

Abstract
SlyD belongs to the FK506-binding protein (FKBP) family with both peptidylprolyl isomerase (PPIase) and chaperone activities, and is considered to be a ubiquitous cytosolic protein-folding facilitator in bacteria. It possesses a histidine- and cysteine-rich C-terminus binding to selected divalent metal ions (e.g., Ni2+, Zn2+), which is important for its involvement in the maturation processes of metalloenzymes. We have determined the solution structure of C-terminus-truncated SlyD from Helicobacter pylori (HpSlyDΔC). HpSlyDΔC folds into two well-separated, orientation-independent domains: the PPIase-active FKBP domain and the chaperone-active insert-in-flap (IF) domain. The FKBP domain consists of a four-stranded antiparallel β-sheet with an α-helix on one side, whereas the IF domain folds into a four-stranded antiparallel β-sheet accompanied by a short α-helix. Intact H. pylori SlyD binds both Ni2+ and Zn2+, with dissociation constants of 2.74 and 3.79 μM respectively. Intriguingly, binding of Ni2+ instead of Zn2+ induces protein conformational changes around the active sites of the FKBP domain, implicating a regulatory role of nickel. The twin-arginine translocation (Tat) signal peptide from the small subunit of [NiFe] hydrogenase (HydA) binds the protein at the IF domain. Nickel binding and the recognition of the Tat signal peptide by the protein suggest that SlyD participates in [NiFe] hydrogenase maturation processes.
Electronic supplementary material
The online version of this article (doi:10.1007/s00775-011-0855-y) contains supplementary material, which is available to authorized users.
Keywords: Chaperone, Hydrogenase, NMR, Peptidylprolyl isomerase, SlyD
Introduction
The Gram-negative, motile, microaerophilic, flagellate bacterium Helicobacter pylori is one of the most successful pathogenic bacteria, and colonizes the human gastric mucosa and has infected more than half of the global population. H. pylori is now regarded as the leading causes of chronic gastritis, peptic and duodenal ulcers, and even gastric cancers [1]. Its survival in a high acidic environment [1] and successful pathogenesis greatly rely on its capability to produce two important enzymes, i.e., urease [2] and [NiFe] hydrogenase [3], which serve as the essential colonization factors [1]. The efficient acquisition of nickel is crucial for the enzymes to exert their proper functions. Although it is not clear how nickel enters H. pylori and is enriched under the condition of very low concentration in human serum, the NixA protein and ABC transporter system were proposed to be responsible for the nickel transport in H. pylori [1]. It has been shown that many urease- and hydrogenase-related accessory proteins play an important role in the nickel insertion into urease and hydrogenase. UreE was demonstrated to be important for the maturation of urease, but can be complemented by hydrogenase accessory proteins HypA and HypB [4], which are also critical in the maturation of hydrogenase [5]. Previous functional studies as well as an in vivo interactomics study on H. pylori revealed that both HypA and HypB together with SlyD are involved in the nickel incorporation into both urease and hydrogenase [5–7]. However, there appears to be lack of characterization of SlyD in H. pylori (HpSlyD) in spite of its counterparts being extensively studied in other species [8–11].
SlyD, as a multifaceted protein, belongs to a peptidylprolyl isomerase (PPIase) (EC 5.2.1.8) FK506-binding protein (FKBP) family, catalyzing the intrinsically slow cis–trans isomerization of peptidylprolyl bonds (Xaa-Pro) to facilitate the protein folding process [12, 13], but its role as a PPIase in vivo is not well understood. There are no other significant phenotype changes after the deletion of the sensitivity to lysis D gene (slyD) in either Escherichia coli [14] or H. pylori [15], but overexpression of SlyD can interfere with cell division and cell wall synthesis, leading to the filamentation phenotype [14]. SlyD from E. coli (EcSlyD) was found as a prominent contaminant during purification by immobilized metal ion affinity chromatography (IMAC) because of its possession of a histidine- and cysteine-rich metal-binding domain at the C-terminus [16], which is also present in HpSlyD. Within the N-terminal PPIase domain, there is an insert-in-flap (IF) chaperone domain, compared with the sequence of an archaeal FKBP from Methanothermococcus thermolithotrophicus (MtFKBP17) [11] (Scheme 1). The histidine- and cysteine-rich C-terminus of SlyD renders it able to bind various divalent metal ions, which results in the inhibition of its PPIase activity [17], but the enhancement of nucleotide binding [18]. EcSlyD has been demonstrated to bind to various twin-arginine translocation (Tat) signal sequences both in vivo and in vitro, which prevents the export of premature proteins targeted by the Tat system [19], a system that is ubiquitously present among bacteria, including H. pylori [20], and plants [21]. This will facilitate the export of fully folded proteins from the cytoplasm across the inner/cytoplasmic membrane into the periplasm in bacteria [22]. It has been hypothesized that HpSlyD might play a dual role in the maturation process of H. pylori [NiFe] hydrogenase through interaction with both HypB and the Tat signal sequence of HydA (small subunit of hydrogenase) to timely coordinate the nickel insertion with the export of holohydrogenase into the periplasm of H. pylori [6]. But there appears to be lack of direct evidence to support this hypothesis.
