

Short abstract
The first comprehensive analysis of human processed pseudogenes (PPs) using all known human genes as queries is presented. The data suggest a nearly simultaneous burst of PP and Alu formation in the genomes of ancestral primates.
Abstract
Background
Abundant pseudogenes are a feature of mammalian genomes. Processed pseudogenes (PPs) are reverse transcribed from mRNAs. Recent molecular biological studies show that mammalian long interspersed element 1 (L1)-encoded proteins may have been involved in PP reverse transcription. Here, we present the first comprehensive analysis of human PPs using all known human genes as queries.
Results
The human genome was queried and 3,664 candidate PPs were identified. The most abundant were copies of genes encoding keratin 18, glyceraldehyde-3-phosphate dehydrogenase and ribosomal protein L21. A simple method was developed to estimate the level of nucleotide substitutions (and therefore the age) of PPs. A Poisson-like age distribution was obtained with a mean age close to that of the Alu repeats, the predominant human short interspersed elements. These data suggest a nearly simultaneous burst of PP and Alu formation in the genomes of ancestral primates. The peak period of amplification of these two distinct retrotransposons was estimated to be 40-50 million years ago. Concordant amplification of certain L1 subfamilies with PPs and Alus was observed.
Conclusions
We suggest that a burst of formation of PPs and Alus occurred in the genome of ancestral primates. One possible mechanism is that proteins encoded by members of particular L1 subfamilies acquired an enhanced ability to recognize cytosolic RNAs in trans.
Background
The abundance of pseudogenes is a remarkable feature of mammalian genomes. Aptly named, pseudogenes are copies of specific genes and are present in every mammalian chromosome [1-5]. In general, pseudogenes are thought to be nonfunctional [2] as they have accumulated vast numbers of mutations during evolution and have lost the ability to be transcribed. Pseudogenes fall into two distinct categories depending on the mechanism by which they are generated: processed pseudogenes (PPs) are reverse transcribed from mRNAs (and thus do not contain introns) whereas nonprocessed pseudogenes arise from duplications of genomic DNA [2,4]. Among the abundant PPs, there are a substantial number of 'processed genes' or 'retrogenes' of novel function that also derive from mRNAs of various intron-containing genes [6-8].
In addition to PPs, mammalian genomes contain a large number of retrotransposons (retroposons) that represent a reverse flow of genetic information via RNA [9-13]. In humans, short interspersed elements (SINEs) and long interspersed elements (LINEs) occupy over 30% of the genome [14]. Progress in LINE1 (L1) molecular biology has enabled L1 'retrotransposition' studies in cultured HeLa cells [15,16]. Recent work [17-21] shows that mammalian L1-encoded proteins may have been involved in the reverse transcription of PP and Alu [22-26]. Furthermore, L1-encoded proteins predominantly mobilize the RNA in which they are encoded [18,19]. This so-called 'cis preference' explains the fact that, among the overwhelming number of nonfunctional L1 RNAs, recent mutagenic L1 insertions in humans and mice are derived from a progenitor L1 RNA that contained intact open reading frames (ORFs) [16]. In fact, Moran's group estimated that a functional L1 mobilizes nonfunctional L1 RNAs and other cellular mRNAs in trans at frequencies of only 0.2%-0.9% and 0.01%-0.05%, respectively, relative to processes involving cis RNA [19]. This finding also raised the question of how human Alu repeats could have been amplified in trans to their present level of approximately 10% of the human genome, given that L1-encoded proteins preferentially mobilize their own transcripts. Boeke proposed that Alu RNA secondary structure could have positioned this RNA on the ribosome in a manner that promoted effective interactions with L1-encoded proteins [21,27].
The initial analysis of the human genome draft sequence by the International Human Genome Sequencing Consortium provided the first comprehensive view of retroposons such as LINEs and SINEs, although the description of PPs was largely ignored [14]. The Celera report briefly described a preliminary analysis of PPs [28]. Here, we present the first comprehensive analysis of human PPs using all known human genes as queries. These PPs were derived from 6% of all annotated human genes, and our data suggest a possible burst of PP genesis early in primate evolution.
Results
Whole-genome screening for human PPs and their content
We initially searched for PPs that exhibit sequence similarity to any of the transcripts from the 21,921 genes annotated by the Ensembl project [29]. The fact that PPs contain few if any introns enabled our search to generate 3,664 PP candidates (Table 1 and Additional data file 1; pseudogenes generated by DNA duplication contained many introns and were eliminated). These candidate PPs represented a minimum set because not all human genes have yet been annotated [30] and the search included only those PPs whose length is more than 90% of the respective mRNA. If the estimated 35,000 human genes [14,28] had been used in the search and shorter PPs included in the analysis, over 7,000 PPs would have been expected.
Table 1.
Processed pseudogene content of the human genome
| Gene class* | Genes that generated PPs | PPs |
| Annotated genes† | ||
| Enzymes | ||
| Kinase | 24 | 37 |
| Dehydrogenase | 16 | 80 |
| Transferase | 15 | 25 |
| Peptidase | 10 | 15 |
| Phosphatase | 9 | 13 |
| Synthase | 8 | 20 |
| Synthetase | 5 | 23 |
| Translocase | 4 | 7 |
| Protease | 4 | 4 |
| Reductase | 3 | 3 |
| Phospholipase | 2 | 5 |
| RNA polymerase | 2 | 3 |
| Others | 46 | 63 |
| Total | 148 | 298 |
| Structural proteins | ||
| Ribosomal proteins | 31 | 416 |
| Actin-related proteins | 9 | 23 |
| Keratin | 5 | 57 |
| Ribosomal proteins (mitochondrial) | 4 | 7 |
| Tubulin | 4 | 6 |
| Histone | 2 | 5 |
| Myosin | 2 | 4 |
| Dynein | 2 | 3 |
| Kinesin | 1 | 1 |
| Total | 60 | 522 |
| Others | ||
| Ligand-binding proteins‡ | 30 | 56 |
| Transcription factor‡ | 11 | 23 |
| RNA-binding proteins‡ | 11 | 15 |
| Translation initiation/termination | 9 | 21 |
| Proteasome | 9 | 19 |
| Heat-shock protein | 8 | 29 |
| Solute carrier | 7 | 14 |
| Zinc finger protein‡ | 7 | 11 |
| Ring finger protein‡ | 7 | 10 |
| Nuclear ribonucleoprotein‡ | 6 | 19 |
| Autoantigen | 6 | 12 |
| Receptor | 6 | 8 |
| Splicing factor‡ | 5 | 7 |
| DEAD/H box polypeptide | 4 | 4 |
| Carcinoma-associated antigen | 4 | 4 |
| Channel | 3 | 11 |
| Thioredoxin | 3 | 5 |
| Others | 295 | 464 |
| Total | 431 | 732 |
| Total annotated genes | 639 | 1,552 |
| Hypothetical genes§ | 660 | 2,112 |
| Grand total | 1,299 | 3,664 |
*The functional annotation of NCBI Reference Sequence (RefSeq) collection (v2003.01.06) was used for this classification [61]. Respective genes were classified into only one category. †Ensembl gene transcripts (v1.1.0) which are correspond to the RefSeq collection (v2003.01.06). ‡These seven gene classes were classified as 'Ligand binding' in Figure 1a,b for simplicity. §Ensembl gene transcripts (v1.1.0) that do not correspond to the RefSeq collection (v2003.01.06).
Parental genes of human PPs are of various types, including those for enzymes, structural proteins and regulatory proteins such as ligand-binding proteins and transcription factors (Table 1). Of the total PPs analyzed, the relative frequency of those derived from genes encoding enzymes, structural proteins and ligand-binding proteins was 19%, 34%, and 9% (Figure 1b), respectively, whereas the PP parental genes for structural proteins constituted only 9% of the total parental genes (Figure 1a). Among 1,299 parental genes identified in this study, kinases, ribosomal proteins and ligand-binding proteins were predominant.
Figure 1.
