

Abstract
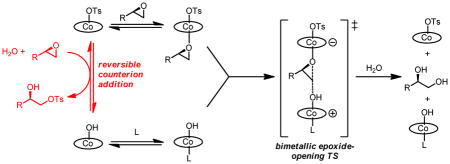
The (salen)Co(III)-catalyzed hydrolytic kinetic resolution (HKR) of terminal epoxides is a bimetallic process with a rate controlled by partitioning between a nucleophilic (salen)Co–OH catalyst and a Lewis acidic (salen)Co–X catalyst. The commonly used (salen)Co–OAc and (salen)Co–Cl precatalysts undergo complete and irreversible counterion addition to epoxide during the course of the epoxide hydrolysis reaction, resulting in quantitative formation of weakly Lewis acidic (salen)Co–OH, and severely diminished reaction rates in the late stages of HKR reactions. In contrast, (salen)Co–OTs maintains high reactivity over the entire course of HKR reactions. We describe here an investigation of catalyst partitioning with different (salen)Co–X precatalysts, and demonstrate that counterion addition to epoxide is reversible in the case of the (salen)Co–OTs. This reversible counterion addition results in stable partitioning between nucleophilic and Lewis acidic catalyst species, allowing highly efficient catalysis throughout the course of the HKR reaction.
Introduction
The (salen)Co(III)-catalyzed hydrolytic kinetic resolution (HKR) of terminal epoxides1 has become an important method for the synthesis of enantiomerically pure epoxides (Scheme 1), with widespread application in both academic and industrial settings.2 Hydrolysis of a wide range of terminal epoxides occurs at room temperature and neutral pH with krel > 50 for almost all substrates examined,1b making this system intrinsically interesting from a mechanistic standpoint. Our interest in elucidating the mechanism of catalysis is further motivated by practical considerations, as such understanding may point the way to improved catalysts with even greater reactivity and scope, thereby providing access to useful chiral building blocks at lower costs. In addition, (salen)metal(III) complexes promote many related reactions with high stereoselectivity—including epoxide ring-opening with other nucleophiles,3,4 oxetane ring-opening,5 epoxide polymerization,6 and epoxide/CO2 co-polymerization7—and thus the mechanistic principles acquired from the study of epoxide hydrolysis may also shed light on the mechanisms of other important reactions.
Scheme 1.

Hydrolytic kinetic resolution of terminal epoxides catalyzed by (salen)Co(III) complexes
A preliminary mechanistic investigation of the HKR reported in 2004 concluded with the proposal outlined in Scheme 2.8 The central feature of this mechanism is the cooperative action of two different (salen)Co(III) units in the rate-determining epoxide ring-opening event. This proposal was based on several experimental observations. The reaction displays a second-order kinetic dependence on total (salen)Co(III) concentration (Figure 1).9 Reactions promoted by (salen)Co–X precatalysts (X = OAc, Cl) display a discernible induction period, and epoxide ring-opening products corresponding to H–X addition to the epoxide are generated in high enantiomeric excess during the early stages of the reaction. These observations are consistent with (salen)Co–X alone being inactive for epoxide hydrolysis, but undergoing conversion to nucleophilic (salen)Co–OH through a mechanism that involves counterion addition to epoxide followed by hydrolysis of the resulting (salen)Co(III) alkoxide (Scheme 2). Using (salen)Co–Cl as a precatalyst, counterion addition also proceeds rapidly in the absence of water,10 and the resulting complex forms the catalytically active (salen)Co–OH complex 1b quantitatively upon addition of water.11
Scheme 2.

Proposed mechanism of catalysis for HKR reactions catalyzed by mixtures of (salen)Co–X and (salen)Co–OH
Figure 1.

Rate dependence of epoxide hydrolysis on [cat]tot 2 and %-conversion. Plots of the rate of hydrolysis of (R)-1,2-epoxyhexane ([epoxide]i = 5.63 M) versus [(S,S)-(salen)Co–OH]2 in 1,2-hexanediol ([diol]i = 2.18 M) at different %-conversion of water ([H2O]i = 2.82 M). The catalyst was generated by aging (S,S)-(salen)Co–Cl in epoxide for 1 h prior to addition of water. The black curves represent least-squares fits to f(x) = a x, 20 % conversion, a = 1.92 ± 0.01 M−1 s−1; 50 % conversion, a = 1.314 ± 0.009 M−1 s−1; 80 % conversion, a = 0.597 ± 0.006 M−1 s−1.
Both the length of the induction period and the amount of time required for full conversion depend on the identity of the counterion X on the (salen)Co(III) precatalyst (Figure 2).12 In the case of (salen)Co–Cl precatalyst 1c, a short induction period is observed, consistent with rapid generation of (salen)Co–OH. The rate of the HKR reaches a maximum within 2 minutes, but then diminishes rapidly and the overall reaction requires nearly 12 h before reaching 95% conversion. In the case of (salen)Co–OAc precatalyst 1a, the induction period is longer and the rate diminishes more slowly; still, low reactivity is observed within 1 hour and 95% conversion is achieved only after 8 h.13 In sharp contrast, (salen)Co–OTs precatalyst 1d displays an induction period qualitatively similar to that observed with 1c, but the rate remains high throughout the course of the reaction and the reaction is complete in well under 1 h.
Figure 2.