Scheme 1.

Domain organization of SlyD in Helicobacter pylori (HpSlyD). Cyan boxes FK506-binding protein (FKBP) domains, red box insert-in-flap (IF) domain, green line metal-binding domain (MBD), black lines linkers
To understand the Ni2+ flow in H. pylori, we have systematically studied structurally and/or functionally a series of metalloproteins from H. pylori, including Ni2+ storage proteins, e.g., Hpn [23–25] and Hpnl [26], and metallochaperone proteins, e.g., HspA [27] and HypA/HypB [28]. In the work reported here, we overexpressed and purified HpSlyD and its variants. The solution structure of HpSlyD (1–151, C-terminus-truncated HpSlyD, HpSlyDΔC) showed an overall topology similar to that of its homologues with two independent domains (FKBP and IF). We also performed biochemical and physical studies to explore the metal binding properties of HpSlyD and its potential roles in the maturation of [NiFe] hydrogenase of H. pylori.
Materials and methods
Molecular cloning
PCR amplification was performed using Phusion® high-fidelity DNA polymerase (Finnzymes). All restriction endonucleases were purchased from New England BioLabs. E. coli XL-1 Blue (Stratagene) was used for cloning. H. pylori slyD and all its variant genes were inserted into the plasmid pET-32a(+) (Novagen) for expression. All primers and plasmids used and constructed in this study are listed in Table S1.
H. pylori slyD gene was amplified from H. pylori 26695 genomic DNA using the primer pair HpSlyD-for/HpSlyD-rev. slyDΔC gene was amplified using HpSlyD-for/HpSlyDΔC-rev. To generate IF-domain-deleted HpSlyD (HpSlyDΔIF), the IF domain of HpSlyD was replaced by the hFKBP12 loop (ATGHPGIIPPHAT) [29] and the primer pairs HpSlyD-for/ΔIF-1 and ΔIF-2/HpSlyD-rev were used for overlap PCR.
To express and purify the Tat signal peptide with the sequence taken from the [NiFe] hydrogenase small subunit (pre-HydA) of H. pylori (HpHydA SP), a signal-peptide-less maltose-binding protein (MBP) (SP-less MalE) expression vector was constructed in a manner similar to that in a previous description [30] by amplifying malEΔSP from pMAL-p2x (New England BioLabs) using the primer pair MalE-SPless/MalE-Xa and inserted into pET-32a(+), generating pETMalE. pETMalE was then digested by EcoRI and XhoI, blunted by T4 DNA polymerase (New England BioLabs), and circulated again with T4 DNA ligase (Invitrogen), generating pETMalEHis, which encodes the MalE protein with a factor Xa cleavage site, a flexible extension, and a hexahistidine tag at the C-terminus (ΦMalE::H6). The coding sequence of HpHydA SP was PCR-amplified from H. pylori 26695 genomic DNA using HydA-SP-up/HydA-SP-down, digested by EcoRI and XhoI, and cloned into pETMalEHis, generating pETMalE-HydASP (product ΦMalE::HydASP-H6).
Protein expression and purification
E. coli strain KMl603 [BL21(DE3) ΔslyD::kan (a gift from A.R. Davidson, University of Toronto)] was used for expressing all proteins in this study to exclude the contamination of SlyD from E. coli. The proteins were overexpressed in 1 L LB medium at 30 °C for 3–5 h after the addition of 0.2 mM isopropyl β-d-1-thiogalactopyranoside. For isotopically (15N/13C) labeled proteins, proteins were overexpressed in 1 L M9 minimal medium, supplemented with either 1 g 15NH4Cl or and 4 g d-[U-13C6]glucose (Cambridge Isotope) at 25 °C for 22 h.
