 Å (Figure 4). On the other hand, (RN)-2 represents a cis-pyrrolidine conformation that is nearly eclipsed; (RN)-2 does not fit the electron density map, with a corresponding distance of only 2.6 Å (Figure 2). The bound structure of (SN)-2 thus supports the destabilization of substrates by Pin1 indicated by the trans-prolyl conformation of the bound ketone inhibitors.(35) Destabilization of substrates has been proposed as a common mechanism for single-substrate enzymes, of which the PPIases are examples.(17)
Bruice has proposed that single-substrate enzymes bind their substrates close to the transition state conformation in a “near-attack complex” (NAC), destabilizing the bound substrate.(17) Both substrate destabilization, and transition state stabilization thus contribute to lowering the ΔG‡ barrier for the enzymatic reaction. In such cases, it may be difficult to attain the binding energies expected of transition-state analogues with small molecule inhibitors. As Bruice said, “TS in E·TS may or may not be bound tighter than NAC in E·NAC.”(17) Inhibitors that mimic substrate in a conformation similar to the NAC could be just as viable as TS analogues, yet neither substrate nor transition-state small molecule analogues would produce very tight-binding affinities. This may be one reason that reduced amide 1 was only a 4.5-fold better inhibitor of Pin1 than the similarly substituted ground-state analogue, Fmoc-pSer–Ψ[(Z)CH=C]Pro–2-(indol-3-yl)-ethylamine (IC50 = 28.3 μM).(39)
Several other factors may contribute to the unexpectedly low inhibition of Pin1 by the reduced amide inhibitors: 1) an entropic penalty upon binding due to the flexibility of the reduced-amide backbone, and 2) fewer flanking residues than more potent peptidic inhibitors of Fischer and coworkers.(37) In addition, the groups flanking the pSer–[CH2N]–Pro of 1 have not yet been optimized for inhibition of Pin1. Our inhibitors do not include the N-terminal or C-terminal extensions, nor the d-pThr that gave the tightest binding inhibitors in Fischer’s peptides.(37) We sought to mimic the native substrate as closely as possible, while probing the transition-state conformation. Although the inhibition of Pin1 by 1 was not as potent as expected of a transition-state analogue,(34) the crystal structure of 2 reveals details of binding that give us the best glimpse yet of what a twisted-amide Pin1 transition state might look like.
Significantly for the proposed mechanism of Pin1, hydrogen bonding was not detected between the prolyl nitrogen and active-site residues, including His59, as previously proposed (Figure 4).(18) His157 was ideally positioned for hydrogen bonding, orthogonal to the ring on the backside of the prolyl nitrogen, yet the distance between the prolyl nitrogen and any active-site hydrogen-bond donors are beyond hydrogen-bonding range (Figure 4). The amine of 2 is pyramidalized with the lone pair away from the active site. This allows the prolyl ring to adopt the observed trans conformation; a hydrogen bond to His157 would necessitate a cis-pyrrolidine conformation. The latter appears to be disfavored by strong binding of the phosphate and prolyl ring groups. Since the pKa of a tertiary amine is about 11, these inhibitors are likely to be protonated; they may not be acidic enough to give up a proton and accept a hydrogen bond from any of the nearby enzymatic residues. It remains an open question whether the transition state of the enzymatic reaction has a transient hydrogen bond involved in the twisted-amide mechanism, as expected from studies of cyclophilin and FKBP.(15-17)
The Cys113 sulfur was 4.6 Å from the reduced amide methylene carbon (Figure 4). We have previously shown that highly electrophilic ketoamides, and ketone substrate analogues, which have carbonyls ideally placed for nucleophilic addition from the Pin1 Cys113 residue, were quite poor Pin1 inhibitors, suggesting that this mechanism is unlikely.(35)
Structure-Activity Relationships (SAR)
Compound 1 was found to be the most potent inhibitor among these reduced amides. The IC50 value of 2 for Pin1 was 12 μM, only 2-fold less potent than inhibitor 1 (Table 1). Although replacement of the Fmoc at the N-terminus in compound 1 with an Ac to give compound 2 improved the water solubility, this modification reduced the inhibitory activity by 2-fold. This is likely due to non-specific hydrophobic interactions of Fmoc with Pin1.