Rates of epoxide hydrolysis with different (salen)Co(III) precatalysts. Plot of the rates of hydrolysis of (S)-1,2-epoxyhexane ([epoxide]i = 6.0 M) in 1,2-hexanediol versus time in 1,2-hexanediol ([H2O]i = 3.4 M). In each experiment, (R,R)-(salen)Co–X (0.15 mol %) was added to a reaction mixture containing epoxide, diol, and water as an 83.0 mM CH2Cl2 solution. The indicated times represent the length of time required to achieve 95% conversion of water with each precatalyst.
This remarkable counterion effect clearly demonstrates that (salen)Co–OH is not the only catalytically active species in the HKR, and is consistent with the two distinct mechanistic roles proposed for (salen)Co(III) in Scheme 2: that of nucleophile delivery agent as (salen)Co–OH, and as Lewis acid for epoxide activation. The Lewis acidity of the catalyst depends on the identity of the counterion, X, with highest reactivity expected with the least coordinating counterions. Because hydroxide is more coordinating than the other counterions (i.e., Cl, OAc, OTs), reactions catalyzed by (salen)Co–OH alone are relatively slow. However, a much faster rate of epoxide hydrolysis can occur during the stages of the reaction before counterion addition is complete, when both (salen)Co–OH and (salen)Co–X (X = Cl, OAc, OTs) are present. The rate of counterion addition controls the rate of (salen)Co–OH formation, thereby influencing both the length of the induction period and of the period where both (salen)Co–OH and (salen)Co–X coexist and can react in a cooperative manner.
This analysis led to an experimentally testable hypothesis: for a given total (salen)Co(III) concentrations, maximal overall rates of epoxide hydrolysis are expected when the nucleophilic (salen)Co–OH catalyst and a highly Lewis acidic (salen)Co–X are present in a 1:1 ratio for the entire course of the reaction. Constant ratios of the two catalysts could be established by employing mixtures of (salen)Co(III) complexes in which the counterion of one of the complexes was sufficiently nonnucleophilic so that counterion addition did not occur. This scenario was achieved using (salen)Co–SbF6, which contains a non-transferable counterion, in combination with (salen)Co–OH generated quantitatively from (salen)Co–Cl. In this manner, ratios of the two catalysts could be varied, with the total concentration of (salen)Co(III) held constant. In accord with the mechanism outlined in Scheme 2, the reaction rate displays a parabolic dependence on catalyst partitioning, and the 1:1 mixture of (salen)Co–OH and (salen)Co–SbF6 represents the most active catalyst combination (Figure 3). This experiment demonstrated that optimal catalyst partitioning—a 1:1 mixture of a nucleophilic and a Lewis acidic catalyst—is the key to achieving optimal reactivity in the HKR.14
Figure 3.

Plot of the maximum rate hydrolysis of (S)-1,2-epoxyhexane as a function of different ratios of (salen)Co–OH and (salen)Co–SbF6, at a constant total [(salen)Co(III)]. The (salen)Co–OH is generated by premixing (salen)Co–Cl and epoxide followed by addition of water. The curve represents a least-squares fit to f(x) = a x2 + b x + c, a = −1620 ± 60, b = 1540 ± 60, c = 30 ± 10 mJ s−1.
The behavior of the (salen)Co–Cl and (salen)Co–OAc precatalysts is readily explained according to this analysis and the mechanism in Scheme 2. The rate of epoxide hydrolysis increases until a 1:1 ratio of (salen)Co–OH and (salen)Co–X is attained, but then decreases until ultimately all the catalyst exists as (salen)Co–OH. The reaction then continues in the much slower all (salen)Co–OH manifold until consumption of the limiting reagent (water, in these experiments) is complete. The behavior of the (salen)Co–OTs precatalyst is strikingly different, however, in that the slower, all (salen)Co–OH manifold is never reached. In this paper we describe a detailed kinetic study to probe mechanistic aspects of (salen)Co(III) partitioning by counterion addition, and conclude that the basic mechanism of partitioning by (salen)Co–OTs is fundamentally different compared with those of other precatalysts: the tosylate counterion undergoes reversible rather than irreversible counterion addition. This conclusion has an important practical consequence: the use of (salen)Co–OTs allows for the in situ generation of an approximately 1:1 mixture of nucleophilic and Lewis acidic catalysts that is maintained over the entire course of the reaction, thereby avoiding the need for two different precatalysts or high loadings of a single less efficient catalyst.
Results
A. Evaluation of catalyst partitioning: delayed addition experiments
To probe the effect of catalyst partitioning in the HKR, we sought to quantify the rate of counterion addition with different (salen)Co–X precatalysts. Because the catalyst loading in the HKR is typically very low (e.g., 0.15 mol% in the experiments described here), the counterion addition product represents only a small component of the reaction mixture (eq 1), and is thus difficult to quantify accurately via spectroscopic methods. Although isolation of the counterion addition product from HKR reaction mixtures is possible,8 doing so requires separation from large excess of epoxide and diol, and is therefore not practical for kinetic analysis.
 |
(1) |
Instead, we devised an indirect approach to study the rate of counterion addition in HKR reactions, in which the rate of epoxide hydrolysis could be used as a probe for the amount of counterion addition (Scheme 3). In these experiments, a solution of enantiomerically pure 1,2-epoxyhexane and racemic 1,2-hexanediol was combined with a solution of (salen)Co–X in CH2Cl2 (0.15 mol % relative to epoxyhexane).15 The resulting solution was aged for a specific time period of between 0 and 180 min, during which time counterion addition to generate alkoxide complexes 1f–1h and the analogous diolate complex 1i is assumed to occur to some extent. Water was then added directly to the reaction mixture, thereby initiating epoxide hydrolysis by generating (salen)Co–OH. The rate of epoxide hydrolysis was monitored by reaction calorimetry until reaction was complete, as evidenced by the heat flow returning to near-background level.16
Scheme 3.

Delayed-addition of water to probe catalyst partitioning.
The raw heat flow versus time data were then converted to rate versus conversion by integration of the experimental data.17 Analysis of the data in this format allows for direct rate comparisons between different reaction conditions: at any given amount of conversion, the concentrations of epoxide, diol and water are identical for all curves, so rate differences are due entirely to differences in catalyst concentrations. Because the total concentration of (salen)Co(III) is the same in each experiment as well, the only difference between these experiments that is expected to have an impact on reaction rate is the precise partitioning of the catalyst between (salen)Co–X and (salen)Co–OH. Thus, if two rate versus conversion curves are not superimposable for all or part of the course of an HKR experiment, then the composition of the catalyst mixture can be concluded to be different in the two experiments for all or part of the reaction. Conversely, if two curves are superimposable for the entire course of an HKR experiment, then the catalysts in the two experiments are kinetically indistinguishable—and likely of identical composition—during the course of the experiment.18,19
Control experiments reveal that addition of CH2Cl2 is accompanied by a small but reproducible positive heat of mixing, whereas addition of water is accompanied by a small but reproducible negative heat of mixing (Figure 4). Because these heats of mixing are negligible relative to the overall enthalpy of epoxide hydrolysis,20 we have not corrected for this effect. However, the positive or negative spikes in heat observed during the first few minutes of each experiment may be ascribed to heat of mixing rather than to epoxide hydrolysis.21
Figure 4.