HpSlyD and its variants were purified on an ÄKTAfplc™ system (GE Healthcare). Briefly, cultured cells were lysed in a lysis buffer [20 mM tris(hydroxymethyl)aminomethane (Tris)/HCl, pH 8.0, 1 mM phenylmethanesulfonyl fluoride, 1 mM dl-dithiothreitol (DTT), and 1 mM EDTA] and loaded onto a HiPrep DEAE FF 16/10 column. Proteins were eluted by a gradient of 0–500 mM NaCl. Targeted protein-containing fractions were pooled together, concentrated, and loaded onto a HiLoad 16/10 Superdex 75 pg column preequilibrated with 20 mM Tris/HCl buffer, pH 7.6, 1 mM DTT, 1 mM EDTA, and 150 mM NaCl. Eluted proteins were further purified using a RESOURCE Q or Mono Q column with a gradient of 0–500 mM NaCl in 20 mM Tris/HCl, pH 7.6, 1 mM DTT. Prior to each experiment, HpSlyD was treated with EDTA and tris(2-carboxyethyl)phosphine hydrochloride (TCEP) overnight and desalted into the buffer for the experiment. The purity of the proteins was more than 90% as judged by sodium dodecyl sulfate polyacrylamide gel electrophoresis and the identities of the proteins were verified by matrix-assisted laser desorption/ionization time-of-flight tandem mass spectrometry analyses.
ΦMalE::H6 and ΦMalE::HydASP-H6 were purified by IMAC using a HisTrap HP column followed by size-exclusion chromatography. Protein concentration was determined by a bicinchoninic acid protein assay kit (Novagen) or UV–vis spectroscopy according to the predicated absorption coefficient (ε 280) using ProtParam (http://expasy.org/tools/protparam.html) (only for apoprotein samples) or the previously reported values [8, 31]. The molar absorption coefficients (ε 278) for reduced and carboxymethylated (Ser54Gly, Pro55Asn) RNase T1 (RCM-T1) and reduced and carboxymethylated α-lactalbumin (RCM-La) are 21,060 and 28,340 M−1 cm−1, respectively, and ε 280 for HpSlyD (reduced) and HpSlyDΔC is 12,950 M−1 cm−1 and for HpSlyDΔIF (reduced) and C-terminus-truncated HpSlyDΔIF (HpSlyDΔIFΔC) is 8,480 M−1 cm−1.
Equilibrium dialysis
The dissociation constants of HpSlyD–metal complexes were determined by equilibrium dialysis using a 1-kDa cutoff mini dialysis kit (GE Healthcare). Known amounts of the protein (200 μL, 20 μM) were loaded into the dialysis tubes and dialyzed against 30 mL dialysis buffer (20 mM Tris/HCl and 150 mM NaCl, pH 7.6, 200 μM TCEP) containing different concentrations of Ni2+ (as NiSO4, from 0.5 to 80 μM) or Zn2+ (as ZnSO4, from 0.5 to 80 μM) for overnight incubation at 15 °C. The metal concentrations inside and outside the dialysis tubes were determined by inductively coupled plasma mass spectrometry (Agilent 7500 spectrometer). The maximal binding capacity (B max) and dissociation constants (K d) were obtained and averaged from three independent experiments using a one-site binding equation: Y = B max X/(K d + X), where X is the concentration of free metal ions and Y is the molar ratio of bound metal ions to protein.
UV–vis spectroscopy
All UV–vis spectra were collected with a Varian Cary 50 spectrophotometer using a 1-cm quartz cuvette at room temperature. Apo-HpSlyD samples (30 μM) were freshly prepared in 10 mM N-(2-hydroxyethyl)piperazine-N′-ethanesulfonic acid/NaOH buffer containing 50 mM NaCl, pH 7.4, and prior to titration, an aliquot of a concentrated solution of TCEP was added to give 10 molar equiv of the protein. The spectrum of apo-HpSlyD containing TCEP was recorded after equilibration for 30 min. Aliquots of stock solution of Ni2+ (as NiSO4) were then titrated into the sample, and UV–vis absorption spectra were recorded in the range from 230 to 600 nm. Binding of Ni2+ was monitored by the increase in absorbance at 246, 278, 333, and 420 nm.
NMR spectroscopy
All spectra were acquired at 25 °C with a Bruker Avance 600 spectrometer operated at a frequency of 600.13 MHz for 1H and equipped with a triple resonance (TCI) CryoProbe. Spectra for assignment of HpSlyDΔC were recorded in an about 1.3 mM 15N/13C doubly labeled sample in the NMR buffer (20 mM [bis(2-hydroxyethyl)amino-tris(hydroxymethyl)methane/HCl buffer and 100 mM NaCl, pH 6.8, containing 10% (v/v) D2O, 0.02% NaN3). All NMR data were processed with either NMRPIPE [32] or XWINNMR and were analyzed by SPARKY [33]. Distance restraints were obtained on the basis of two 3D 15N/13C-edited nuclear Overhauser effect (NOE) spectroscopy experiments. Hydrogen-bond amide protons were identified by acquiring a series of 2D [1H–15N] heteronuclear single quantum coherence (HSQC) spectra at a range of temperatures (10–35 °C). Backbone dihedral angles were derived on the basis of the chemical shifts of Cα, Cβ, and C′ from TALOS [34]. T 1, T 2, and {1H}–15N NOE measurements were performed by collecting a time series of [1H–15N] HSQC spectra with sensitivity enhancement and analyzed by NMRPIPE.
