

Summary
Purpose
The mammalian target of rapamycin (mTOR) pathway has been implicated in contributing to progressive epileptogenesis in models of chronic epilepsy. Conversely, seizures themselves may directly cause acute activation of the mTOR pathway. To isolate the direct effects of seizures on the mTOR pathway, the time course and mechanisms of mTOR activation were investigated with acute seizures induced by pentylenetetrazole (PTZ), which does not lead to chronic epilepsy.
Methods
Western blot analysis was used to assay phosphorylation of Akt and S6, as measures of activation of the phosphoinositide 3-kinase (PI3K)/Akt and mTOR pathways, respectively, at various time points after PTZ seizures in rats. The ability of wortmannin, a PI3K inhibitor, to inhibit PTZ seizure-induced activation of the mTOR pathway was tested.
Key Findings
PTZ seizures produced an immediate, transient mTOR activation lasting several hours, but no later, more chronic activation over days to weeks. This acute stimulation of the mTOR pathway by PTZ seizures was mediated by upstream PI3K/Akt pathway activation and was blocked by a PI3K inhibitor.
Significance
Compared with models of chronic epilepsy which exhibit biphasic (acute and chronic) mTOR pathway activation, PTZ seizures only produce acute, but not chronic, mTOR activation. These results in the PTZ seizure model highlight potential differences in the involvement of the mTOR pathway between self-limited seizures and progressive epileptogenesis. These findings also suggest a potential therapeutic role of PI3K inhibitors in epilepsy.
Keywords: epilepsy, seizure, pentylenetetrazole, kainate, rapamycin, rat
Introduction
The mammalian target of rapamycin (mTOR) signaling pathway has been implicated in promoting mechanisms of epileptogenesis in various animal models of epilepsy. In mouse models of the genetic epilepsy, tuberous sclerosis complex (TSC), Tsc1 or Tsc2 gene inactivation causes dysregulated mTOR activity and epilepsy. mTOR inhibitors prevent the development of seizures and associated cellular and molecular abnormalities that promote epileptogenesis in these models, such as glial proliferation, neuronal hypertrophy, and deficient glutamate transporters (Meikle et al., 2008; Zeng et al., 2008; Zeng et al., 2011). Similarly, in the kainate and pilocarpine models of acquired temporal lobe epilepsy, an initial episode of status epilepticus induces abnormal mTOR activation, and mTOR inhibitors reduce mossy fiber sprouting (Buckmaster et al., 2009) and chronic epilepsy (Zeng et al., 2009; Huang et al., 2010). Thus, there has been intense interest in the general role of the mTOR pathway in epileptogenesis and the potential utility of mTOR inhibitors as treatments for multiple types of epilepsy (Wong, 2010).
Determining the pathophysiological significance of the mTOR pathway in epilepsy is complicated by the fact that seizures themselves appear to activate mTOR, thus potentially triggering a circular, progressive process. In the kainate and pilocarpine models, prolonged seizures (status epilepticus) induced acutely by these convulsant drugs serve as the initial stimulus for pathological processes (e.g. mossy fiber sprouting, neuronal death) that may promote chronic, spontaneous epilepsy days to weeks later. In the kainate model, a biphasic activation of the mTOR pathway occurs (Zeng et al., 2009). First, an initial acute phase of mTOR activation lasts a few hours and appears to be directly driven by the status epilepticus. Then, a second, more chronic phase of mTOR activation evolves over days to weeks and correlates with the process of epileptogenesis and eventual development of spontaneous seizures. In contrast to the kainate model, penetylenetetrazole (PTZ) is an antagonist of gamma-aminobutyric acid receptors that induces acute seizures in rodents, but, without repetitive applications of PTZ (kindling), does not appear to lead to significant pathological changes or spontaneous epilepsy (Holmes et al., 1984; Wong et al., 2003). Thus, the PTZ model offers an opportunity to isolate the direct effects of seizures on the mTOR pathway, in the absence of chronic epileptogenesis.
Materials and Methods
Animals and drug injection
Care and use of animals were conducted according to an animal protocol approved by the Washington University Animal Studies Committee. Six-week old male Sprague Dawley rats were injected with a single dose of PTZ (Sigma, St. Louis, MO, 75 mg/kg, i.p.) to induce generalized clonic or tonic seizures. The latency and duration of seizure activity were observed behaviorally. Rats were sacrificed at various times intervals (ranging from 5 min to 3 wk) after seizure onset to harvest brains for western blotting. In some experiments, the PI3 kinase inhibitor, wortmannin (Sigma, 0.6-2.4 mg/kg, i.p.), was administered 30 minutes before PTZ injection and brains were collected 3 hours after seizures induced by PTZ. Saline-injected rats served as controls.