Heat of mixing experiments showing the heat flow spikes due to addition of H2O (85 μL) and CH2Cl2 (150 μL) to a solution of 1,2-epoxyhexane (1.00 mL) and 1,2-hexanediol (300 μL). Nearly identical effects are observed when the order of addition is reversed.
Results from delayed-addition experiments with (salen)Co–Cl, (salen)Co–OAc, and (salen)Co–OTs are depicted in Figures 5–7. The delay times shown on each plot are identical: 0, 15, 30, 60, 120, and 180 min.22,23 Direct comparison between the three precatalysts with delay times of 0 min and 180 min are provided in Figures 8 and 9.24
Figure 5.

Rate dependence on the time between addition of (salen)Co–Cl precatalyst and water to a solution of epoxide. Plot of the rates of hydrolysis of (S)-1,2-epoxyhexane ([epoxide]i = 5.4 M) in 1,2-hexanediol versus conversion of water ([H2O]i = 3.1 M) in 1,2-hexanediol. In each experiment, (R,R)-(salen)Co–Cl (0.15 mol %) was added to the reaction mixture as an 83 mM CH2Cl2 solution; water was added subsequently after the indicated delay time.
Figure 7.

Rate dependence on the time between addition of (salen)Co–OTs and water to a solution of epoxide. Plot of the rates of hydrolysis of (S)-1,2-epoxyhexane ([epoxide]i = 5.4 M) in 1,2-hexanediol versus conversion of water ([H2O]i = 3.1 M) in 1,2-hexanediol. In each experiment, (R,R)-(salen)Co–OTs (0.15 mol %) was added to the reaction mixture as an 83 mM CH2Cl2 solution; water was added subsequently after the indicated delay time.
Figure 8.

Rate dependence on the nature of the counterion, X, in HKR experiments in which catalyst was added as the last reagent (i.e., 0 min delay time). Plot of the rates of hydrolysis of (S)-1,2-epoxyhexane ([epoxide]i = 5.4 M) in 1,2-hexanediol versus time in 1,2-hexanediol ([H2O]i = 3.1 M). In each experiment, (R,R)-(salen)Co–X (0.15 mol %) was added to a reaction mixture containing epoxide, diol, and water as an 83 mM CH2Cl2 solution.
Figure 9.

Rate dependence on the nature of the counterion, X, with a 180 min delay time between catalyst and water addition. Plot of the rates of hydrolysis of (S)-1,2-epoxyhexane ([epoxide]i = 5.4 M) in 1,2-hexanediol versus conversion of water ([H2O]i = 3.1 M) in 1,2-hexanediol. In each experiment, (R,R)-(salen)Co–OTs (0.15 mol %) was added to the reaction mixture as an 83 mM CH2Cl2 solution; water was added subsequently after the indicated delay time.
Analysis of rate versus conversion plots derived from experiments using (salen)Co–Cl confirms that this catalyst is initially highly active, but that it rapidly loses most of its activity, both during the course of the HKR reaction or by aging with epoxide for 15 min (Figure 5). Experiments using delay times of 60–180 min yield rate versus conversion curves that are superimposable for the entire course of the reaction, and all six curves are superimposable from approximately 40–100 % conversion. These data conform to the mechanistic model proposed in Scheme 2, in which the epoxide undergoes rapid and quantitative counterion addition to form (salen)Co–OH.25
A slightly different picture emerges from an analysis of experiments using (salen)Co–OAc (Figures 6 and 8). In experiments with short delay times, the maximum rate is achieved only after a substantial amount of water has been consumed. Nevertheless, experiments using delay times of 120 and 180 min yield rate versus conversion curves that are superimposable for the entire course of the reaction, and all curves are superimposable from approximately 80–100 % conversion. In addition, the curves derived from 180 min delay times of (salen)Co–Cl and (salen)Co–OAc are nearly superimposable for the entire course of the reaction (Figure 9). These data are thus also in agreement with the mechanistic model proposed in Scheme 2, in which the epoxide undergoes complete counterion addition to form (salen)Co–OH upon addition of water. However, this process is somewhat slower than with (salen)Co–Cl, requiring approximately 120 min in the absence of water.26
Figure 6.

Rate dependence on the time between addition of (salen)Co–OAc and water to a solution of epoxide. Plot of the rates of hydrolysis of (S)-1,2-epoxyhexane ([epoxide]i = 5.4 M) in 1,2-hexanediol versus conversion of water ([H2O]i = 3.1 M) in 1,2-hexanediol. In each experiment, (R,R)-(salen)Co–OAc (0.15 mol %) was added to the reaction mixture as an 83 mM CH2Cl2 solution; water was added subsequently after the indicated delay time.
As illustrated in Figures 8 and 9, the kinetic behavior of (salen)Co–OTs in delayed-addition experiments differs substantially from those of the (salen)Co–Cl and (salen)Co–OAc precatalysts. Epoxide hydrolysis with (salen)Co–OTs induced with a 0 min delay time before addition of water reaches its maximum rate at approximately 50% conversion, but the curve derived from this experiment never becomes superimposable with the analogous curves derived from (salen)Co–Cl or (salen)Co–OAc (Figure 8). In addition, this catalyst retains most of its activity with long delay times, and remains highly active for the entire course of the reaction in all cases (Figure 7).27 These data provide strong evidence that (salen)Co–OTs never undergoes complete conversion to (salen)Co–OH during the course of epoxide hydrolysis reactions.
B. Effect of tosylate addition product on the rate of epoxide hydrolysis
There are two alternative, straightforward explanations for the observation that complete counterion addition occurs with (salen)Co–Cl and (salen)Co–OAc, but not with (salen)Co–OTs: either tosylate counterion addition might simply occur at a much slower rate than chloride or acetate counterion addition, or tosylate counterion addition might be reversible under the reaction conditions (eq 2). If counterion addition of (salen)Co–OTs to epoxide is reversible, then it should be possible to approach this equilibrium from the either direction. In other words, reaction of (salen)Co–OH and the counterion addition product 3d would be expected to generate (salen)Co–OTs and 1,2-epoxyhexane.
 |
(2) |
We were able to obtain clear evidence for the reversibility of tosylate addition by indirect methods. The data described above indicate that (salen)Co–OH catalyzes epoxide hydrolysis by a relatively slow second-order pathway in the absence of a more Lewis acidic co-catalyst, so any measurable increase in reaction rate due to added tosylate addition product 3d28 can be ascribed to in situ formation of (salen)Co–OTs via the equilibrium depicted in eq 2. In one set of experiments, (salen)Co–Cl was aged with (R)-1,2-epoxyhexane and 1,2-hexanediol for approximately 45 min to induce quantitative counterion addition (Scheme 4). Water was then added directly to the reaction mixture, thereby initiating formation of (salen)Co–OH. After a given time period, 0.15 mol % of 3d was added as a solution in CH2Cl2. The rate of epoxide hydrolysis was monitored by reaction calorimetry as described above.
Scheme 4.