For the Ni2+ titration experiment, a 2D [1H–15N] HSQC spectrum of 0.3 mM 15N-labeled full-length HpSlyD was recorded and compared with the spectrum of HpSlyDΔC, and most of the backbone amides of full-length HpSlyD were therefore assigned. A series of [1H–15N] HSQC spectra were acquired before and after addition of 40 mM NiSO4 in NMR buffer at molar ratios of Ni2+ to the protein of 0.3, 0.6, 1.0, 1.5, and 2.0, respectively. Solutions of 0.1–0.3 mM 15N-labeled HpSlyDΔC were used for titrations of FK506, rapamycin, and permanently unfolded protein and peptide substrates, which were added stepwise at molar ratios of substrates to the protein of 0.5, 1, and 1.25. The mean weighted variations of 15N and 1H chemical shifts upon titrations of Ni2+ and substrates were calculated as reported previously [35] using the following equation:
Structure calculation
The solution structure of HpSlyDΔC was calculated by torsion angle dynamics using CYANA 2.1 [36] on the basis of NOE-derived distance constraints, hydrogen-bond constraints, and TALOS angle constraints. A set of 200 structures was initiated and a standard protocol of an automated NOE assignment approach was applied. A set of 50 structures with the lowest target functions and no NOE violation larger than 4.0 Å and torsion angle violation larger than 5° was chosen and subjected to further energy minimization using AMBER 7 [37]. The final 20 energy-minimized and best-converged structures were selected to represent the ensemble, and the quality of the structures was assessed by PROCHECK-NMR [38] and analyzed by MOLMOL [39]. The structure was displayed by MOLMOL and PyMOL [40].
Results
Secondary structure of HpSlyD
HpSlyD and its C-terminus-truncated variants (HpSlyDΔC) were successfully expressed and purified to apparent homogeneity (Fig. S1). The purified HpSlyD was subjected to gel filtration chromatography on a Superdex 10/300 GL column (GE Healthcare) and was calibrated as an apparent molecular mass of approximately 33 kDa in the presence of TCEP using an LMW gel filtration calibration kit (GE Healthcare) (Fig. 1). A larger molecular mass observed by both size-exclusion chromatography and sodium dodecyl sulfate polyacrylamide gel electrophoresis (approximately 25 kDa, Fig. S1) in comparison with the calculated one (approximately 20 kDa) was also seen previously for SlyD from other species [18, 41], probably attributable to the elongated shape of the protein. We therefore subjected the purified HpSlyD to electrospray ionization mass spectrometry and obtained a molecular mass of 19,995.84 Da (Fig. S2), confirming that HpSlyD exists as a monomer in solution under the conditions used. Deconvolution of the circular dichroism spectrum (190–260 nm) of HpSlyD (Fig. S3) gave rise to secondary structure contents of about 7% α-helix, 43% β-sheet, 17% turn, and 33% random coil. Addition of selected metal ions (i.e., Ni2+ and Zn2+) to a solution of HpSlyD did not change the examined circular dichroism spectrum (190–260 nm) (data not shown), indicating there were no significant effects on the secondary structure of the protein.
Fig. 1.