Compounds 3, 4a, and 4b, based on potent nanomolar peptidomimetic inhibitors, were made to further investigate SAR.(22, 37) Compound 1 showed 5-fold better inhibition than compound 3, with pThr substituted for pSer (Table 1). So, at least for reduced amides, Pin1 prefers pSer–Pro to pThr–Pro in the catalytic site. Compound 4a, with Pip substituting for Pro, had a comparable IC50 value to 2, indicating that the 6-membered Pip ring was not necessarily a better fit than its 5-membered Pro counterpart in the case of reduced amides (Table 1). Therefore, we decided not to expend more effort to determine the stereochemistry of 4a and 4b at the Pip α-carbon. Yet, since compound 4a was approximately 12-fold more potent than its diastereomer 4b, the configuration of the α-carbon on the Pip ring is important. The stereochemistry at the α-carbon of Pip was probably (S), analogous to l-Pro, since 2 and 4a had comparable IC50 values (Table 1). Similar stereochemical results were obtained for ketone substrate analogue inhibitors.(35)
The reduced amide, Fmoc–pSer–Ψ[CH2N]-Pro–2-(indol-3-yl)-ethylamine, 1 was among the most potent small molecule Pin1 inhibitors we have synthesized to date.(21) Reduced amide 2 was 6-fold better than the similarly substituted α-ketoamide inhibitor, Ac–pSer–Ψ[COCON]-Pro–2-(naphth-2-yl)-ethylamine (IC50 = 100 μM),(40) and 5-fold better than the similarly substituted ketone, Ac–pSer–Ψ[COCH]-Pip–2-(indol-3-yl)-ethylamine (IC50 = 61 μM).(35) The most potent Pin1 inhibitors known to date are the d-Thr–Pip containing peptide analogues of Wildemann et al.(37) The octapeptide, Ac–Lys(N-biotinoyl)–Ala–Ala–Bth–d-Thr(PO3H2)–Pip–Nal–Gln–NH2 had a Ki value of 0.21 nM.(37)
Conclusions
The X-ray crystal structure of reduced amide inhibitor 2 bound in the active site of Pin1 reveals a conformation similar to the proposed twisted-amide transition state of Pin1.(18) The conformation of 2 bound to Pin1 has a trans-pyrrolidine ring, supporting the proposal that Pin1 destabilizes substrates by stretching the prolyl ring conformation to initiate catalysis.(35) No hydrogen bond was observed between any active-site residue and the tertiary amine nitrogen of the prolyl ring.(18) Inhibitors 1 and 2 represent a new application of reduced-amide peptide isosteres as Pin1 PPIase inhibitors with improved potency over similarly substituted alkene isosteres,(39) α-ketoamides,(40) or ketone substrate analogues.(35) These are among the best small molecule inhibitors we have synthesized to date, indicating that reduced-amide structures may be optimized to give potent inhibitors of Pin1. The synthesis of these isosteres was facile, so the reduced-amide motif will be exploited as a core design element for future Pin1 inhibitors.
Supplementary Material
Acknowledgments
We thank J. P. Noel (Chemical Biology, The Salk Institute) for training, use of facilities, and eminently helpful discussions, N. Vogelaar (Virginia Tech), J. Chrzas and A. Fu (SER-CAT beamline, Advanced Photon Source, Argonne National Lab) for technical assistance with data collection, and F. Schubot and Q. Han (Department of Biochemistry, Virginia Tech) for advice on data processing.
We thank the NIH for financial support from Grant No. R01 CA110940 (FAE), S10 RR16658 (FAE) for the LC-MSMS, and the Welch Foundation for Grant F-1778 (YZ).
Abbreviations used
- PPIase
peptidyl-prolyl isomerase
- CyP
cyclophilin
- FKBP
FK506 binding protein
- CDK1
cyclin-dependent kinase 1
- Fmoc
9-fluorenylmethoxycarbonyl
- Ac
acetyl
- Pip
pipecolate
- DMSO
dimethylsulfoxide
- HEPES
4-(2-hydroxyethyl)-1-piperazine-ethanesulfonic acid
- PEG
polyethylene glycol
- pNA
p-nitroanilide
- RMS
root-mean-square
- SAR
structure-activity relationships
- IC50
inhibitory concentration at 50%
- Nal
naphthylalanine
Footnotes
Accession Codes: Coordinates and structure factors have been deposited in the Protein Data Bank. PDB accession code: Pin1–inhibitor 2 complex, 3NTP.