Experiment to probe the viability of the equilibrium in eq 2.
In experiments in which no 3d is added, epoxide hydrolysis is slow (Figure 10, black curve), as is expected for the pure (salen)Co–OH pathway on the basis of the delayed addition experiments with (salen)Co–Cl described above (Figure 5).29 Addition of 3d 1 min after addition of water leads to a rate versus conversion curve that closely resembles the analogous curve in which (salen)Co–OTs is added as the last reagent (compare the 0 min curve in Figure 7 with the 1 min curve in Figure 10). In experiments in which 3d was added 40 min after addition of water, the rate of epoxide hydrolysis is also observed to increase immediately (Figure 10). The increase in reaction rate upon addition of 3d either near the beginning or during the course of the reaction may be ascribed to the in situ formation of (salen)Co–OTs via the equilibrium described by eq 2. Interestingly, both the maximal rate under these conditions (≈ 20 × 104 M s−1) and the time to reach the maximal rate (≈ 15 min) is the same as when (salen)Co–OTs is added as the last reagent (blue curve in Figure 8), indicating that near-optimal catalyst partitioning is achieved over the same time frame by entering the equilibrium in eq 2 from the left, using (salen)Co–OTs as the precatalyst; or from the right, using (salen)Co–OH as the precatalyst.
Figure 10.
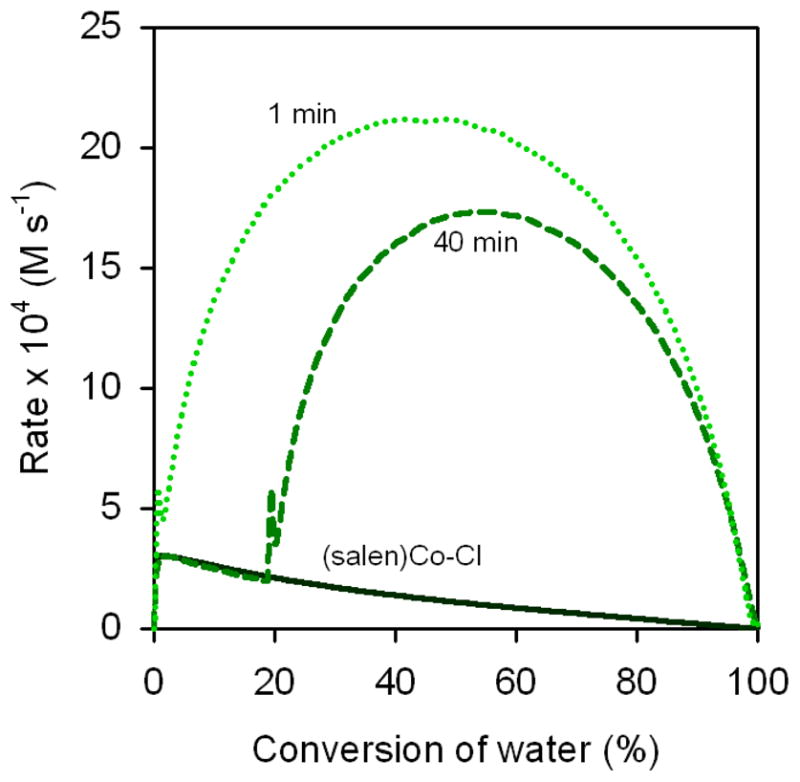
Rate dependence on delay time between addition of water and 3d. Plot of the rates of hydrolysis of (R)-1,2-epoxyhexane ([epoxide]i = 5.4 M) in 1,2-hexanediol versus conversion of water ([H2O]i = 3.1 M) in 1,2-hexanediol. In each experiment, (S,S)-(salen)Co–Cl (0.15 mol %) was added to the reaction mixture and aged for 45 min, followed by water; 3d (0.15 mol %) was added as a solution in CH2Cl2 subsequently after the indicated delay time. The black curve is derived from an experiment in which neither 3d nor CH2Cl2 was added.
The experiment described above provides evidence that an equilibrium between (salen)Co–OH and (salen)Co–OTs is established under the HKR reaction conditions in the presence of water (eq 2). In addition, the experiments described in Figure 7 suggest that a closely related equilibrium involving (salen)Co–OR (1h or 1i, Scheme 3) and (salen)Co–OTs might also be established before water is added. To test this idea, the effect of added tosylate addition product 3d on the rate of HKR reactions was analyzed using delayed-addition experiments analogous to the ones described above, but in the absence of water. In these studies, 0.15 mol % (salen)Co–Cl was aged with epoxide and diol for approximately 45 min to induce quantitative counterion addition (Scheme 5). At this point, 0.15 mol % of 3d was added as a solution in CH2Cl2, and the reaction mixture was aged for measured periods. Water was then added directly to the reaction mixture, thereby initiating epoxide hydrolysis. The rate of epoxide hydrolysis was monitored by reaction calorimetry as described above.
Scheme 5.