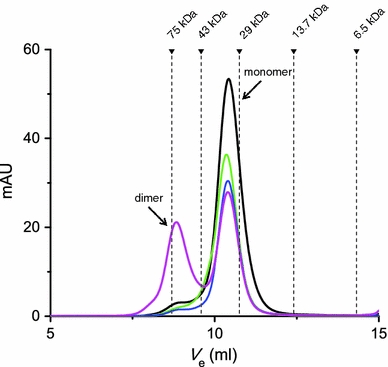
Gel filtration chromatography profiles of 10 μM HpSlyD in the absence of metal ions (black) and in the presence of 5 molar equiv of Ni2+ (green) and Zn2+ (blue) without incubation and Zn2+ with overnight incubation (magenta). The experiments were performed using a Superdex 10/300 GL column (GE Healthcare) with 20 mM tris(hydroxymethyl)aminomethane/HCl buffer, 150 mM NaCl, pH 7.6, at 4 °C. Similar profiles were also observed when 2 molar equiv of metal ions was added. Apo-HpSlyD was calibrated as an apparent molecular mass of about 33 kDa, larger than the calculated value, i.e., 20 kDa. The dimer was calibrated as about 65 kDa
Solution structure determination of HpSlyDΔC
The [1H–15N] HSQC spectra of both full-length HpSlyD (1–185) and HpSlyDΔC (1–151) were compared and nearly all the resonances from HpSlyDΔC can be overlaid with those from the full-length protein, indicating that the C-terminal residues have little effect on the structure of the protein. Backbone resonances of 13C/15N doubly labeled HpSlyDΔC were assigned using standard triple-resonance experiments such as HNCACB, CBCA(CO)NH, HNCO, and HNCA, whereas HCCH–total correlation spectroscopy, HCCH–correlation spectroscopy, and HBHA(CO)NH were used for the aliphatic side chain assignment. The solution structure of HpSlyDΔC was determined using CYANA 2.1 on the basis of 2,525 NOE distance constraints derived from 15N/13C-edited NOE spectroscopy and 164 TALOS angle constraints derived from chemical shifts of Cα, Cβ, and C′ and 68 hydrogen-bond constraints derived from both experimental data and NOE patterns. Structural statistics are summarized in Table 1. An ensemble of the 20 best converged energy-minimized structures exhibits high convergence within the FKBP domain (Fig. 2a) and the IF domain (Fig. 2b). The structure close to the average one was chosen to represent HpSlyDΔC and is shown in a ribbon model (Fig. 2c).
Table 1.
Summary of structural statistics
| Total number of NOE cross-peaks | 4,546 |
| NOE-derived distance restraints | 2,525 |
| Short range |i–j| ≤ 1 | 1,373 |
| Medium range 1 < |i–j| < 5 | 288 |
| Long range |i–j| ≥ 5 | 864 |
| H-bond constrains | 68 |
| ϕ/ψ dihedral angle constraints (TALOS) | 164 |
| Residual NOE target function (Å2) | 1.61 |
| Deviations from the experimental restraints | |
| RMS distance restraint violation (Å) | 0.0082 ± 0.0007 |
| Number of distance restraint violations greater than 0.4 Å | 0 |
| Maximum distance restraint violations (Å) | 0.37 |
| RMS dihedral angle restraint violations (°) | 0.12 ± 0.06 |
| Number of dihedral angle restraint violations greater than 3° | 1 |
| AMBER energy (kJ/mol) | −23,804.34 ± 88.62 |
| Pairwise atomic RMSD from the mean (Å) | |
| PPIase domaina | |
| Backbone heavy atoms | 0.46 ± 0.09 |
| All heavy atoms | 1.16 ± 0.16 |
| IF domainb | |
| Backbone heavy atoms | 0.70 ± 0.19 |
| All heavy atoms | 1.41 ± 0.25 |
| Ramachandran analysis (%) | |
| Most favored region | 83.0 |
| Additionally allowed region | 17.0 |
| Generously allowed region | 0.0 |
| Disallowed region | 0.0 |
| Overall G factor | −0.25 ± 0.03 |
| H-bond energy (kJ/mol) | 2.93 |
NOE nuclear Overhauser effect, RMS root mean square, RMSD root mean square deviation, PPIase peptidylprolyl isomerase, IF insert-in-flap
aResidues 12–21, 27–30, 37–40, 48–54, 63–67, and 135–145
bResidues 81–89, 99–104, 108–118, and 121–126
Fig. 2.
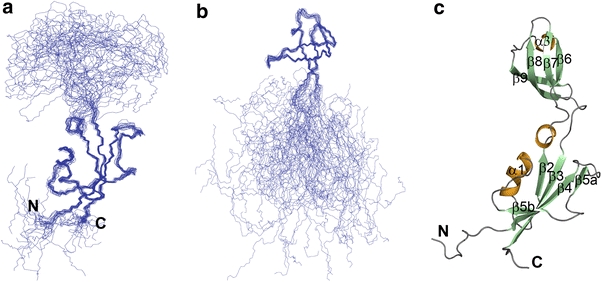
The 20 lowest-energy solution structures of C-terminus-truncated HpSlyD (HpSlyDΔC) superimposed over the backbone coordinates of well-defined regions for the FKBP domain (a) and the IF domain (b) and ribbon diagram of the mean structure of HpSlyDΔC with the helices shown in orange and β-sheets shown in pale green (c)
Structural description and comparison with other homologues
As shown in Fig. 2c, HpSlyDΔC folds into two separated globular domains, i.e., the FKBP domain and the IF domain, linked by a deep cleft. The FKBP domain is formed by two polypeptide segments consisting of 73 N-terminal redidues (1–73) and 17 C-terminal residues (135–151), and the IF domain comprises residues 80–126. The connecting segments span residues 74–79 and 127–134, forming a pair of antiparallel strands and hinging the bending motion of the domains. These segments are relatively less well defined owing to the limited numbers of NOEs assigned.