Supporting Information Available. Schemes, experimental procedures for the synthesis and spectroscopic data for compounds 1–28, HPLC for 1–4, and 7–15, and inhibition data for 1, 2, 3, 4a, and 4b. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lu KP, Hanes SD, Hunter T. A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature. 1996;380:544–547. doi: 10.1038/380544a0. [DOI] [PubMed] [Google Scholar]
- 2.Fischer G, Wittamann-Liebold B, Lang K, Kiefhaber T, Schmid FX. Cyclophilin and peptidyl-prolyl cis-trans isomerase are probably identical proteins. Nature. 1989;337:476–478. doi: 10.1038/337476a0. [DOI] [PubMed] [Google Scholar]
- 3.Harding MW, Galat A, Uehling DE, Schreiber SL. A receptor for the immuno-suppressant FK506 is a cis-trans peptidyl-prolyl isomerase. Nature. 1989;341:758–760. doi: 10.1038/341758a0. [DOI] [PubMed] [Google Scholar]
- 4.Wiederrecht G, Etzkorn FA. Immunophilins. In: Sigal N, Wyvratt M, editors. Perspectives in Drug Discovery and Design. ESCOM Science Publishers, B.V; 1994. pp. 57–84. [Google Scholar]
- 5.Lu KP. Pinning down cell signaling, cancer and Alzheimer’s disease. Trends Biochem Sci. 2004;29:200–209. doi: 10.1016/j.tibs.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 6.Yaffe MB, Schutkowski M, Shen M, Zhou XZ, Stukenberg PT, Rahfeld JU, Xu J, Kuang J, Kirschner MW, Fischer G, Cantley LC, Lu KP. Sequence-specific and phosphorylation-dependent proline isomerization: a potential mitotic regulatory mechanism. Science. 1997;278:1957–1960. doi: 10.1126/science.278.5345.1957. [DOI] [PubMed] [Google Scholar]
- 7.Stukenberg PT, Kirschner MW. Pin1 acts catalytically to promote a conformational change in Cdc25. Mol Cell. 2001;7:1071–1083. doi: 10.1016/s1097-2765(01)00245-3. [DOI] [PubMed] [Google Scholar]
- 8.Satyanarayana A, Kaldis P. Mammalian cell-cycle regulation: several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene. 2009;28:2925–2939. doi: 10.1038/onc.2009.170. [DOI] [PubMed] [Google Scholar]
- 9.Ranganathan R, Lu KP, Hunter T, Noel JP. Structural and functional analysis of the mitotic rotamase Pin1 suggests substrate recognition is phosphorylation dependent. Cell. 1997;89:875–886. doi: 10.1016/s0092-8674(00)80273-1. [DOI] [PubMed] [Google Scholar]
- 10.Joseph JD, Yeh ES, Swenson KI, Means AR, Winkler The peptidyl-prolyl isomerase Pin1. Prog Cell Cycle Res. 2003;5:477–487. [PubMed] [Google Scholar]
- 11.Xu GG, Etzkorn FA. Pin1 as an anticancer drug target. Drug News Perspect. 2009;22:399–407. doi: 10.1358/dnp.2009.22.7.1381751. [DOI] [PubMed] [Google Scholar]
- 12.Fischer G. Peptidyl-Prolyl cis/trans Isomerases and Their Effectors. Angew Chem Int Ed Engl. 1994;33:1415–1436. [Google Scholar]
- 13.Rosen MK, Standaert Robert F, Galat A, Nakatsuka M, Schreiber SL. Inhibition of FKBP Rotamase Activity by Immunosuppressant FK506: Twisted Amide Surrogate. Science. 1990;248:863–866. doi: 10.1126/science.1693013. [DOI] [PubMed] [Google Scholar]
- 14.Harrison RK, Caldwell CG, Rosegay A, Melillo D, Stein RL. Confirmation of the Secondary Deuterium Isotope Effect for the Peptidyl Prolyl Cis-Trans Isomerase Activity of Cyclophilin by a Competitive, Double-Label Technique. J Am Chem Soc. 1990;112:7063–7064. [Google Scholar]
- 15.Zydowsky LD, Etzkorn FA, Chang H, Ferguson SB, Stolz LA, Ho SI, Walsh CT. Active Site Mutants of Human Cyclophilin A Separate Peptidyl-Prolyl Isomerase Activity from Cyclosporin A Binding and Calcineurin Inhibition. Protein Sci. 1992;1:1092–1099. doi: 10.1002/pro.5560010903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischer S, Michnick S, Karplus M. A Mechanism for Rotamase Catalysis by the FK506 Binding Protein (FKBP) Biochemistry. 1993;32:13830–13837. doi: 10.1021/bi00213a011. [DOI] [PubMed] [Google Scholar]
- 17.Hur S, Bruice TC. The Mechanism of Cis-Trans Isomerization of Prolyl Peptides by Cyclophilin. J Am Chem Soc. 2002;124:7303–7313. doi: 10.1021/ja020222s. [DOI] [PubMed] [Google Scholar]
- 18.Schroeder OE, Carper E, Wind JJ, Poutsma JL, Etzkorn FA, Poutsma JC. Theoretical and experimental investigation of the energetics of cis-trans proline isomerization in peptide models. J Phys Chem A. 2006;110:6522–6530. doi: 10.1021/jp060642u. [DOI] [PubMed] [Google Scholar]