Accessing the (salen)Co–OTs/(salen)Co–OH equibrium from tosylate addition complex 1h.
As described above, addition of 3d as the last reagent water leads to a rate versus conversion curve with a 15 min induction period (Figure 11, 0 min curve).30 Addition of 3d with longer delay times decreases the length of the induction period; in each case, substantial rate acceleration is observed compared with the reaction catalyzed by (salen)Co–OH alone (Figure 11). These observations are consistent with generation of (salen)Co–OTs by addition of 3d in the absence of water. The maximal rates obtained from the two independent methods of generating mixtures of (salen)Co–OH and (salen)Co–OTs in situ are similar under these reaction conditions (≈ 25 × 10−4 M s−1), indicating that similar, nearly optimal catalyst partitioning can be achieved via either method.31,32
Figure 11.

Rate dependence on delay time between addition of 3d and water. Plot of the rates of hydrolysis of (R)-1,2-epoxyhexane ([epoxide]i = 5.4 M) in 1,2-hexanediol versus conversion of water ([H2O]i = 3.1 M) in 1,2-hexanediol. In each experiment, (S,S)-(salen)Co–Cl (0.15 mol %) was added to the reaction mixture and aged for 45 min, followed by 3d (0.15 mol %) as a solution in CH2Cl2; water was added subsequently after the indicated delay time. The black curve is derived from an experiment in which CH2Cl2 is added, but not 3d.
Discussion
The mechanism outlined in Scheme 2, together with the data presented in Figure 3, reveal that at a given total (salen)Co(III) concentration, optimal rates of epoxide hydrolysis are achieved when (salen)Co–OH and a more Lewis acid complex (salen)Co–X are present in equimolar concentrations. The (salen)Co–X precatalyst does not promote epoxide hydrolysis by itself, but rather is converted to the active nucleophile for hydrolysis, (salen)Co–OH, during the course of the reaction. However, epoxide hydrolysis catalyzed by (salen)Co–OH alone is relatively slow, a result of the relatively poor Lewis acidity of (salen)Co–OH. If generation of (salen)Co–OH from (salen)Co–X is irreversible and occurs before epoxide hydrolysis is complete, then dramatic decreases in reaction rate result. The practical consequences of this phenomenon are profound: under the conditions described in this paper, hydrolysis of 1,2-epoxyhexane catalyzed by (salen)Co–OAc requires 25 min to reach 50% conversion, but over 8 h to reach 95% conversion (Table 1).33 In other words, the second half of the reaction takes almost twenty times as long as the first half.
Table 1.
Epoxide hydrolysis reaction times required to achieve 50, 80, and 95% conversion of water with different (salen)Co(III) precatalysts, in experiments in which catalyst was added as the last reagent.
| Conversion of water (%) | Reaction time (min)
|
||
|---|---|---|---|
| (salen)Co–Cl | (salen)Co–OAc | (salen)Co–OTs | |
| 50 | 117 | 25 | 17 |
| 80 | 377 | 114 | 25 |
| 95 | 715 | 504 | 33 |
In kinetic resolutions in which recovery of starting material in high enantiomeric excess is desired, achieving high conversion is critical,34 and thus inefficient catalyst partitioning in reactions starting with (salen)Co–OAc presents a serious practical limitation. Nonetheless, this precatalyst has seen widespread use over the past decade,2 and the reactivity problems have been addressed simply by using increased catalyst loadings or long reaction times. This strategy introduces practical limitations, especially on large scales. One approach to increasing the efficiency of this reaction is suggested by Scheme 2: by using a mixture of precatalysts—one that undergoes fast counterion addition, and another that does not undergo counterion addition at all—it is possible to maintain a highly active catalyst for the entire course of the reaction. This approach has been successful (Figure 3), but it requires the synthesis and introduction of two separate catalysts.
The results described in this paper reveal a fundamentally different—and unexpected—solution to the problem of preparing a catalyst that remains highly active for the entire course of the reaction: (salen)Co–OTs undergoes rapid but reversible counterion addition to generate a balanced mixture of nucleophilic (salen)Co–OH and (salen)Co–OTs that is maintained over the entire course of the reaction (Figure 12). The epoxide hydrolysis reaction catalyzed by (salen)Co–OTs achieves 50% conversion of water in 17 min, and 95% conversion of water in 33 min. In other words, the first 50% conversion requires the same amount of time as the second 45% conversion (Table 1). Indeed, (salen)Co–OTs is the optimal monomeric catalyst for the HKR of a range of terminal epoxides.1d,8
Figure 12.