The relative orientation of the two domains is poorly defined and no unambiguous contacts (NOEs) were identified between the two domains. When the orientation of the two domains was fixed by superimposing the structures over the well-defined residues of entire molecules, a very large root mean square deviation (RMSD) (larger than 5 Å) was noted. In comparison, small RMSDs were obtained when each domain was overlaid over well-defined residues separately (Table 1). The average overall rotational correlation time for the two domains was estimated on the basis of the backbone dynamic data (Fig. S4) to be 13.1 and 15.2 ns for the IF and FKBP domains, respectively, in line with the two-domain structure. The poorly defined orientation between the FKBP and IF domains was also seen for other SlyDs, e.g., EcSlyD, as well as the structural and functional homologue MtFKBP17.
The FKBP domain consists of a canonical four-stranded antiparallel β-sheet accompanied by α1 on one side (Fig. 2c), with a topology of β4–β5–α1–β2–β3, which closely resembles the structures of the FKBP domains of other FKBPs. The secondary structure elements were named according to the convention used for other FKBPs. α1 is amphipathic with apolar residues pointing to the β-sheet to make hydrophobic contacts and polar residues that are solvent-exposed. Similar to the structures of MtFKBP17 and other SlyDs, HpSlyDΔC lacks an N-terminal β1, as observed in human FKBP (HsFKBP12) [42]. β5 of HpSlyDΔC is split into two halves separated by a five-residue bulge loop as in EcSlyD and HsFKBP12, but different from MtFKBP17 and SlyD from Thermus thermophilus (TtSlyD), whereas a unique helix, α2, is found to separate strand β5 to β5a and β5b in MtFKBP17 and no splitting of β5 occurs in TtSlyD. An additional helix, α4, found in EcSlyD and TtSlyD was not observed in HpSlyD, probably owing to the truncation point selected in the present study. α4 of EcSlyD is formed by residues 144–149, corresponding to residues 148–153 of HpSlyD. Nevertheless, our bioinformatic studies demonstrated indeed the presence of α4, suggesting that α4 is likely to be the common feature for the structures of the SlyD family. This is distinct from other FKBPs and it may therefore serve an important biological function (vide infra). A small helix at the end of β2 is formed by residues 69–73 in HpSlyDΔC, which is also observed in the two crystal structures of the non-substrate-peptide-bound form of TtSlyD. However, this helix was not observed in the structures of EcSlyD, MtFKBP17, and substrate-peptide-bound TtSlyD; instead, a reversed turn was found in these structures. The pairwise structural comparison of HpSlyDΔC with other homologues, e.g. HsFKBP12 (1FKF) [42], EcSlyD (2K8I) [9], and TtSlyD (3CGM) [8], was made with the aid of DaliLite [43], and the aligned structures are shown representatively in Fig. 3. The FKBP domain of HpSlyDΔC can be superimposed well with that of HsFKBP12, EcSlyD, and TtSlyD, with RMSDs of 2.5, 3.4, and 3.2 Å over the Cα atoms of 81, 88, and 80, respectively, identified by DaliLite.
Fig. 3.
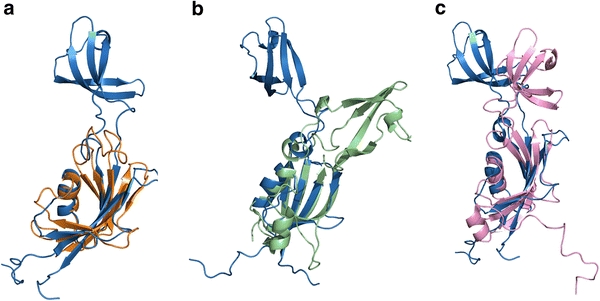
Superimposition of the FKBP domain of HpSlyDΔC (sky blue) with that of a human FKBP (HsFKBP12) (orange), b SlyD from Thermus thermophilus (TtSlyD) (pale green), and c SlyD from Escherichia coli (EcSlyD) (pink) was shown. The structures were aligned over Cα residues identified using DaliLite [43] and the images were generated using PyMOL [40]
The IF domain displays a topology of β6–α3–β9–β8–β7 and folds into a four-stranded antiparallel β-sheet and a short helix, α3, which connects β6 and a partially flexible loop leading to β9 (Fig. 2c). The side chains of Phe-89 and Arg-86 from α1 interact with the side chains of Val-84, Val-100, Ile-114, Phe-117, Val-122, and Val-124 from the β-sheet and Ile-92 and Leu-94 from the flexible loop, forming a hydrophobic core to stabilize the structure. Extremely high similarity was noted among the structures of IF domains of SlyD or its homologues from different species, and RMSDs of 1.8 and 2.2 Å were found when the structural alignment was made for the IF domain of HpSlyD and EcSlyD or TtSlyD via DaliLite. Importantly, a large proportion of apolar residues (50%) is noted for this domain, which gives rise to large exposed hydrophobic surfaces. Moreover, the backbone dynamic data (Fig. S4) demonstrated that the IF domain generally has a a higher degree of mobility in comparison with the FKBP domain, in agreement with its counterpart EcSlyD. The characteristics of high flexibility and large exposed hydrophobic surfaces may allow it to rapidly adopt different conformations, resulting in facilitating association with or dissociation from unfolded or partially folded proteins or substrates.