Western blotting
Western blotting was performed using standard methods as described previously (Zeng et al., 2008). Briefly, neocortex and hippocampus were dissected and homogenized separately. Equal amounts of total protein extract were separated by gel electrophoresis and transferred to nitrocellulose membranes. Primary antibodies to P-S6(Ser240/244), P-S6(Ser235/236), S6, P-Akt(Ser473), P-Akt(Thr308), or Akt (1:1,000, Cell Signaling Technology, Danvers, MA) were used. The membranes were then reacted with a peroxidase-conjugated secondary antibody. Signals were detected by enzyme chemiluminescence (Pierce, Rockford, IL) and quantitatively analyzed with ImageJ software. The ratios of P-S6 to total S6 and P-Akt to total Akt were used as measures of activation of the mTOR and phosphoinositide 3-kinase (PI3K)/Akt pathways, respectively. Normalization to total S6 and total Akt also served as a control for loading. Data are reported as mean ± SEM. Differences in protein expression were compared by one-way ANOVA, with p<0.05 considered significant.
Results
A single injection of PTZ (75 mg/kg) induced acute clonic or tonic seizure activity within a couple minutes (latency=101.2±25.2s; duration=56.4±2.8s) in all rats. In addition, most rats exhibited brief episodes of isolated myoclonic jerks or freezing, but these generally resolved within five minutes after the clonic or tonic seizure. PTZ injection caused a corresponding activation of the mTOR pathway in both hippocampus and neocortex of rats, as reflected by an increase in the P-S6 to total S6 ratio (Fig. 1). This activation of S6 protein involved phosphorylation of both the Ser240/244 (Fig. 1A,B) and Ser235/236 (Fig. 1C,D) regulatory sites. In initial time course experiments, mTOR activation was increased by 1 hour, remained elevated at 3-6 h and returned to baseline by 16 h after PTZ seizure onset (Fig. 1), demonstrating that the duration of mTOR activation greatly outlasted the period of clinical seizure activity. Additional, more acute studies also showed evidence of an initial small, but significant, increase in mTOR activation as soon as 5 minutes after seizure onset (Fig. 2A,B), which subsequently peaks by 1 hour (Fig. 1). In contrast to the kainate model (Zeng et al. 2009), there was no secondary chronic increase in mTOR activation observed over days to weeks following PTZ-induced seizures in both hippocampus and neocortex (Fig. 2C,D).
Figure 1.

PTZ-induced seizures cause acute activation of the mTOR pathway. A single injection of PTZ (75 mg/kg) in rats induced acute convulsive seizure activity within a few minutes and caused a corresponding activation of the mTOR pathway, as reflected by an increase in the P-S6 to total S6 ratio at both the Ser240/244 (A, B) and Ser235/236 (C, D) phosophorylation sites. mTOR activation increased by 1 h, remained elevated at 3-6 h and returned to baseline by 16 h after seizure onset in both hippocampus (A, C) and neocortex (B, D) of rats. (*p<0.05, **p<0.01, compared to saline (SA); n = 4-6 per group).
Figure 2.

PTZ-induced seizures cause hyperacute, but not chronic, activation of the mTOR pathway. (A, B) In a separate set of studies from Fig. 1, a single injection of PTZ (75 mg/kg) in rats induced acute seizure activity followed by a small but significant increase in mTOR activation within five minutes, as reflected by an increase in the P-S6 to total S6 ratio at Ser240/244. (C,D) Beyond the initial phase of mTOR activation, PTZ-induced seizures had no longer-term effects on mTOR activation over days to weeks. (*p<0.05, compared to saline (SA); n = 4-6 per group).
The PI3K/Akt pathway is a potential upstream activator of the mTOR pathway. As reflected by the P-Akt (at Thr308, as well as at Ser473) to total Akt ratio, PTZ-induced seizures also triggered PI3K-Akt pathway activation with a time course over several hours parallel to mTOR pathway activation (Fig. 3). Furthermore, pretreatment with the PI3K inhibitor, wortmannin, thirty minutes prior to PTZ injection had no effect on the properties of PTZ seizures themselves (latency=101.2±25.2s for PTZ versus 106.4±21.4s for wortmannin + PTZ; duration=56.4±2.8 s for PTZ versus 53.8±4.3s for wortmannin + PTZ; p>0.05), but wortmannin blocked the PTZ-induced PI3K/Akt (Fig. 4A) and mTOR pathway (Fig. 4B) activation in a dose-dependent fashion, indicating that the PI3K-Akt pathway is an upstream activator of the mTOR pathway with PTZ seizures.