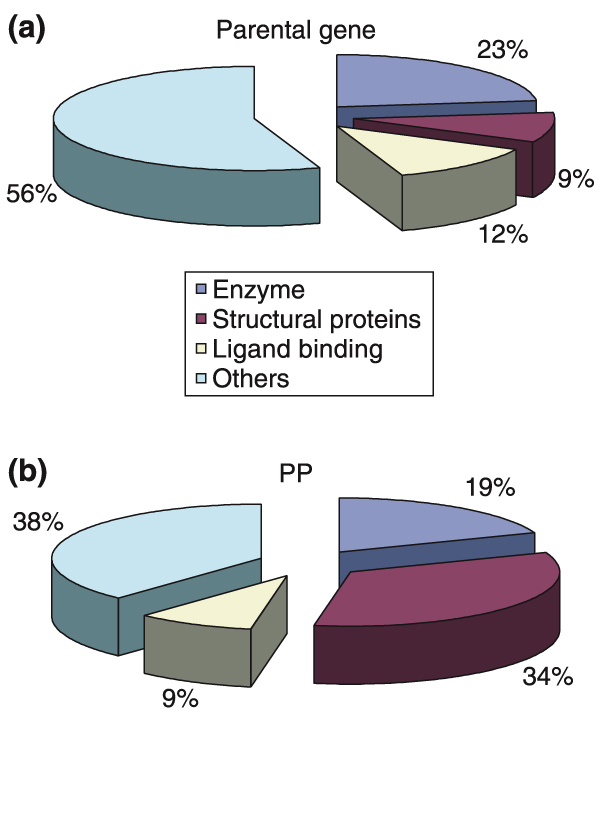
Difference between the profiles of the PP parental genes and PPs in the human genome. (a) Classifications of the PP parental genes. (b) Classifications of the PPs. Gene classes were based on the functional annotation of the NCBI Reference Sequence collection [61] for the respective genes (see Table 1) and were further integrated into four main classes. Ligand-binding proteins, transcription factors, RNA-binding proteins, zinc finger protein, ring finger proteins, nuclear ribonucleoproteins and splicing factors were classified as 'Ligand binding'.
Table 2 shows a compilation of the abundant PPs in the human genome (see Additional data file 2). The three most abundant types of human PPs were derived from the genes for keratin 18, glyceraldehyde-3-phosphate dehydrogenase (GAPD) and ribosomal protein L21 (RP L21). These genes generated at least 52, 43 and 38 copies of PPs, respectively, in the genome. Keratin 18 is commonly expressed in internal epithelia and is one of the earliest intermediate filament proteins expressed during embryogenesis [31]. The genes for GAPD and ribosomal proteins are housekeeping genes. These data suggest that mRNAs for keratin 18, GAPD and RPL21 were highly expressed or stable in either the germline cells or at an early stage of development, as heritable copies of these genes must have been reverse transcribed in one of those two instances.
Table 2.
The most abundant PPs in the human genome
| PP number* | Ensembl ID | RefSeq ID | Gene name | mRNA (bases)† | GC content‡ | Chromosome |
| 52 | ENST00000228652 | NM_000224 | Keratin 18 (KRT18) | 1,311 | 0.59 | 12 |
| 43 | ENST00000229239 | NM_002046 | Glyceraldehyde-3-phosphate dehydrogenase (GAPD) | 975 | 0.55 | 12 |
| 38 | ENST00000241454 | NM_000982 | Ribosomal protein L21 (RPL21) | 623 | 0.41 | 13 |
| 36 | ENST00000264258 | NM_000993 | Ribosomal protein L31 (RPL31) | 412 | 0.46 | 2 |
| 32 | ENST00000226734 | NM_000995 | Ribosomal protein L34 (RPL34) | 382 | 0.44 | 4 |
| 31 | ENST00000256818 | NM_001019 | Ribosomal protein S15a (RPS15A) | 440 | 0.45 | 16 |
| 23 | ENST00000202773 | NM_000970 | Ribosomal protein L6 (RPL6) | 861 | 0.47 | 12 |
| 23 | ENST00000241929 | NM_000969 | Ribosomal protein L5 (RPL5) | 951 | 0.43 | 1 |
| 21 | ENST00000255320 | NM_002128 | High-mobility group box 1 (HMGB1) | 971 | 0.41 | 13 |
| 20 | ENST00000245458 | NM_001032 | Ribosomal protein S29 (RPS29) | 195 | 0.53 | 14 |
| 18 | ENST00000260896 | NM_001026 | Ribosomal protein S24 (RPS24) | 390 | 0.44 | 10 |
| 17 | ENST00000009589 | NM_001023 | Ribosomal protein S20 (RPS20) | 504 | 0.47 | 8 |
| 16 | ENST00000225430 | NM_000981 | Ribosomal protein L19 (RPL19) | 667 | 0.52 | 17 |
| 14 | ENST00000230050 | NM_001016 | Ribosomal protein S12 (RPS12) | 493 | 0.49 | 6 |
| 12 | ENST00000253004 | NM_054012 | Argininosuccinate synthetase (ASS) | 1,245 | 0.56 | 9 |
| 12 | ENST00000216296 | NM_004500 | Heterogeneous nuclear ribonucleoprotein C (C1/C2) (HNRPC) | 1,588 | 0.43 | 14 |
| 12 | ENST00000211372 | NM_022551 | Ribosomal protein S18 (RPS18) | 494 | 0.51 | 6 |
| 11 | ENST00000263097 | NM_004368 | Calponin 2 (CNN2) | 882 | 0.61 | 19 |
| 11 | ENST00000253788 | NM_000988 | Ribosomal protein L27 (RPL27) | 450 | 0.46 | 17 |
| 11 | ENST00000259689 | NM_001010 | Ribosomal protein S6 (RPS6) | 784 | 0.46 | 9 |
| 11 | ENST00000260379 | NM_001003 | Ribosomal protein, large, P1 (RPLP1) | 510 | 0.56 | 15 |
| 11 | ENST00000011649 | NM_007104 | Ribosomal protein L10a (RPL10A) | 682 | 0.51 | 6 |
| 10 | ENST00000255477 | NM_003295 | Tumor protein, translationally-controlled 1 (TPT1) | 829 | 0.45 | 13 |
| 10 | ENST00000227378 | NM_006597 | Heat shock 70 kDa protein 8 (HSPA8) | 1,938 | 0.46 | 11 |
| 10 | ENST00000218437 | NM_001007 | Ribosomal protein S4, X-linked (RPS4X) | 853 | 0.48 | X |
| 9 | ENST00000220072 | NM_001021 | Ribosomal protein S17 (RPS17) | 453 | 0.49 | 15 |
| 9 | ENST00000265385 | NM_000883 | IMP (inosine monophosphate) dehydrogenase 1 (IMPDH1) | 1,425 | 0.59 | 7 |
| 9 | ENST00000265264 | NM_000986 | Ribosomal protein L24 (RPL24) | 447 | 0.48 | 3 |
| 9 | ENST00000216146 | NM_000967 | Ribosomal protein L3 (RPL3) | 1,265 | 0.54 | 22 |
| 8 | ENST00000196551 | NM_001009 | Ribosomal protein S5 (RPS5) | 720 | 0.58 | 19 |
| 8 | ENST00000228140 | NM_001017 | Ribosomal protein S13 (RPS13) | 495 | 0.45 | 11 |
| 7 | ENST00000221267 | NM_003333 | Ubiquitin A-52 residue ribosomal protein fusion product 1 (UBA52) | 384 | 0.53 | 19 |
| 7 | ENST00000233609 | NM_001018 | Ribosomal protein S15 (RPS15) | 469 | 0.62 | 19 |
| 7 | ENST00000257522 | NM_030940 | Hypothetical protein MGC4276 similar to CG8198 (MGC4276) | 255 | 0.38 | 9 |
| 7 | ENST00000265333 | NM_003374 | Voltage-dependent anion channel 1 (VDAC1) | 1,498 | 0.45 | 5 |
| 7 | ENST00000236900 | NM_001028 | Ribosomal protein S25 (RPS25) | 426 | 0.45 | 11 |
| 7 | ENST00000264254 | NM_024065 | Hypothetical protein MGC3062 (MGC3062) | 955 | 0.42 | 2 |
| 7 | ENST00000246201 | NM_003908 | Eukaryotic translation initiation factor 2, subunit 2 beta (EIF2S2) | 1,300 | 0.39 | 20 |
| 6 | ENST00000245206 | NM_002080 | Glutamic-oxaloacetic transaminase 2, mitochondrial (GOT2) | 2,331 | 0.49 | 16 |