- 19.Szelke M, Leckie B, Hallett A, Jones DM, Sueiras J, Atrash B, Lever AF. Potent new inhibitors of human renin. Nature. 1982;299:555–557. doi: 10.1038/299555a0. [DOI] [PubMed] [Google Scholar]
- 20.Zivec M, Jakopin Z, Gobec S. Recent advances in the synthesis and applications of reduced amide pseudopeptides. Curr Med Chem. 2009;16:2289–2304. doi: 10.2174/092986709788453113. [DOI] [PubMed] [Google Scholar]
- 21.Wang XJ, Xu B, Mullins AB, Neiler FK, Etzkorn FA. Conformationally locked isostere of phosphoSer-cis-Pro inhibits Pin1 23-fold better than phosphoSer-trans-Pro isostere. J Am Chem Soc. 2004;126:15533–15542. doi: 10.1021/ja046396m. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y, Daum S, Wildemann D, Zhou XZ, Verdecia MA, Bowman ME, Lucke C, Hunter T, Lu KP, Fischer G, Noel JP. Structural basis for high-affinity peptide inhibition of human Pin1. ACS Chem Biol. 2007;2:320–328. doi: 10.1021/cb7000044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 24.Navaza J. AMoRe: an automated package for molecular replacement. Acta Crystallogr, Sect A: Found Crystallogr. 1994;A50:157–163. [Google Scholar]
- 25.Anonymous. The CCP4 suite: programs for protein crystallography. Acta Crystallogr, Sect D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 26.Brunger AT. Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 27.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr, Sect D. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 28.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 29.Roeske RW, Weitl FL, Prasad KU, Thompson RM. Selective Reduction of the Amide Carbonyl Group in Dipeptides by Borane. J Org Chem. 1976;41:1260–1261. doi: 10.1021/jo00869a041. [DOI] [PubMed] [Google Scholar]
- 30.Cushman M, Oh Y-I. Development of Methodology for the Synthesis of Stereochemically Pure Phe[CH2N]Pro Linkages in HIV Protease Inhibitors. J Org Chem. 1991;56:4161–4167. [Google Scholar]
- 31.Verdecia MA, Bowman ME, Lu KP, Hunter T, Noel JP. Structural basis for phosphoserine-proline recognition by group IV WW domains. Nat Struct Biol. 2000;7:639–643. doi: 10.1038/77929. [DOI] [PubMed] [Google Scholar]
- 32.Lu PJ, Zhou XZ, Shen M, Lu KP. Function of WW domains as phosphoserine- or phosphothreonine-binding modules. Science. 1999;283:1325–1328. doi: 10.1126/science.283.5406.1325. [DOI] [PubMed] [Google Scholar]
- 33.SYBYL. Tripos International; 1699 South Hanley Rd., St. Louis, MO, 63144, USA: 2008. [Google Scholar]
- 34.Schramm VL. Enzymatic transition states and transition state analogues. Curr Opin Struct Biol. 2005;15:604–613. doi: 10.1016/j.sbi.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 35.Xu G. Pin1 Inhibitors: Towards Understanding the Enzymatic Mechanism. Virginia Polytechnic Institute and State University; Blacksburg, VA: 2010. [Google Scholar]
- 36.Etzkorn FA, Noel JP, Zhang Y, Wang XJ. Pin1: Inhibitors and Mechanism. In: Blondelle SE, editor. Understanding Biology Using Peptides, Proc 19th Am Pept Symp. Springer, Inc.; San Diego: 2006. pp. 759–762. [Google Scholar]
- 37.Wildemann D, Erdmann F, Alvarez BH, Stoller G, Zhou XZ, Fanghanel J, Schutkowski M, Lu KP, Fischer G. Nanomolar Inhibitors of the Peptidyl Prolyl Cis/Trans Isomerase Pin1 from Combinatorial Peptide Libraries. J Med Chem. 2006;49:2147–2150. doi: 10.1021/jm060036n. [DOI] [PubMed] [Google Scholar]
- 38.Namanja AT, Wang XJ, Xu B, Mercedes-Camacho AY, Wilson BD, Wilson KA, Etzkorn FA, Peng JW. Toward Flexibility-Activity Relationships by NMR Spectroscopy: Dynamics of Pin1 Ligands. J Am Chem Soc. 2010;132:5607–5609. doi: 10.1021/ja9096779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao S, Etzkorn FA. A phosphorylated prodrug for the inhibition of Pin1. Bioorg Med Chem Lett. 2007;17:6615–6618. doi: 10.1016/j.bmcl.2007.09.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu GG, Etzkorn FA. Convergent Synthesis of α-Ketoamide Inhibitors of Pin1. Org Lett. 2010;12:696–699. doi: 10.1021/ol9027013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA, USA: 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.