Figure 3.

PTZ-induced seizures cause activation of the PI3K-Akt pathway. A single injection of PTZ (75 mg/kg) in rats induced acute seizure activity and corresponding activation of the PI3K-Akt pathway, as reflected by an increase in the P-Akt to total Akt ratio. Parallel to P-S6 expression (Fig. 1), P-Akt expression increased in the first few hours after the onset of PTZ-induced seizure, and returned to baseline within 16 hours after the seizures in neocortex of rats. P-Akt at both Thr308 (upstream from mTOR) (A,B) and Ser473 (downstream from mTOR) (C,D) were increased by PTZ-induced seizures. (*p< 0.05 compared to saline (SA); n = 4-6 per group).
Figure 4.
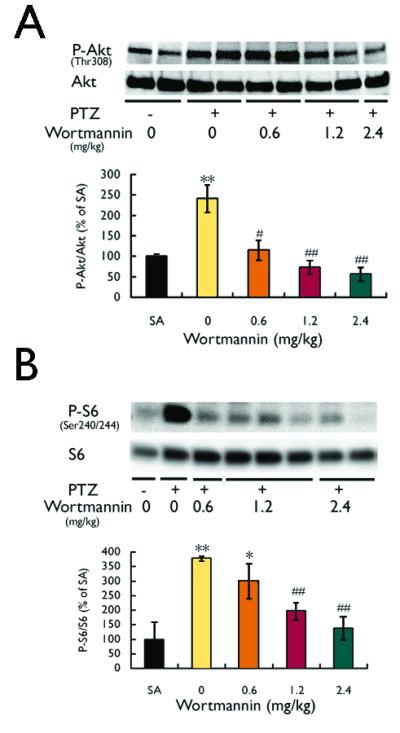
PTZ-induced seizures activate mTOR via the PI3K-Akt pathway. (A) The PI3K inhibitor, wortmannin, administered 30 min before PTZ injection, caused a dose-dependent inhibition of the PTZ seizure-induced PI3K-Akt pathway activation, as reflected by the P-Akt (Thr308) to total Akt ratio assayed 3 hours after PTZ injection. (B) Wortmannin, administered 30 min before PTZ injection, also caused a dose-dependent inhibition of the PTZ seizure-induced mTOR pathway activation, as reflected by the P-S6 to total S6 ratio assayed 3 hours after PTZ injection, indicating that PTZ seizures activate the mTOR pathway first via upstream PI3K-Akt activation (*p<0.05, **p<0.01, compared to saline (SA); #p<0.5, ##p<0.01, compared to PTZ + 0 mg/kg wortmannin; n = 4-6 per group). Data shown in both panels are from neocortex. Similar results were obtained in hippocampus (not shown).
Discussion
In this study, the direct effects of PTZ-induced seizures on the mTOR pathway were examined. PTZ seizures caused acute, transient mTOR activation lasting several hours, but no chronic or secondary phase over days to weeks. This transient mTOR pathway activation was paralleled by upstream activation of Akt and could be blocked by a PI3K inhibitor, indicating that the PI3K-Akt pathway mediates the effects of seizures on mTOR activation. While a number of cortical and subcortical structures have been implicated in mediating PTZ-induced seizures (Andre et al., 1998), this study focused on the hippocampus and cortex to compare with our previous work in the kainate model (Zeng et al., 2009). Overall, these results have mechanistic and therapeutic implications for the role of the mTOR pathway in epilepsy.
Abnormal, dysregulated mTOR activity has been implicated in promoting epileptogenesis in several types or models of epilepsy (Wong, 2010). A primary role of the mTOR pathway in the pathophysiology of epilepsy is most strongly supported in genetic disorders involving mutations of genes that directly regulate the mTOR pathway, such as the tuberous sclerosis complex (TSC) or phosphatase and tensin homolog (PTEN) genes. In addition to inhibiting tumor growth, there is evidence in both mouse models and patients with TSC that mTOR inhibitors have beneficial effects in preventing or decreasing seizures (Meikle et al., 2008; Zeng et al., 2008; Ljungberg et al., 2009; Krueger et al., 2010; Sunnen et al., 2011; Zeng et al., 2011). In animal models of acquired epilepsy secondary to brain injury, there is also evidence that the mTOR pathway is abnormally activated and that mTOR inhibitors may limit the development of potential epileptogenic pathological changes, such as mossy fiber sprouting (Buckmaster et al., 2009), and spontaneous seizures (Zeng et al., 2009; Huang et al., 2010), although effects on epileptogenesis are less clear (Buckmaster and Lew, 2011).