| 6 | ENST00000238591 | NM_015962 | CGI-35 protein (CGI-35) | 1,019 | 0.37 | 14 |
| 6 | ENST00000249380 | NM_005000 | NADH dehydrogenase 1 alpha subcomplex, 5, 13 kDa (NDUFA5) | 339 | 0.41 | 7 |
| 6 | ENST00000228825 | NM_005719 | Actin-related protein 2/3 complex, subunit 3, 21 kDa (ARPC3) | 786 | 0.41 | 12 |
| 6 | ENST00000261565 | NM_003187 | TATA box binding protein (TBP)-associated factor, 32 kDa (TAF9) | 833 | 0.34 | 5 |
| 6 | ENST00000227157 | NM_005566 | Lactate dehydrogenase A (LDHA) | 1,589 | 0.43 | 11 |
| 6 | ENST00000264221 | NM_006452 | Phosphoribosylaminoimidazole carboxylase, (PAICS) | 1,385 | 0.41 | 4 |
| 6 | ENST00000037869 | NM_032822 | Hypothetical protein FLJ14668 (FLJ14668) | 414 | 0.56 | 2 |
| 6 | ENST00000235094 | NM_001688 | ATP synthase, mitochondrial F0 complex, subunit b (ATP5F1) | 1,104 | 0.43 | 1 |
| 6 | ENST00000234875 | NM_000983 | Ribosomal protein L22 (RPL22) | 541 | 0.41 | 1 |
| 6 | ENST00000005593 | NM_001152 | Solute carrier family 25, member 5 (SLC25A5) | 894 | 0.52 | X |
| 5 | ENST00000216252 | NM_032758 | PHD finger protein 5A (PHF5A) | 330 | 0.48 | 22 |
*The number of PPs that were derived from respective genes. The top 50 genes are shown. †Length of the Ensembl gene transcripts (v1.1.0). ‡GC content of the Ensembl gene transcripts (v1.1.0). The list of all the genes is available as Additional data file 2.
As shown in Figure 1a and 1b, structural-protein PPs constitute the largest class (34%). The 50 most prolific PP parental genes include 25 ribosomal protein genes (Table 2) which contribute substantially to the high incidence of structural proteins among the total number of PPs presented in Figure 1b.
GC content in PP parental genes
Human PPs are derived from mRNAs that exhibit a wide range of GC content. We examined the possible relationship between the number of PPs derived from a gene and the GC content of its mRNA (Figure 2). The rates of PP generation from parental genes within each GC group show no significant statistical difference except for genes of high GC content (> 0.62). This result differs from that of a previous study in which an inverse correlation between the number of ribosomal protein PPs and the GC content of the parental genes was observed [32]. Because we analyzed a wide variety of PPs, including those of ribosomal protein genes, the correlation observed in this previous study probably reflects a specific correlation between GC content and either expression level or stability of ribosomal protein mRNAs.
Figure 2.
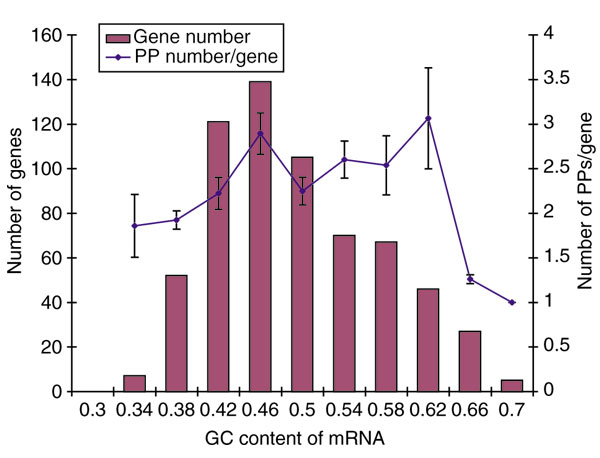
GC content of the PP parental genes and the number of PP copies of those genes. The total number of PP parental genes having a given GC content is shown as individual bars in increments of 4%. The PP-generation rate (the PP number/gene) is shown as a line that connects averages for respective groups. The vertical error bars indicate standard error of the mean.
Chromosomal distribution of human PPs
The 3,664 PP candidates are distributed throughout all 24 chromosomes and were derived from genes on various chromosomes (Table 3 and Figure 3). No stringent bias of gene 'projections' (that is the insertion of the PP of a gene in a specific chromosomal location) toward specific chromosomes was observed. In some chromosomes, however, (for example chromosome 19), the ratios of self-projection are relatively high. Interestingly, the PP density within each chromosome roughly parallels gene density (Figure 4). For example, chromosomes that are gene-rich, such as 19, 17 and 11 [14,28], tend to be relatively PP-rich. On the other hand, chromosomes that are gene-poor, such as Y, 21, 13 and 4 [14,28], tend to also be poor in PPs. As human gene density shows strong positive correlation with local GC content [14,28], this result suggests that the integration of PPs into chromosomes in general may be dependent on aspects of the genomic environment that are strictly related to chromosomal gene density [14,28], such as local GC content and an open chromatin structure that facilitates transcription.
Table 3.
Chromosomal distribution and density of human PPs
| Chromosome | PPs | Genes that generated PPs | Number of genes (Ensembl 4.28.1) | Genes/Mb* | PPs/Mb |
| All | 3,664 | 1,299 | 23,863 | 7.33 | 1.12 |
| 1 | 359 | 117 | 2,482 | 8.90 | 1.28 |
| 2 | 241 | 84 | 1,550 | 6.31 | 0.98 |
| 3 | 225 | 76 | 1,277 | 5.94 | 1.04 |
| 4 | 163 | 57 | 868 | 4.33 | 0.81 |
| 5 | 193 | 72 | 1,093 | 5.61 | 0.99 |
| 6 | 207 | 64 | 1,297 | 7.07 | 1.12 |
| 7 | 176 | 72 | 1,251 | 7.59 | 1.06 |
| 8 | 144 | 47 | 787 | 5.23 | 0.95 |
| 9 | 150 | 49 | 934 | 6.57 | 1.05 |
| 10 | 178 | 45 | 939 | 6.56 | 1.24 |
| 11 | 238 | 79 | 1,506 | 9.98 | 1.57 |
| 12 | 234 | 78 | 1,212 | 8.25 | 1.59 |
| 13 | 94 | 24 | 425 | 3.61 | 0.80 |
| 14 | 146 | 45 | 785 | 7.33 | 1.36 |
| 15 | 114 | 41 | 770 | 7.65 | 1.13 |
| 16 | 95 | 54 | 1,040 | 10.15 | 0.92 |
| 17 | 126 | 63 | 1,272 | 14.44 | 1.43 |
| 18 | 74 | 20 | 370 | 4.43 | 0.88 |
| 19 | 123 | 78 | 1,504 | 20.80 | 1.70 |
| 20 | 59 | 27 | 640 | 10.15 | 0.93 |
| 21 | 34 | 11 | 232 | 5.20 | 0.76 |
| 22 | 62 | 34 | 577 | 12.14 | 1.30 |
| X | 207 | 51 | 922 | 5.84 | 1.31 |
| Y | 22 | 11 | 130 | 2.53 | 0.42 |
*The number of Ensembl genes per megabases.
Figure 3.