Catalyst partitioning in the (salen)Co–OTs-catalyzed HKR. The green and blue arrows represent two different methods of generating a highly active catalyst mixture.
During the course of our studies of (salen)Co(III)-catalyzed epoxide hydrolysis, we have identified and characterized four types of catalyst partitioning:35
Highly nucleophilic precatalysts such as (salen)Co–Cl lead to catalyst partitioning that rapidly reaches 100% (salen)Co–OH, leading to relatively inefficient epoxide ring-opening, because (salen)Co–OH is only weakly Lewis acidic.
Moderately nucleophilic precatalysts such as (salen)Co–OAc undergo slower conversion to (salen)Co–OH. This results in a longer period where both (salen)Co–OAc and (salen)Co–OH are present together and efficient cooperative catalysis takes place. However, complete counterion addition occurs eventually, and from that point slow epoxide hydrolysis due to catalysis by (salen)Co–OH alone is observed.
Mixtures of nucleophilic precatalysts such as (salen)Co–Cl with highly Lewis acidic and non-nucleophilic catalysts such as (salen)Co–SbF6 lead to an unchanging catalyst partitioning and to highly efficient catalysis throughout the course of the reaction
The weakly nucleophilic precatalyst (salen)Co–OTs never undergoes complete conversion to (salen)Co–OH because an equilibrium between (salen)Co–OTs and (salen)Co–OH is established. This scenario results in high rates throughout the course of the reaction.
Conclusions
Our efforts to elucidate the details of counterion addition from (salen)Co–X complexes to terminal epoxides have not only served to clarify key elementary steps in the HKR, but have also led to substantial practical improvements in this synthetically important method.1d,8 We have shown that attaining optimum reactivity in epoxide hydrolysis requires maintaining a balanced concentration of (salen)Co–OH and (salen)Co–X, where X is a weakly associated counterion that imparts high levels of Lewis acidity to the (salen)Co(III) complex. This balance can be achieved by combining two different precatalysts in equal amounts, one of which undergoes facile transformation to (salen)Co–OH—such as (salen)Co–Cl—and the other that does not undergo any counterion addition under the HKR conditions—such as (salen)Co–SbF6. However, a more straightforward solution is obtained using (salen)Co–OTs. In this study we have found that the exceptionally high reactivity of (salen)Co–OTs in the HKR can be traced to a remarkable phenomenon in which this complex enters into an epoxide-mediated equilibrium with (salen)Co–OH, thereby ensuring that both complexes are present in relatively steady concentrations throughout the entire course of the reaction.36
This work has revealed important subtleties in the cooperative bimetallic mechanism in epoxide ring-opening event and thereby sheds light on how (salen)Co(III) complexes can induce facile epoxide hydrolysis under mild conditions. However, this analysis does not address how this cooperative mechanism results in the exquisite levels of stereoselectivity observed across a range of terminal epoxide substrates in the HKR. This question is addressed in the following paper.
Experimental Section
Representative calorimetry experiment: An 16-mL glass vial equipped with a 3 mm × 12 mm stir bar was charged with (S)-1,2-epoxyhexane (1.00 mL, 8.30 mmol, 1.0 equiv) and 1,2-hexanediol (300 μL). The vial was capped with a black phenolic cap fitted with a PTFE septum, and was inserted into the calorimeter and allowed to thermally equilibrate to 25.0 °C with stirring for approximately 45 min. A solution of (salen)Co(III) complex (150 μL of an 83 mM solution) was added via syringe, and the reaction mixture was stirred in the calorimeter for the specified time period. Water (85 μL, 4.73 mmol, 0.57 equiv) was then added via syringe. The reaction was monitored by reaction calorimetry at a data collection rate of 10 points/min. The maximum heat flow observed under these conditions is approximately 300 mW. The reaction was monitored until the change in heat flow became negligible (generally, < 0.01 mW change in heat flow per min). At this point, the observed heat flow was within 1–3 mW of the background heat flow prior to addition of water. The raw calorimetry data were then τ-corrected and imported into Microsoft Excel. These data were converted from the form heat versus time to rate versus conversion using methods described previously.8 In several cases, the reaction mixtures were analyzed by 1H NMR spectroscopy to provide an independent estimate of %-conversion. In most cases, the enthalpy of hydrolysis calculated using this protocol was 20–21 kcal/mol. In the slowest reactions, the enthalpy was slightly lower (18.5–20 kcal/mol); although the basis for this deviation is not known, an error in rate measurement of this magnitude does not affect any of our conclusions.
Synthesis of tosylate addition product (R)-2-hydroxyhexyl 4-methylbenzenesulfonate (3d)
An oven-dried 25-mL round-bottomed flask was charged with (R)-1,2-hexanediol (1.0 g, 8.5 mmol, 1.0 equiv), anhydrous CH2Cl2 (12 mL), and toluenesulfonyl chloride (1.78 g, 9.3 mmol, 1.1 equiv). The flask was capped with a rubber septum, and pyridine (1.37 mL, 17.0 mmol, 2.0 equiv) was added via syringe under N2. The reaction mixture was stirred at room temperature for 16 h, and then transferred to a separatory funnel, rinsing with 30 mL of CH2Cl2. The reaction mixture was washed with water (25 mL), 1 N HCl (25 mL), saturated NaHCO3 (25 mL), and brine (25 mL). The organic layer was dried over Na2SO4 and concentrated under reduced pressure to yield a viscous white solid. The residue was subjected to purification by flash column chromatography (gradient elution, 9:1 → 1:2 hexanes/diethyl ether, 50 g silica gel) to yield a mixture of mono- and bis-tosylated diols, in which the desired regioisomer was the major product. This residue was subjected again to purification by flash column chromatography (gradient elution, 9:1 → 1:2 hexanes/ethyl acetate, 100 g silica gel) to yield the product as a clear, colorless oil (562 mg, 2.1 mmol, 24% yield). (c 0.9, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.80 (2H, d, J = 8.5 Hz), 7.34 (2H, d, J = 8.0 Hz), 4.01 (1H, dd, J = 10.0, 3.0 Hz), 3.88 (1H, dd, J = 10.5, 7.0 Hz), 3.81 (1H, m), 2.34 (3H, s), 2.19, (1H, br s), 1.42–1.23 (6H, m), 0.86 (3H, apparent t, J = 7.5 Hz). 13C{1H}NMR (125 MHz, CDCl3): δ 145.2, 132.8, 130.0, 128.0, 74.1, 69.5, 32.4, 27.4, 22.6, 21.7, 14.0. IR (cm−1): 3148 (br m), 2956 (m), 2933 (m), 2871 (m), 1598 (m), 1495 (w), 1455 (m), 1354 (s), 1173 (s), 1096 (m), 968 (s), 813 (s), 667 (s). LRMS (ESI): 295.1 (100 %) [M + Na]+.