Defining the specific role of abnormal mTOR signaling in epileptogenesis is complicated by the fact that seizure activity in itself appears to activate the mTOR pathway. In the kainate and pilocarpine models of acquired epilepsy, the initial episode of status epilepticus directly triggers an acute activation of mTOR (Zeng et al., 2009; Huang et al., 2010), similar to the present results with PTZ. The consistent pattern of acute mTOR activation in several different chemiconvulsant models confirms that the seizure activity itself is likely the trigger for mTOR activation, not other pharmacological effects of the various seizure-inducing drugs. However, there are some interesting differences between the PTZ model, which involves self-limited seizures, and the kainate and pilocarpine models of status epilepticus. As behaviorally-observed PTZ seizures only lasted minutes in this study and previous EEG studies also documented a limited period of epileptiform spike discharges following a single PTZ injection (Wong et al., 2003), the duration of mTOR activation of at least several hours greatly outlasts the period of seizure activity. Furthermore, a remarkable difference is apparent between PTZ and the other models with regard to later, more chronic mTOR activation. While in the kainate and pilocarpine models, a second, chronic phase of mTOR activation occurs over days to weeks following the initial seizures (Huang et al., 2010; Zeng et al., 2009), there is no such secondary, late mTOR activation following PTZ seizures. As PTZ seizures do not typically lead to neuropathological injury or spontaneous epilepsy (Wong et al., 2003; Holmes et al, 1984), this suggests that the acute phase of seizure-induced mTOR activation is not sufficient to trigger epileptogenesis. By comparison, after resolution of the acute seizures in the kainate and pilocarpine models, subsequent inhibition of the later phase of mTOR activation has been implicated in suppressing pathological changes (Buckmaster et al., 2009) and spontaneous seizures (Huang et al., 2010; Zeng et al., 2009) that develop in these other models. Thus, future studies dissecting differences in the timing and mechanisms of mTOR activation between the PTZ and kainate/pilocarpine models may be key in understanding necessary conditions that lead to epileptogenesis.
While upstream molecular events causing mTOR activation are clearly delineated in specific genetic models of epilepsy, such as TSC, the mechanisms triggering mTOR activation following acquired brain injury are poorly understood. Among numerous upstream signaling pathways that may regulate mTOR, the PI3K-Akt pathway represents a logical candidate for mediating the effects of seizures, because seizures result in massive glutamate release and glutamate can stimulate PI3K (Sutton and Chandler, 2002; Zhu et al. 2002). Our present results with PTZ, as well as previous findings in the kainate model (Zeng et al., 2010), support an intermediary role of the Akt-PI3K pathway in seizure-induced mTOR activation. Feedback pathways and both upstream and downstream interactions may exist between the PI3K-Akt and mTOR pathways, as supported by the PTZ seizure-induced activation of the different Akt phosphorylation sites, Thr308 (upstream from mTOR) and Ser473 (downstream from mTOR). Although PI3 kinase inhibitors, such as wortmannin, may have other non-specific effects, the inhibition of the PTZ seizure-induced activation of both Akt and S6 by wortmannin indicates that the PI3K-Akt pathway mediates the effects of PTZ seizures on mTOR pathway activation. While the mTOR pathway has been implicated in epileptogenesis, independent effects of seizure-induced PI3K-Akt activation on cell survival might relate to the lack of cell death reported in the PTZ model. In either case, these results suggest the potential utility of PI3K inhibitors as therapeutic agents in epilepsy.
Acknowledgements
This work was supported by NIH R01 NS056872 (MW). Neither of the authors has any conflict of interest to disclose.
Footnotes
The authors have read the Journal’s position on issues involved in ethical publications and affirm that this report is consistent with those guidelines.