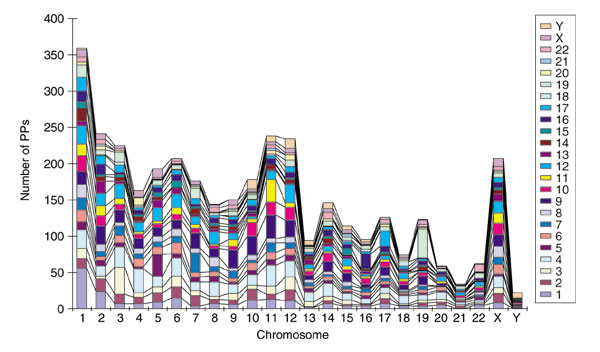
Chromosomal origins of human PPs. Individual bars indicate the total number of PPs in each chromosome. The different colors represent the chromosomal origins of the PPs.
Figure 4.
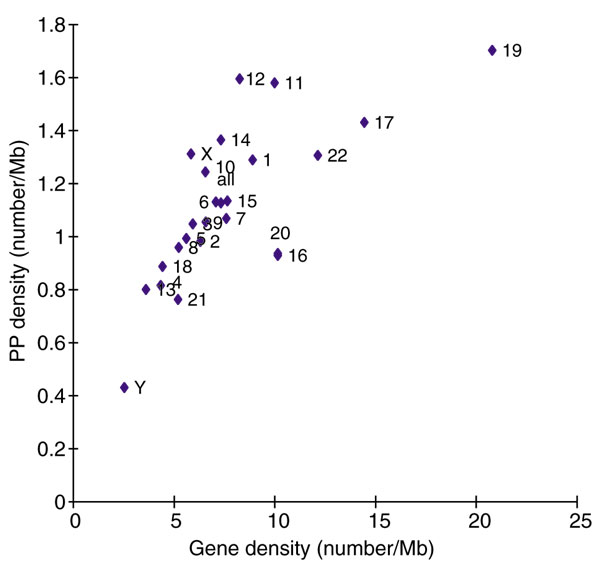
PP and gene density within each chromosome. For each chromosome, the number of PPs per megabase is plotted against the number of genes per megabase.
A simple method for estimating the level of nucleotide substitutions in PPs
To approximate the age of each PP, we developed a method for estimating the level of nucleotide substitutions relative to the parental gene. Initially, this method corrected for the sequence divergence value (a consequence of nucleotide-substitution processes) by removing the contribution of mutations at CpG sites. The C-to-T transition rate in CpG pairs is around 12-fold higher than the rate for other transitions [33] and causes distortions when comparing different genomic elements of high (for example, Alus) or low (for example, L1s) CpG content. Assuming that CpG frequency (ι) in a genomic element that was generated by duplication of a functional gene of high CpG content decreases over time (t) and reaches a state of equilibrium (ε) (approximately 20% of the frequency [14,33] expected from the local fraction of cytosines and guanosines [14]), the time since the duplication (T) was calculated (see Materials and methods) from the given sequence divergence (D) and the neutral mutation rate (μ) of primates:
D = ∫0Tμ(1 + 11((ι - ε)/((0.01ι/(0.99ι-ε))t + 1) + ε))dt
Next, the quantity Σ (=μT) was corrected for multiple substitutions at the same site using the Jukes-Cantor model [34], giving the average number of substitutions per 100 base-pairs (bp), (K). For PPs, sequence divergences were defined as the mismatch rates of respective PPs relative to the current parental gene sequences. Finally, the levels of substitution that accumulated only in PPs were estimated (see Materials and methods). The estimated levels of substitutions in PPs (K(ψ)) were then calculated as K(ψ) = 0.705 K.
Simultaneous burst of processed pseudogenes and Alu repeats in ancestral primates
Using the levels of nucleotide substitution in PPs estimated by K(ψ), we next evaluated the total number of PPs having the same substitution value, thus approximating the age distribution of PPs. We initially presumed that if PPs were generated at a roughly constant rate during primate evolution, their age distribution would be nearly flat. Surprisingly, a Poisson-like distribution was obtained (Figure 5a). This result indicates that PPs in general may have been generated at extraordinarily high rates during some periods. If the rate of nucleotide substitution is assumed to be 1.5 × 10-9 per nucleotide per year [34,35], then our data estimate that the peak of PP generation occurred approximately 40 million years ago, coincident with the onset of the radiation of the higher primates [36,37].
Figure 5.
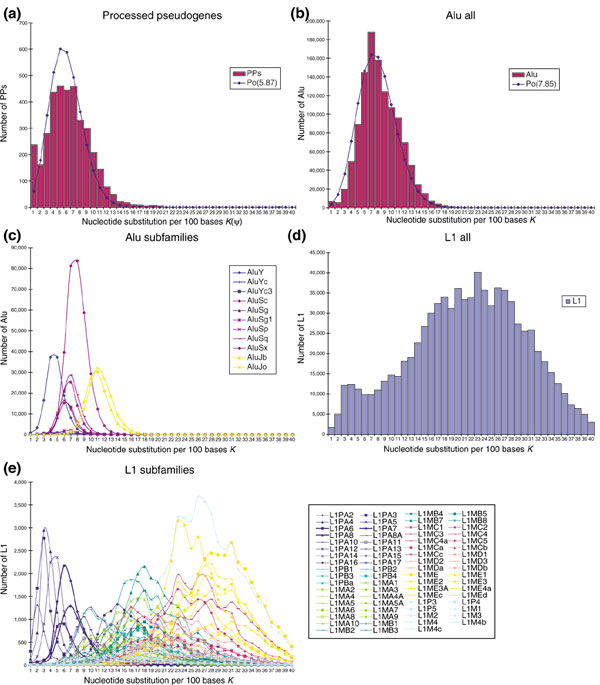
Age distribution of human retroposons represented by the level of nucleotide substitutions. (a) Human PPs. The number of nucleotide substitutions per 100 bases (except CpG sites) was calculated for each PP, and the total number of PPs having a given number of substitutions is shown as individual bars in one-nucleotide increments. For comparison, the line shows a Poisson distribution of the same average values for PPs. (b) Alu repeats, calculated and presented as in (a). The line shows a Poisson distribution of the same average values for Alus. (c) Alu subfamilies, calculated as in (a). The curves connect apices of respective bars calculated as in (a). For simplicity, subfamilies that contain less than 5,000 Alus, such as Alu Ya and Yb, are not shown. (d) L1s, calculated and presented as in (a). (e) L1 subfamilies, calculated and presented as in (c). For simplicity, subfamilies that contain less than 1,000 L1s, such as L1PA1 (L1Hs) and L1P1, are not shown. L1PA6, L1PA7 and L1PA8 are shown as bold blue lines.
The above results are reminiscent of the amplification profile of Alu repeats. Alu elements comprise approximately 10% of the human genome [14] and are restricted to primates [22-26]. It has been proposed that the average age of Alu repeats is around 40 million years and that the majority of Alus were generated around this time [14,22,23,26]. We confirmed these previous results by re-estimating the age distribution of all human Alu repeats (Figure 5b). The Alus also showed a Poisson-like distribution with a sharp peak. Alus are classified into distinct subfamilies that can be identified on the basis of mutations shared among subfamily members [23,26]. Alu subfamilies were derived from a small number of source or master genes. Accordingly, a consensus sequence constructed from members of each subfamily represents each subfamily's source gene(s) [23,26]. To evaluate the contribution of each subfamily to the entire distribution of Alus, we estimated the age distribution of respective Alu subfamilies (Figure 5c). The peaks for respective subfamilies are grouped closely, and the subfamily Alu Sx strongly influences the overall distribution of Alus (compare with Figure 5b). Therefore, the Sx subfamily (and thus Alus in general) appears to have been amplified intensively over a relatively short period. To the best of our knowledge, many previous discussions of Alu amplification reflect this viewpoint of Alu evolution [14,22,23,26]. However, our results show that the intensive generation of two distinct elements, PPs and Alus, occurred almost simultaneously suggesting that an unknown change in either the cellular environment or the proliferation mechanism itself enhanced the proliferation of such retroposons in ancestral primates 40-50 million years ago.