Supplementary Material
Acknowledgments
This work was supported by the NIH (GM-43214) and by fellowship support to L. P. C. N. from the Hertz Foundation and to S. J. Z. from the American Chemical Society and Roche. We thank Dr. Jason Hong for important preliminary studies, and Prof. Donna G. Blackmond for extensive, valuable discussions.
Footnotes
Supporting Information Available: General experimental procedures, NMR spectra of 3d, kinetic data in tabular format, and mass spectral analysis of epoxide oligomerization. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Tokunaga M, Larrow JF, Kakiuchi F, Jacobsen EN. Science. 1997;277:936–938. doi: 10.1126/science.277.5328.936. [DOI] [PubMed] [Google Scholar]; (b) Schaus SE, Brandes BD, Larrow JF, Tokunaga M, Hansen KB, Gould AE, Furrow ME, Jacobsen EN. J Am Chem Soc. 2002;124:1307–1315. doi: 10.1021/ja016737l. [DOI] [PubMed] [Google Scholar]; (c) Larrow JF, Hemberger KE, Jasmin S, Kabir H, Morel P. Tetrahedron: Asymmetry. 2003;14:3589–2592. [Google Scholar]; (d) Stevenson CP, Nielsen LPC, Jacobsen EN, McKinley JD, White TD, Couturier MA, Ragan J. Org Synth. 2006;83:162–169. [Google Scholar]
- 2.For recent reviews of applications of the HKR reaction in industrial and natural products synthesis, see: Kumar P, Naidu V, Gupta P. Tetrahedron. 2007;63:2745–2785.Furukawa Y, Suzuki T, Mikami M, Kitaori K, Yoshimoto H. J Synth Org Chem Japan. 2007;65:308–319.Kumar P, Gupta P. Synlett. 2009:1367–1382.
- 3.Phenols: Ready JM, Jacobsen EN. J Am Chem Soc. 1999;121:6086–6087.Carbamates: Bartoli G, Bosco M, Carlone A, Locatelli M, Melchiorre P, Sambri L. Org Lett. 2004;22:3973–3975. doi: 10.1021/ol048322l.Azides: Martínez LE, Leighton JL, Carsten DH, Jacobsen EN. J Am Chem Soc. 1995;117:5897–5898.Anilines: Bartoli G, Bosco M, Carlone A, Locatelli M, Massaccesi M, Melchiorre P, Sambri L. Org Lett. 2004;6:2173–2176. doi: 10.1021/ol049372t.Indoles: Bandini M, Cozzi PG, Melchiorre P, Umani-Ronchi A. Angew Chem Int Ed. 2004;43:84–87. doi: 10.1002/anie.200352073.
- 4.For reviews see: Jacobsen EN. Acc Chem Res. 2000;33:421–431. doi: 10.1021/ar960061v.Nielsen LPC, Jacobsen EN. In: In Aziridines and Epoxides in Organic Synthesis. Yudin AK, editor. Chapter 7 Wiley-VCH; Weinheim: 2006.
- 5.Loy RN, Jacobsen EN. J Am Chem Soc. 2009;131:2786–2787. doi: 10.1021/ja809176m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hirahata W, Thomas RM, Lobkovksy EB, Coates GW. J Am Chem Soc. 2008;130:17658–17659. doi: 10.1021/ja807709j. [DOI] [PubMed] [Google Scholar]
- 7.Reviews: Coates GW, Moore DR. Angew Chem, Int Ed. 2004;43:6618–66396. doi: 10.1002/anie.200460442.Darensbourg DJ. Chem Rev. 2007;107:2388–2410. doi: 10.1021/cr068363q.
- 8.Nielsen LPC, Stevenson CP, Blackmond DG, Jacobsen EN. J Am Chem Soc. 2004;126:1360–1362. doi: 10.1021/ja038590z. [DOI] [PubMed] [Google Scholar]
- 9.In the preliminary report (ref 8), this conclusion was drawn from an analysis of reaction rates over a relatively limited, two-fold range of catalyst concentrations (see the Supporting Information in ref 8). The data provided in Figure 1 represent a more extensive analysis over a 5-fold range of catalyst concentrations.
- 10.A crystal structure of the catalyst-bound chloride addition product derived from propylene oxide and (salen)Al–Cl has been reported: Chen P, Chisholm MP, Gallucci JC, Zhang X, Zhou Z. Inorg Chem. 2005;44:2588–2595. doi: 10.1021/ic048597x.For a crystal structure of an analogous azide-addition product bound to (salen)Cr(III), see: Hansen KB, Leighton JL, Jacobsen EN. J Am Chem Soc. 1996;118:10924–10925.
- 11.Isolation and direct characterization of complex 1b has not been achieved. For characterization of a related compound proposed to be a (salen)Co–OH complex: Hutson GE, Dave AH, Rawal VH. Org Lett. 2007;9:3869–3872. doi: 10.1021/ol071342d.In addition, (salen)Co–OPh derivatives of 1 have been isolated and demonstrated to act as catalytically active nucleophile delivery agents in phenolic kinetic resolutions of epoxides (ref 3a).
- 12.A similar figure generated from experiments run under slightly different conditions was provided in ref 8. All experiments described in this paper were carried out under the conditions described in Figure 2.
- 13.It may appear counterintuitive that the reaction catalyzed (salen)Co–Cl requires longer to proceed to full conversion than the reaction catalyzed by (salen)Co–OAc, given that the rate of the former reaction is greater for most of the course of the experiment (after approximately 1 h). However, reaction rate depends both on the relative concentrations of catalysts and on the concentrations of epoxide and H2O. Plotting the data in Figure 2 in the form rate versus conversion of water (Figure 8) reveals that the reaction catalyzed by (salen)Co–OAc is faster than the reaction catalyzed by (salen)Co–Cl for most of the course of the experiment (approximately 10–100 % conversion of water).
- 14.In accord with the idea that (salen)Co(III) complexes act as both Lewis acids and nucleophile delivery agents, dimeric or oligomeric (salen)Co complexes have been found to be substantially more active than their monomeric counterparts: Ready JM, Jacobsen EN. J Am Chem Soc. 2001;123:2687–2688. doi: 10.1021/ja005867b.and refs therein. For a recent overview, see: Haak RM, Wezenberg SJ, Kleij AW. Chem Commun. 2010;46:2713–2723. doi: 10.1039/c001392g.
- 15.Because water has low solubility in 1,2-epoxyhexane, reaction mixtures that do not include 1,2-hexanediol are heterogeneous at low %-conversion, rendering them difficult to study using kinetic methods. We have demonstrated previously that 1,2-hexanediol functions as an inert solvent in the HKR, having no effect on the rate law for epoxide hydrolysis (ref 8).
- 16.Reaction completion was also verified by 1H NMR spectroscopy.
- 17.The application of this methodology to analysis of the HKR was described in the preliminary communication (ref 8).
- 18.This experiment is conceptually related to the “same excess” experiment, but differs in that the variable examined is catalyst aging time rather than reaction time. See: Blackmond DG. Angew Chem, Int Ed. 2005;44:4302–4320. doi: 10.1002/anie.200462544.
- 19.Two rate versus conversion curves of two experiments would also be superimposable if they contained a combination of precatalysts that produced kinetically equivalent but structurally distinct catalyst mixtures. For example, a static mixture of 3:1 (salen)Co–OH and (salen)Co–X would be expected to be kinetically equivalent to a static mixture of ≈ 1:3 (salen)Co–OH and (salen)Co–X. This situation is not expected to arise with precatalysts that undergo counterion addition.