References
- Andre V, Pineau N, Motte JE, Marescaux C, Nehlig A. Mapping of neuronal networks underlying generalized seizures induced by increasing doses of pentylenetetrazol in the immature and adult rat: a c-Fos immunohistochemical study. Eur J Neurosci. 1998;19:2094–2106. doi: 10.1046/j.1460-9568.1998.00223.x. [DOI] [PubMed] [Google Scholar]
- Buckmaster PS, Lew FH. Rapamycin suppresses mossy fiber sprouting but not seizure frequency in a mouse model of temporal lobe epilepsy. J Neurosci. 2011;9:2337–2347. doi: 10.1523/JNEUROSCI.4852-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckmaster PS, Ingram EA, Wen X. Inhibition of the mammalian target of rapamycin signaling pathway suppresses dentate granule cell axon sprouting in a rodent model of temporal lobe epilepsy. J Neurosci. 2009;29:8259–8269. doi: 10.1523/JNEUROSCI.4179-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes GL, Albala BJ, Moshe SL. Effect of a single brief seizure on subsequent seizure susceptibility in the immature rat. Arch Neurol. 1984;41:853–855. doi: 10.1001/archneur.1984.04050190059014. [DOI] [PubMed] [Google Scholar]
- Huang X, Zhang H, Yang J, Wu J, McMahon J, Lin Y, Cao Z, Gruenthal M, Huang Y. Pharmacological inhibition of the mammalian target of rapamycin pathway suppresses acquired epilepsy. Neurobiol Dis. 2010;40:193–199. doi: 10.1016/j.nbd.2010.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P, Wilson KA, Byars A, Sahmoud T, Franz DN. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. 2010;363:1801–1811. doi: 10.1056/NEJMoa1001671. [DOI] [PubMed] [Google Scholar]
- Ljungberg MC, Sunnen CN, Lugo JN, Anderson AE, D’Arcangelo G. Rapamycin suppresses seizures and neuronal hypertrophy in a mouse model of cortical dysplasia. Dis Model Mech. 2009;2:389–398. doi: 10.1242/dmm.002386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meikle L, Pollizzi K, Egnor A, Kramvis I, Lane H, Sahin M, Kwiatkowski DJ. Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J Neurosci. 2008;28:5422–5432. doi: 10.1523/JNEUROSCI.0955-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunnen CN, Brewster AL, Lugo JN, Vanegas F, Turcios E, Mukhi S, Parghi D, D’Arcangelo G, Anderson AE. Inhibition of the mammalian target of rapamycin blocks epilepsy progression in NS-Pten conditional knockout mice. Epilepsia. 2011. Epub ahead of print. [DOI] [PMC free article] [PubMed]
- Sutton G, Chandler LJ. Activity-dependent NMDA receptor-mediated activation of protein kinase B/Akt in cortical neuronal cultures. J Neurochem. 2002;82:1097–1105. doi: 10.1046/j.1471-4159.2002.01031.x. [DOI] [PubMed] [Google Scholar]
- Wong M, Wozniak DF, Yamada KA. An animal model of generalized nonconvulsive status epilepticus: immediate characteristics and long-term effects. Exp Neurol. 2003;183:87–99. doi: 10.1016/s0014-4886(03)00099-2. [DOI] [PubMed] [Google Scholar]
- Wong M. Mammalian target of rapamycin (mTOR) inhibition as a potential antiepileptogenic therapy: from tuberous sclerosis to common acquired epilepsies. Epilepsia. 2010;51:26–36. doi: 10.1111/j.1528-1167.2009.02341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng LH, McDaniel S, Rensing NR, Wong M. Regulation of cell death and epileptogenesis by the mammalian target of rapamycin (mTOR): a double-edged sword? Cell Cycle. 2010;9:2281–2285. doi: 10.4161/cc.9.12.11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng LH, Rensing NR, Wong M. The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J Neurosci. 2009;29:6964–6972. doi: 10.1523/JNEUROSCI.0066-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng LH, Rensing NR, Zhang B, Gutmann DH, Gambello MJ, Wong M. Tsc2 gene inactivation causes a more severe epilepsy phenotype than Tsc1 inactivation in a mouse model of tuberous sclerosis complex. Hum Mol Genet. 2011;20:445–454. doi: 10.1093/hmg/ddq491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng LH, Xu L, Gutmann DH, Wong M. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol. 2008;63:444–453. doi: 10.1002/ana.21331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu D, Lipsky RH, Marini AM. Coactivation of the phosphatidylinositol-3-kinase/Akt signaling pathway by N-methyl-D-aspartate and TrkB receptors in cerebellar granule cell neurons. Amino Acids. 2002;23:11–17. doi: 10.1007/s00726-001-0103-9. [DOI] [PubMed] [Google Scholar]