Metal binding properties
The full-length HpSlyD was used to study its interaction with metal ions such as Ni2+ and Zn2+. When Ni2+ was titrated in a step of 0.25 molar equiv into 30 μM HpSlyD, pH 7.4, in the presence of 300 μM TCEP, four new peaks centered at approximately 246, 278, 333, and 420 nm appeared gradually in the difference electronic absorption spectra, indicative of the binding of Ni2+ to the protein (Fig. 4a). According to a previous study on nickel binding to small molecules [44], the absorption bands at 278 and 333 nm are due to σ(S)(Cys) → Ni(II) and π(S)(Cys) → Ni(II) ligand-to-metal charge transfer transitions, whereas the one at 246 nm is due to a σ(N)(His) → Ni(II) ligand-to-metal charge transfer transition. The 420-nm band may be due to a d–d transition of Ni2+ [45], as for other metal-binding proteins [24, 27, 28, 46]. These bands increased in intensity further upon addition of metal ions and the intensity leveled off at a molar ratio of Ni2+ to HpSlyD of approximately 2 (Fig. 4a inset). The binding of Ni2+ and Zn2+ was further investigated by equilibrium dialysis (Fig. 4b), which revealed stoichiometry of 2.4 ± 0.1 nickel ions per protein and 3.3 ± 0.2 zinc ions per protein with dissociation constants (K d) of 2.74 ± 0.26 and 3.79 ± 0.92 μM for Ni2+ and Zn2+, respectively. No oligomerization of the protein was observed upon addition of 2 or even 5 molar equiv of Ni2+. In contrast, dimerization was found upon incubation of the protein with 2 and 5 molar equiv of Zn2+ for an extended period of time (longer than 2 h) (Fig. 1).
Fig. 4.
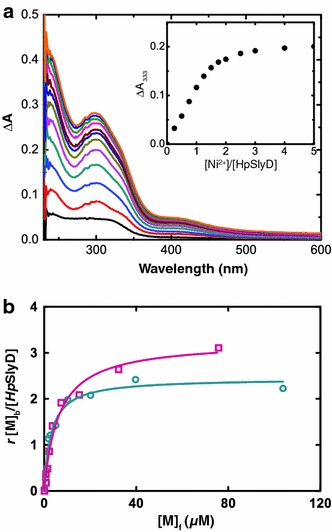
a UV difference spectra of 30 μM HpSlyD with addition of 0.25–5 molar equiv of Ni2+. The titration curve plotted with ΔA 333 is shown in the inset. b Metal binding affinity of HpSlyD determined by equilibrium dialysis: Ni2+ (circles) and Zn2+ (squares)
NMR spectroscopy was subsequently applied to monitor Ni2+ binding to HpSlyD as well as potential metal-induced structural changes. The 2D [1H–15N] HSQC spectrum of the full-length HpSlyD was assigned on the basis of that of HpSlyDΔC. Addition of Ni2+ at a molar ratio of Ni2+ to protein of 0.6:1 led to several cross-peaks decreasing in intensity and new peaks appearing. The latter are assigned as the bound forms. The observation of two sets of peaks indicated that the apo and Ni2+-bound forms are in slow exchange on the NMR timescale. On further addition of Ni2+ to a molar ratio of Ni2+ to protein of 1:1, the cross-peaks for the apo form disappeared completely, whereas those of the bound form further increased in intensity. No apparent changes were observed upon further titration of Ni2+ to a ratio of 2:1 Ni2+ to protein. The chemical shift perturbation upon Ni2+ binding was quantified and changes larger than 0.03 ppm were noticed for Glu-18, Asp-29, Ile-46, Ile-47, Gly-49, Lys-52, Val-65, and Arg-138. Significantly, these residues can be mapped exclusively into the PPIase domain of the protein (Fig. 5a) and are either very close to or belong to the active sites. Some residues likely from α4 are also perturbed upon Ni2+ binding, suggesting the importance of this helix. As a control, 15N-labeled HpSlyDΔC was also titrated with Ni2+ (up to 2 molar equiv), resulting in no significant chemical shift perturbation, indicating that Ni2+ itself neither binds to HpSlyDΔC nor changes its chemical environment. Surprisingly, addition of Zn2+ to the protein caused no chemical shift perturbation in the PPIase domain (data not shown), clearly indicating the difference in either coordinating ligands or geometry between these two metal ions. Ni2+-induced structural changes were also noted previously for EcSlyD [10], and recent crystal structures of both apo-TtSlyD and Ni2+-bound TtSlyD [8] further illustrate such metal-induced structural changes, which might be potentially important in the accomplishment of their biological functions.