- 20.The enthalpy of hydrolysis of 1,2-epoxyhexane under these reaction conditions is 20–21 kcal/mol, whereas the overestimation of ΔHrxn due to CH2Cl2 mixing is 0.20 kcal/mol and the underestimation of ΔHrxn due to H2O mixing is 0.17 kcal/mol.
- 21.The maximum heat flow in typical HKR experiments under these conditions is 100–300 mW, and thus the heats of mixing shown in Figure 4 represent a substantial portion of the heat flow during the first three minutes of an HKR experiment.
- 22.For the sake of clarity, not all delay times are labeled on the plots. Longer delay times are indicated by the use of darker colors and longer dashes in the lines.
- 23.In all cases described in this paper, the “0 min” delay time experiments were carried out by adding water to an equilibrated solution of 1,2-epoxyhexane and 1,2-hexanediol, followed by catalyst or 3d within 1–5 min (i.e., water and catalyst or 3d were not added simultaneously).
- 24.The same experimental data were used to construct Figures 2 and 8. These two figures differ only in whether they are plotted in the form rate versus time or rate versus conversion.
- 25.The curve with a delay time of 0 min becomes superimposable with the other curves after approximately 40% conversion, which is achieved in 70 min. This time for complete counterion transfer during the course of an epoxide hydrolysis experiment corresponds closely to the delay time required for the curves to become superimposable for the entire course of the reaction (> 60 min).
- 26.The curve with a delay time of 0 min is superimposable with the other curves after approximately 80% conversion, corresponding to a reaction time of 115 min. This reaction time is similar to the delay time required until the curves are superimposable for the entire course of the reaction (> 120 min).
- 27.This catalyst retains much of its activity with even longer delay times (3–8 h). See the Supporting Information for details.
- 28.Addition product 3d was prepared by treatment of (R)-1,2-hexanediol with TsCl and pyridine in CH2Cl2, followed by purification by flash chromatography to remove the bis-tosylated and the regioisomeric monotosylated byproducts.
- 29.The black curves in Figures 5 and 10 were derived from experiments run under identical conditions, except that the experiments leading to Figure 5 contained 150 μL CH2Cl2 (approximately 10% of the volume of the reaction mixture), whereas the experiment leading to the black curve in Figure 10 contained no CH2Cl2.
- 30.The “0 min” curve in Figure 11 and the “1 min” curve in Figure 10 are identical and are derived from the same experiment.
- 31.The catalyst partitioning established via the equilibria in Schemes 4 and 5 appear to be similar, but not identical. In experiments with long delay times in the absence of water (> 15 min), the reaction rate increases slightly during the first 30% conversion and then remains relatively constant until approximately 60% conversion of water. This observation suggests that catalyst partitioning improves slightly upon addition of water (i.e., becomes slightly closer to 50:50 (salen)Co–OTs/(salen)Co–OH).
- 32.Further small but significant decreases in catalyst activity are observed with longer delay times with both experimental setups. This is due to irreversible consumption of tosylate by formation of tosylateterminated epoxide oligomers under the reaction conditions. Small amounts of epoxide oligomerization or polymerization are observed under the reaction conditions by 1H NMR spectroscopy. (S)- and (R)-1,2-epoxyhexane undergo oligomerization at significantly different rates in the presence of (R,R)-(salen)Co–OTs, and the matched epoxide/catalyst combination for oligomerization is the same as for epoxide hydrolysis. This observation demonstrates that oligomerization is promoted by the chiral catalyst, rather than by adventitious acid or another achiral impurity. Both hydroxide- and tosylateterminated epoxide dimers, trimers, and tetrameters are observed as minor byproducts by mass spectrometric analysis of HKR reaction mixtures. Tosylate-terminated epoxide dimers, trimers, tetrameters, pentamers, hexamers, and heptamers are observed as the dominant product by mass spectrometry when 3d (0.4 equiv), 1,2-epoxyhexane (1.0 equiv), and (salen)Co–OAc (0.15 mol%) in the absence of water. Details and relevant 1H NMR and mass spectra are included in the Supporting Information. Similar observations have been made in (salen)Co(III)-catalyzed copolymerization reactions of epoxides and CO2, in which OCOC6F5 was detected within the polymer by 19F NMR spectroscopy. This observation was rationalized through a mechanism in which (salen)CoOCOC6F5 or an exogenous source of OCOC6F5 anion initiates polymerization by counterion-addition to the terminal epoxide: Cohen CT, Chu T, Coates GW. J Am Chem Soc. 2005;127:10869–10878. doi: 10.1021/ja051744l.
- 33.In order to simplify analysis of the data, the experiments described in this paper have been conducted with water as the limiting reagent and with enantiomerically pure 1,2-epoxyhexane. In the context of a true kinetic resolution, this effect would likely be magnified, as the minor enantiomer acts as a competitive inhibitor by binding to the catalyst but undergoing reaction at 2–3 orders of magnitude times slower rates.
- 34.Keith JM, Larrow JF, Jacobsen EN. Adv Synth Catal. 2001;343:5–26. [Google Scholar]
- 35.These principles also apply to epoxide ring-opening reactions using phenol and carbamate nucleophiles: Nielsen LPC. Ph D Thesis. Harvard University; Cambridge, MA: Nov, 2006. . The complete text of this document is available via the internet through Proquest (http://www.proquest.com).
- 36.The preparation of catalyst 1d from the corresponding (salen)Co(II) precursor is described in an Org. Synth. procedure (ref 1d). Prior to our discovery that (salen)Co–OTs complex 1d is an optimal catalyst for epoxide hydrolysis, we had prepared or obtained > 0.2 kg of (salen)Co–OAc complex 1a. In order to make best use of this supply, we developed a procedure for generating OTs complex 1d in situ using (salen)Co–OAc complex 1a as a precatalyst. In HKR reactions carried out on 0.1 or 0.2-kg scale of (±)-1,2-epoxyhexane using 0.1–0.15 mol% of 1a, 0.05–0.08 mol % of TsOH·H2O was added 30 min after initiation of the reaction. Complete resolution to > 99.5% ee of the epoxide was achieved within 12–20 h. Using 1a alone at this catalyst loading, complete resolution is not achieved on practical time scales, as the reaction stalls within 1 h.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.