Concordant amplification of certain LINE1 subfamilies with PPs and Alus
Recent progress in L1 biology shows that mammalian L1-encoded proteins are likely to have been involved in the reverse transcription of Alus and PPs [17-21]. To elucidate the cause of the elevated retrotransposition of PPs and Alus, we analyzed the age distribution of all human L1s (Figure 5d). Curiously, the rate of amplification (retrotransposition in cells and fixation within a population) of L1s does not peak around 7%, as was the case for PPs and Alus (compare with Figure 5a,b), raising the issue of how the rate of PP/Alu retrotransposition became elevated during a period of moderate change in L1 retrotransposition. To address this problem, L1s were divided into around 80 subfamilies [38], and age distributions for representative subfamilies are shown in Figure 5e. Although the distributions of respective subfamilies overlap, each subfamily has emerged successively during approximately 150 million years of mammalian evolution. Merging the distribution profiles of all the L1s yields a curve that is rather flat (almost equal to the curve that connects the apices of the respective bars in Figure 5d). Among a large number of L1 subfamilies, certain subfamilies, namely L1PA6, L1PA7 and L1PA8, were amplified intensively around 47 million years ago (the time corresponding to the 7% score). These data suggest that only one or a few L1 subfamilies may have contributed to the increased level of Alu and PP amplification (see Discussion).
Figure 6 shows phylogenetic relationships between L1 subfamilies. A considerable number of substitutions are evident that could explain a possible functional change in L1s between these subfamilies and the current L1 subfamily (L1Hs/L1PA1). There are several amino-acid substitutions within evolutionarily conserved domains (for example, 'C (cysteine)-rich domain') that result in altered residue polarity or charge (Figure 6). A key example is the highly variable amino-terminal half of the L1-encoded ORF1 protein, which contains residues that may be critical for interaction with other proteins [39]. There is 41% (54/131) amino-acid divergence between the ORF1 amino-terminal halves of L1PA7 and L1Hs whereas the divergence is only 7% (14/207) in the carboxy-terminal half (data not shown).
Figure 6.
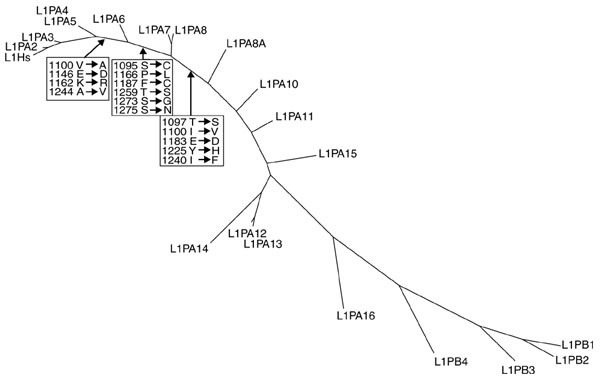
Phylogenetic relationships between L1 subfamilies. Amino-acid substitutions within the 'C domain' at particular stages of L1 evolution are denoted in boxes. The phylogenetic tree was constructed using the neighbor-joining method [62] based on the last 900 bp of the consensus sequences of respective subfamilies.
Discussion
Possible mechanisms of a 'retrotranspositional explosion'
A recent extensive survey of the human genome revealed a large number of ribosomal protein pseudogenes derived from the 79 functional ribosomal protein genes [32]. The discussion of the ages of these pseudogenes is problematic, however, in that ages were calculated by simply dividing sequence divergences by mutation rate. As the sequence divergence of a PP relative to its parental gene (K) is dependent on substitutions in both the PP (K(ψ)) and the gene (K(f)) (see Materials and methods), the ages of the ribosomal protein pseudogenes were overestimated. For example, with respect to RPL21 mRNAs (the most predominant source of ribosomal protein pseudogenes in humans), the sequence divergence between human and mouse or rat is approximately 11% (NM_000982, NM_019647, NM_053330, and [40]). The previous ribosomal protein pseudogene calculations dismissed sequence divergences between the present-day and primordial genes, probably overestimating the ages by around 10 million years (a few percent per 8-10% of divergence). Therefore, it is difficult to compare such values with the ages of Alus/L1s. Our method provides a clear solution to this matter, enabling us to compare the ages of different classes of retroposons. Hence, our method led us to the finding that there was a simultaneous burst of PPs and Alus - a 'retrotranspositional explosion' - in the primate genome.
Regarding the cause of the retrotranspositional explosion, it is worth considering the effect of a 'bottleneck' [34,41] during primate evolution. Only individuals that experienced extensive genomic retrotransposition might have propagated to become a majority within a population of ancestral primates, via a mechanism involving a rapid reduction in the general population. Studies on the molecular phylogeny and demographic history of humans show, however, that the primate lineage leading to humans never experienced an extensive bottleneck, at least since its divergence from the prosimian lineage [42]. Therefore, the effect of a bottleneck can be largely ignored.
The retrotranspositional explosion could be due to a change in the cellular environment of ancestral primates 40-50 million years ago, such as a higher transcriptional potential of parental (master) genes of PPs and Alus. A specific environment of the genome during the period of the retrotranspositional explosion, such as more available target sites of PPs and Alus, might have facilitated this event. Alternatively, a change in the proliferation mechanism of PPs and Alus, such as an increased amount of reverse transcriptase or an enhanced activity of enzymes for retrotransposition, might have promoted the explosion.
Recent studies on the L1 retrotransposons show that mammalian L1-encoded proteins may have been involved in the reverse transcription of Alus and PPs [17-21]. Here, we have shown that the intensive amplification of distinct genetic elements, namely PPs and Alus, seems to have occurred almost simultaneously around 40-50 million years ago, and suggests that only one or a few L1 subfamilies may have contributed to the observed high levels of Alu/PP retrotransposition.
How could a specific L1 subfamily (or subfamilies) have generated Alus and PPs at such an accelerated rate? We propose that L1s within specific subfamilies mobilized RNAs in trans at accelerated rates in ancestral primate genomes. Thus, a specific L1 subfamily may have mediated the Alu/PP retrotranspositional explosion. The age distributions estimated in this study allow the prediction of the most probable L1 subfamilies responsible for the explosion (care must be exercised when comparing ages between distinct genetic elements; see Materials and methods). The most probable candidate subfamilies are L1PA6, L1PA7 and L1PA8 (Figure 5e). As mentioned above, although the youngest L1 subfamily mobilizes cellular RNAs in trans at very low frequencies (0.01-0.05%) in HeLa cells, the frequency is not necessarily intrinsic to L1s. In fact, in cultured feline cells the frequency of L1-mediated PP formation in trans is 5% relative to that of L1 retrotransposition in cis [18]. Moreover, an eel LINE family exhibits a high level of trans retrotransposition (up to 30% [43]), and the frequency of L1-mediated Alu retrotransposition in HeLa cells is 100-1,000 times higher than control mRNAs [21]. Although L1 subfamilies such as L1PA6, L1PA7 and L1PA8 appear to have been extinguished by cumulative mutations, the possibility that an ancient L1 subfamily exhibited an enhanced ability to mobilize RNAs in trans could be verified experimentally in HeLa cells using reconstructed L1 subfamilies [44] as sources for reverse transcription of trans RNAs.
The impact of the retrotranspositional explosion on the ancestral primate genome
Alu insertions mediate many genomic rearrangements, such as unequal crossing over, induction of alternative splicing, and the introduction of new promoters, poly(A) signals and even new exons [6]. Inactivation of CMP-N-acetylneuraminic acid hydroxylase (around 2.8 million years ago) before brain expansion during human evolution occurred by an Alu-mediated inactivating mutation [45], representing yet another example of the impact of the Alu expansion. The current frequency of human endogenous insertional mutations caused by Alu retrotransposition is estimated at around 1 in every 16-200 individuals [26,46]. The frequency of Alu insertion at the time of the retrotranspositional explosion is estimated to have been 30-200 times higher than the frequency over the last 10 million years ([26] and data not shown). This implies that at least one in seven individuals at the time carried new Alu insertions in their genomes (a maximum of 12 insertions per individual). This high Alu insertion rate may have had a much greater impact on ancestral primate genomes compared with the impact of present-day mutations.
Retrotransposition of PPs causes not only insertional mutations but also the propagation of new genes. These 'retrogenes' comprise PPs that inserted themselves next to resident promoter/enhancer elements and thereby escaped transcriptional silencing and PPs that were initially inactive but were reactivated at a later time when flanking regulatory elements became activated by mutation [6]. Retrogenes are often observed in primate genomes [6], one example being the testis-specific human gene CDY (on the Y chromosome), which arose during primate evolution by retrotransposition of the ubiquitous mRNA of the gene CDYL located on chromosome 13 [7]. From the observed distribution of CDY homologs in primates, this event appears to have occurred in the simian lineage after its divergence from prosimians but before the split between Old and New World monkeys [7] during the period of the retrotranspositional explosion. We predict that further studies will demonstrate that many human retrogenes were generated during this period, and postulate that such retrogenes were involved in generating new characteristics that are specific to simian primates [8,47].
Over the course of eukaryotic evolution, the extensive propagation of new genes preceded apparent bursts of new organisms or the emergence of new hierarchies of morphological complexity [48,49]. The time of the retrotranspositional explosion can be estimated at 40-50 million years ago, assuming a nucleotide substitution rate of 1.5 × 10-9 per nucleotide per year [34,35]. Fossil records show that before this period, simian ancestors (monkeys, apes and humans) diverged from prosimians (lemurs and lorises) with the divergence of New World monkeys and the radiation of the remaining primates proceeding immediately thereafter [36,37] (Figure 7). The rapid amplification of Alus to the level of 10% of the primate genome and the creation of numerous replicas of various genes may have provided the molecular basis that led to the radiation of higher primates.
Figure 7.
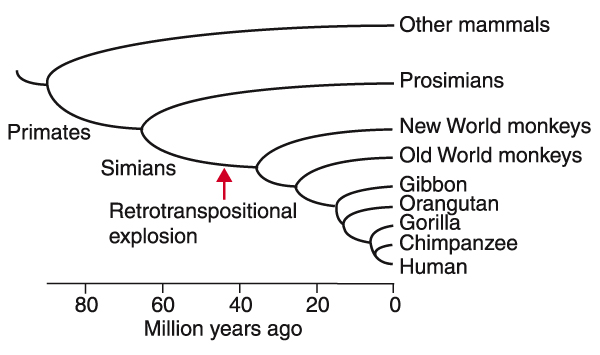
Timing of the retrotranspositional explosion during primate evolution. Phylogenetic relationships among primates and the estimated timeframes are based on data from references [34,36] and [37], and references therein.
Materials and methods
Determining a set of processed pseudogenes
PPs were searched for in an assembled human genome sequence (Human Genome Project Working Draft, April 1 2001) [50] using BLAT [51]. The BLAT setting was as follows: Assembly: April 1, 2001; Query type: DNA; Sort output: query, score; Output type: hyperlink. 'Confirmed cDNAs' (23,929 entries) in Ensembl DB (v1.1.0) [29] were used as queries. The subject with the highest score was regarded as the gene encoding the transcript. Multiple hits were subjected to analysis.
Subjects that contained over 90% of the query length were used. The number of aligning blocks, which usually corresponds to the number of exons, was compared between a gene and other subjects. If the number of aligning blocks was smaller than that of the gene, the subject was further analyzed, thus eliminating pseudogenes generated by DNA duplications. Subjects that were identified by intronless genes (single exon genes) were not included in the analysis to avoid confusing PPs and pseudogenes generated by DNA duplications. To avoid confusing phylogenetic relationships, loci (subjects) that were identified by multiple query hits were not included in the analysis. A series of Perl scripts were designed to analyze the BLAT search results.
Evaluating PP annotation
To evaluate our annotation of PPs, our results for chromosomes 21 and 22 were compared with those from other studies. For chromosome 21, the PP total in this study was 34 whereas previous studies reported 41 [52,53] and 57 [4]. The number of annotations common to two studies totaled 18 (this study and [52]), 14 (this study and [4]) and 21 [4,52]. Annotations common to all studies totaled 10. For chromosome 22, the PP total in this study was 62, whereas previous studies reported 91 [54,55] and 73 [4]. The number of common annotations totaled 37 (this study and [54]), 28 (this study and [4]) and 52 [4,54]. Annotations common to all studies totaled 27. Differences between the numbers appear to derive mainly from differences in the gene sets used for the analyses [30].
Identification of Alus, L1s, and their subfamilies
For each Alu and L1 repeat, the genomic location and sequence divergence was obtained from the output file of the RepeatMasker program applied to the human genome draft sequence (22 December 2001 [56]). Sequence divergences were defined as the mismatch rates of respective repeats relative to the consensus sequence of respective subfamilies.
Analysis of sequence divergence
The level of substitutions that accumulated in a PP (K(ψ)) was estimated using the following method.
First, the sequence divergence value (D) was corrected by removing the contribution of mutations at CpG sites. Sequence divergence (δ) of a sequence (at a given time point) of length (N) including the number of CpG dinucleotides (n) is given as a function of the mutation rate at non-CpG dinucleotides (α) and CpG dinucleotides (β) as follows:
δ = α(1/2 - n/N) + βn/N (1)
From the result of Sved and Bird [33], the ratio of β to α is ∼ 6.5. Therefore, designating α/2 = μ and n/N = ν in Equation 1 gives the following:
δ = μ(1 + 11ν) (2)
Assuming that CpG frequency (ι) in a genomic element that was generated by duplication of a functional gene of high CpG content decreases over time (t) and reaches an equilibrium state (ε) (approximately 20% of the frequency [14,33] expected from the local fraction of cytosines and guanosines [14]), the CpG frequency (ν) at time (t) was calculated as follows:
ν = 1/(At + 1/(ι - ε)) + ε (3)
If we accept the value of 1.5 × 10-9 per nucleotide per year [34,35] as the neutral mutation rate [41] and equate this to the mutation rate at non-CpG dinucleotides (μ) and use a time unit of 1 million years, then the mutation rate at CpG dinucleotides, β/2, will be around 1 per 100 nucleotides per million years (that is, ν will be reduced by 1% every million years). Therefore, ν(t = 1)/ν(t = 0) in Equation 3 gives:
(1/(A + 1/(ι - ε)) + ε)/ι ≈ 0.99
Solving for A gives:
A = 0.01ι/((0.99ι - ε)(ι - ε)) (4)
The sequence divergence value (D) is given as an integral of the sequence divergence (δ) from the present (t = 0) to the time of the duplication (t = T): D = ∫0Tδdt. From Equations 2, 3 and 4,
D = ∫0Tμ(1 + 11((ι - ε)/((0.01ι/(0.99ι - ε))t + 1) + ε))dt (5)
Solving Equation 5 for T gives the time since the duplication. The following ι and ε values (ι, ε, respectively) were used for the retroposons shown [14,57]:
Alu (0.077, 0.020); L1 (0.012, 0.008); PPs (0.015, 0.010)
The substitution level (Σ) at sites other than CpG is given from the time since the duplication (T) and the neutral mutation rate (μ) of primates [41]: Σ = μT. The quantity Σ was corrected for multiple substitutions at the same site using the Jukes-Cantor model [34], giving the average number of substitutions per 100 bp (K): K = - (3/4)ln(1 - (4/3)Σ).
For PPs, sequence divergences were defined as the mismatch rates of respective PPs relative to the current sequences of their parental genes. The mismatch rate of a PP relative to its parental gene (K) consists of the level of substitutions that accumulated only in the PP (K(ψ)) and the level of substitutions that accumulated only in the gene (K(f)): K = K(f) + K(ψ). K(f) and K(ψ) can be further subdivided into the number of synonymous (Ks) and nonsynonymous (Ka) substitutions [58-60]: K(f) = Ks(f) + Ka(f), K(ψ) = Ks(ψ) + Ka(ψ). Kuma and Miyata evaluated the average nucleotide substitution rates of 31 pairs of human PPs and their parental genes using homologs of other species as outgroups (K. Kuma and T. Miyata, personal communication). They used the following genes: ADP-ribosylation factor 1, aldolase A, aldose reductase, alpha-E-catenin, alpha-L-fucosidase, alpha-enolase, arylamine N-acetyltransferase, beta-tubulin, c-Raf protooncogene, cAMP-dependent protein kinase regulatory subunit, calmodulin, ceruloplasmin, creatine kinase, cyclophilin, cytochrome b5, cytochrome c, ferrochelatase, gamma-actin, glucocerebrosidase, glutamine synthetase, glyceraldehyde-3-phosphate dehydrogenase, histone H3.3, hsc70, hsp27, hsp60, lactate dehydrogenase-A, neurotrophin-4, phosphoglycerate kinase, prothymosine alpha, topoisomerase-I, triose phosphate isomerase. They calculated the following ratios: Rs(ψ), the synonymous substitutions in PPs to synonymous substitutions in their parental genes; Ra(f), the ratio of nonsynonymous substitutions in genes to synonymous substitutions in genes; and Ra(ψ), the ratio of nonsynonymous substitutions in PPs to synonymous substitutions in genes. The mean values of Rs(ψ), Ra(ψ) and Ra(f) were:
Rs(ψ) = Ks(ψ)/Ks(f) = 1.40 (6.1)
Ra(ψ) = Ka(ψ)/Ks(f) = 1.13 (6.2)
Ra(f) = Ka(f)/Ks(f) = 0.06 (6.3)
From Equations 6.1-6.3, and Equations K = K(f) + K(ψ), K(f) = Ks(f) + Ka(f), and K(ψ) = Ks(ψ) + Ka(ψ), the estimated level of substitutions in PPs (K(ψ)) is given by:
K(ψ) = 0.705K (7)
Additional data files
A table showing mapping coordinates for human PPs (Additional data file 1) and a list of the human genes that generated PPs (Additional data file 2) are available.
Supplementary Material
A table showing mapping coordinates for human PPs
A list of the human genes that generated PPs
Acknowledgments
Acknowledgements
We thank Katsuhiko Murakami (RIKEN-GSC) for helpful discussions and Kei-ichi Kuma and Takashi Miyata (Kyoto University) for providing the data on the average nucleotide substitution rates of 31 pairs of human PPs. This work was partially supported by the Ministry of Education, Culture, Sports, Science and Technology of Japan, Grant-in-Aid for Scientific Research. This work was also supported by a grant from BIRD of Japan Science and Technology Corporation (JST) for K.O.
References
- Vanin EF. Processed pseudogenes: characteristics and evolution. Annu Rev Genet. 1985;19:253–272. doi: 10.1146/annurev.ge.19.120185.001345. [DOI] [PubMed] [Google Scholar]
- Mighell AJ, Smith NR, Robinson PA, Markham AF. Vertebrate pseudogenes. FEBS Lett. 2000;468:109–114. doi: 10.1016/s0014-5793(00)01199-6. [DOI] [PubMed] [Google Scholar]
- Gonçalves I, Duret L, Mouchiroud D. Nature and structure of human genes that generate retropseudogenes. Genome Res. 2000;10:672–678. doi: 10.1101/gr.10.5.672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison PM, Hegyi H, Balasubramanian S, Luscombe NM, Bertone P, Echols N, Johnson T, Gerstein M. Molecular fossils in the human genome: identification and analysis of the pseudogenes in chromosomes 21 and 22. Genome Res. 2002;12:272–280. doi: 10.1101/gr.207102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Gentles AJ, Jurka J, Karlin S. Genes, pseudogenes, and Alu sequence organization across human chromosomes 21 and 22. Proc Natl Acad Sci USA. 2002;99:2930–2935. doi: 10.1073/pnas.052692099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosius J. RNAs from all categories generate retrosequences that may be exapted as novel genes or regulatory elements. Gene. 1999;238:115–134. doi: 10.1016/s0378-1119(99)00227-9. [DOI] [PubMed] [Google Scholar]
- Lahn BT, Page DC. Retroposition of autosomal mRNA yielded testis-specific gene family on human Y chromosome. Nat Genet. 1999;21:429–433. doi: 10.1038/7771. [DOI] [PubMed] [Google Scholar]
- Betrán E, Wang W, Jin L, Long M. Evolution of the phosphoglycerate mutase processed gene in human and chimpanzee revealing the origin of a new primate gene. Mol Biol Evol. 2002;19:654–663. doi: 10.1093/oxfordjournals.molbev.a004124. [DOI] [PubMed] [Google Scholar]
- Weiner AM, Deininger PL, Efstratiadis A. Nonviral retroposons: genes, pseudogenes, and transposable elements generated by the reverse flow of genetic information. Annu Rev Biochem. 1986;55:631–661. doi: 10.1146/annurev.bi.55.070186.003215. [DOI] [PubMed] [Google Scholar]
- Okada N. SINEs: Short interspersed repeated elements of the eukaryotic genome. Trends Ecol Evol. 1991;6:358–361. doi: 10.1016/0169-5347(91)90226-N. [DOI] [PubMed] [Google Scholar]
- Smit AF. The origin of interspersed repeats in the human genome. Curr Opin Genet Dev. 1996;6:743–748. doi: 10.1016/s0959-437x(96)80030-x. [DOI] [PubMed] [Google Scholar]
- Okada N, Hamada M, Ogiwara I, Ohshima K. SINEs and LINEs share common 3' sequences: a review. Gene. 1997;205:229–243. doi: 10.1016/s0378-1119(97)00409-5. [DOI] [PubMed] [Google Scholar]
- Weiner AM. SINEs and LINEs: the art of biting the hand that feeds you. Curr Opin Cell Biol. 2002;14:343–350. doi: 10.1016/s0955-0674(02)00338-1. [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Moran JV, Holmes SE, Naas TP, DeBerardinis RJ, Boeke JD, Kazazian HH., Jr High frequency retrotransposition in cultured mammalian cells. Cell. 1996;87:917–927. doi: 10.1016/s0092-8674(00)81998-4. [DOI] [PubMed] [Google Scholar]
- Kazazian HH, Jr, Moran JV. The impact of L1 retrotransposons on the human genome. Nat Genet. 1998;19:19–24. doi: 10.1038/ng0598-19. [DOI] [PubMed] [Google Scholar]
- Jurka J. Sequence patterns indicate an enzymatic involvement in integration of mammalian retroposons. Proc Natl Acad Sci USA. 1997;94:1872–1877. doi: 10.1073/pnas.94.5.1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esnault C, Maestre J, Heidmann T. Human LINE retrotransposons generate processed pseudogenes. Nat Genet. 2000;24:363–367. doi: 10.1038/74184. [DOI] [PubMed] [Google Scholar]
- Wei W, Gilbert N, Ooi SL, Lawler JF, Ostertag EM, Kazazian HH, Boeke JD, Moran JV. Human L1 retrotransposition: cis preference versus trans complementation. Mol Cell Biol. 2001;21:1429–1439. doi: 10.1128/MCB.21.4.1429-1439.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlícek A, Paces J, Elleder D, Hejnar J. Processed pseudogenes of human endogenous retroviruses generated by LINEs: their integration, stability, and distribution. Genome Res. 2002;12:391–399. doi: 10.1101/gr.216902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewannieux M, Esnault C, Heidmann T. LINE-mediated retrotransposition of marked Alu sequences. Nat Genet. 2003;35:41–48. doi: 10.1038/ng1223. [DOI] [PubMed] [Google Scholar]
- Britten RJ. Evidence that most human Alu sequences were inserted in a process that ceased about 30 million years ago. Proc Natl Acad Sci USA. 1994;91:6148–6150. doi: 10.1073/pnas.91.13.6148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitonov V, Jurka J. The age of Alu subfamilies. J Mol Evol. 1996;42:59–65. doi: 10.1007/BF00163212. [DOI] [PubMed] [Google Scholar]
- Sarrowa J, Chang DY, Maraia RJ. The decline in human Alu retroposition was accompanied by an asymmetric decrease in SRP9/14 binding to dimeric Alu RNA and increased expression of small cytoplasmic Alu RNA. Mol Cell Biol. 1997;17:1144–1151. doi: 10.1128/mcb.17.3.1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid CW. Does SINE evolution preclude Alu function? Nucleic Acids Res. 1998;26:4541–4550. doi: 10.1093/nar/26.20.4541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batzer MA, Deininger PL. Alu repeats and human genomic diversity. Nat Rev Genet. 2002;3:370–379. doi: 10.1038/nrg798. [DOI] [PubMed] [Google Scholar]
- Boeke JD. LINEs and Alus - the polyA connection. Nat Genet. 1997;16:6–7. doi: 10.1038/ng0597-6. [DOI] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Hubbard T, Barker D, Birney E, Cameron G, Chen Y, Clark L, Cox T, Cuff J, Curwen V, Down T, et al. The Ensembl genome database project. Nucleic Acids Res. 2002;30:38–41. doi: 10.1093/nar/30.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogenesch JB, Ching KA, Batalov S, Su AI, Walker JR, Zhou Y, Kay SA, Schultz PG, Cooke MP. A comparison of the Celera and Ensembl predicted gene sets reveals little overlap in novel genes. Cell. 2001;106:413–415. doi: 10.1016/s0092-8674(01)00467-6. [DOI] [PubMed] [Google Scholar]
- Hesse M, Magin TM, Weber K. Genes for intermediate filament proteins and the draft sequence of the human genome: novel keratin genes and a surprisingly high number of pseudogenes related to keratin genes 8 and 18. J Cell Sci. 2001;114:2569–2575. doi: 10.1242/jcs.114.14.2569. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Harrison P, Gerstein M. Identification and analysis of over 2000 ribosomal protein pseudogenes in the human genome. Genome Res. 2002;12:1466–1482. doi: 10.1101/gr.331902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sved J, Bird A. The expected equilibrium of the CpG dinucleotide in vertebrate genomes under a mutation model. Proc Natl Acad Sci USA. 1990;87:4692–4696. doi: 10.1073/pnas.87.12.4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graur D, Li W-H. Fundamentals of Molecular Evolution. 2. Sunderland, MA: Sinauer Associates; 2000. [Google Scholar]
- Nachman MW, Crowell SL. Estimate of the mutation rate per nucleotide in humans. Genetics. 2000;156:297–304. doi: 10.1093/genetics/156.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoshani J, Groves CP, Simons EL, Gunnell GF. Primate phylogeny: morphological vs. molecular results. Mol Phylogenet Evol. 1996;5:102–154. doi: 10.1006/mpev.1996.0009. [DOI] [PubMed] [Google Scholar]
- Kay RF, Ross C, Williams BA. Anthropoid origins. Science. 1997;275:797–804. doi: 10.1126/science.275.5301.797. [DOI] [PubMed] [Google Scholar]
- Smit AF, Tóth G, Riggs AD, Jurka J. Ancestral, mammalian-wide subfamilies of LINE-1 repetitive sequences. J Mol Biol. 1995;246:401–417. doi: 10.1006/jmbi.1994.0095. [DOI] [PubMed] [Google Scholar]
- Boissinot S, Furano AV. Adaptive evolution in LINE-1 retrotransposons. Mol Biol Evol. 2001;18:2186–2194. doi: 10.1093/oxfordjournals.molbev.a003765. [DOI] [PubMed] [Google Scholar]
- Yoshihama M, Uechi T, Asakawa S, Kawasaki K, Kato S, Higa S, Maeda N, Minoshima S, Tanaka T, Shimizu N, Kenmochi N. The human ribosomal protein genes: sequencing and comparative analysis of 73 genes. Genome Res. 2002;12:379–390. doi: 10.1101/gr.214202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. The Neutral Theory of Molecular Evolution. Cambridge: Cambridge University Press; 1983. [Google Scholar]
- Takahata N. Molecular phylogeny and demographic history of humans. In: Tobias PV, Raath MA, Moggi-Cecchi J, Doyle GA, editor. In Humanity from African Naissance to Coming Millennia. Firenze: Firenze University Press; 2001. pp. 299–305. [Google Scholar]
- Kajikawa M, Okada N. LINEs mobilize SINEs in the eel through a shared 3' sequence. Cell. 2002;111:433–444. doi: 10.1016/s0092-8674(02)01041-3. [DOI] [PubMed] [Google Scholar]
- Ivics Z, Hackett PB, Plasterk RH, Izsvák Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell. 1997;91:501–510. doi: 10.1016/s0092-8674(00)80436-5. [DOI] [PubMed] [Google Scholar]
- Chou H-H, Hayakawa T, Diaz S, Krings M, Indriati E, Leakey M, Paabo S, Satta Y, Takahata N, Varki A. Inactivation of CMP-N-acetylneuraminic acid hydroxylase occurred prior to brain expansion during human evolution. Proc Natl Acad Sci USA. 2002;99:11736–11741. doi: 10.1073/pnas.182257399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazazian HH., Jr An estimated frequency of endogenous insertional mutations in humans. Nat Genet. 1999;22:130. doi: 10.1038/9638. [DOI] [PubMed] [Google Scholar]
- Zhang J, Rosenberg HF, Nei M. Positive Darwinian selection after gene duplication in primate ribonuclease genes. Proc Natl Acad Sci USA. 1998;95:3708–3713. doi: 10.1073/pnas.95.7.3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suga H, Koyanagi M, Hoshiyama D, Ono K, Iwabe N, Kuma K, Miyata T. Extensive gene duplication in the early evolution of animals before the parazoan-eumetazoan split demonstrated by G proteins and protein tyrosine kinases from sponge and hydra. J Mol Evol. 1999;48:646–653. doi: 10.1007/pl00006508. [DOI] [PubMed] [Google Scholar]
- Gu X, Wang Y, Gu J. Age distribution of human gene families shows significant roles of both large- and small-scale duplications in vertebrate evolution. Nat Genet. 2002;31:205–209. doi: 10.1038/ng902. [DOI] [PubMed] [Google Scholar]
- Kent WJ, Haussler D. Assembly of the working draft of the human genome with GigAssembler. Genome Res. 2001;11:1541–1548. doi: 10.1101/gr.183201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ. BLAT - the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori M, Fujiyama A, Taylor TD, Watanabe H, Yada T, Park HS, Toyoda A, Ishii K, Totoki Y, Choi DK, et al. The DNA sequence of human chromosome 21. Nature. 2000;405:311–319. doi: 10.1038/35012518. [DOI] [PubMed] [Google Scholar]
- Human Genome Research Group: Chromosome 21 http://hgp.gsc.riken.go.jp/data_tools/chr21.html
- Dunham I, Shimizu N, Roe BA, Chissoe S, Hunt AR, Collins JE, Bruskiewich R, Beare DM, Clamp M, Smink LJ, et al. The DNA sequence of human chromosome 22. Nature. 1999;402:489–495. doi: 10.1038/990031. [DOI] [PubMed] [Google Scholar]
- Human chromosome 22 project overview http://www.sanger.ac.uk/HGP/Chr22
- UCSC genome bioinformatics http://www.genome.ucsc.edu
- Repbase update http://www.girinst.org/Repbase_Update.html
- Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- Ophir R, Itoh T, Graur D, Gojobori T. A simple method for estimating the intensity of purifying selection in protein-coding genes. Mol Biol Evol. 1999;16:49–53. doi: 10.1093/oxfordjournals.molbev.a026037. [DOI] [PubMed] [Google Scholar]
- Bustamante CD, Nielsen R, Hartl DL. A maximum likelihood method for analyzing pseudogene evolution: implications for silent site evolution in humans and rodents. Mol Biol Evol. 2002;19:110–117. doi: 10.1093/oxfordjournals.molbev.a003975. [DOI] [PubMed] [Google Scholar]
- NCBI Reference sequences http://www.ncbi.nlm.nih.gov/RefSeq/
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A table showing mapping coordinates for human PPs
A list of the human genes that generated PPs