Fig. 5.
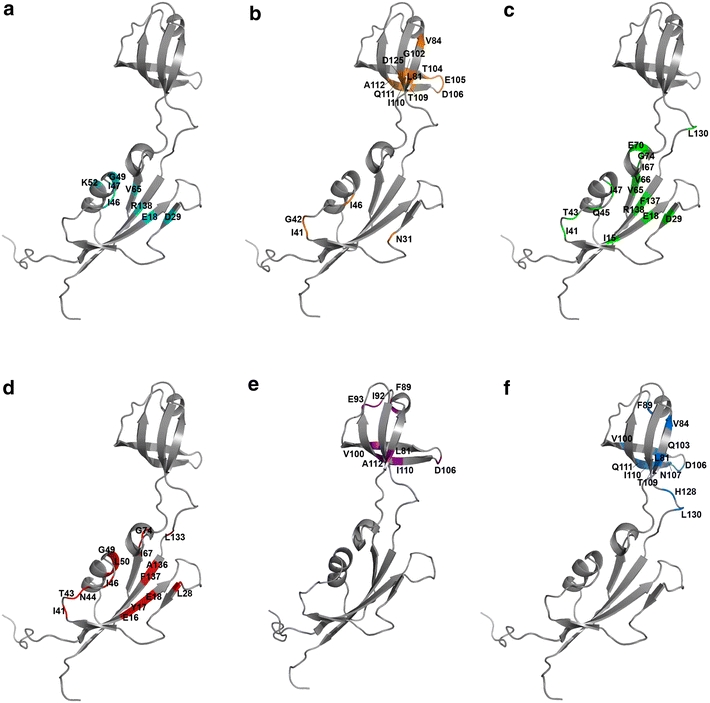
Structural mapping of the residues experiencing relatively large chemical shift perturbations upon interactions of the protein with a Ni2+, b twin-arginine translocation (Tat) signal peptide of HydA from H. pylori, c FK506, d rapamycin, e reduced and carboxymethylated α-lactalbumin, and f insulin. All experiments were conducted using HpSlyDΔC, except for Ni2+ titration, in which full-length HpSlyD was used
IF domain is responsible for binding to Tat signal peptide of HydA from H. pylori
It has been suggested previously that SlyD binds different Tat signal peptides in E. coli such as DnaK as a general chaperone for Tat signal-binding protein [19]. HpSlyD has also been implicated to serve an important role in ensuring the transport of holohydrogenase across the inner membrane into the periplasm by interacting with Tat signal peptide of HydA (small subunit of hydrogenase) of H. pylori [6]. However, there appears lack of direct evidence for such an interaction. Therefore, we constructed HydA Tat signal-fused protein to examine its interaction with the protein by NMR spectroscopy. The sequence of the Tat signal peptide (1–69 amino acids) from pre-HydA of of H. pylori (HP0631) with a longer n-region (positively charged amino-terminal region), which is much longer than normal Tat signal peptides (normally 38 amino acids) [47], was predicted using ScanProsite (http://expasy.org/tools/scanprosite/) with a score of 8.433. The Tat signal peptide was fused to the C-terminus of the signal-peptide-less MBP. Since the Tat signal peptide is located between the MBP and a C-terminal hexahistidine tag, only full-length, signal-peptidase-uncleaved chimera ΦMalE::HydASP-H6 can be purified by IMAC. The MBP-tagged Tat peptide was added at approximately 1:1 ratio to a solution of 15N-labeled HpSlyDΔC. The residues experiencing large chemical shift perturbations (larger than 0.06 ppm) consisted of Asn-31, Ile-41, Gly-42, Ile-46, Leu-81, Val-84, Gly-102, Thr-104, Glu-105, Asp-106, Thr-109, Ile-110, Gln-111, Ala-112, and Asp-125. Most of these residues localized in the IF domain (Fig. 5b). Surprisingly, several residues (Ile-41, Gly-42, Ile-46, and Asn-31) were from the FKBP domain, which is likely due to the binding of the longer n-region of HydA Tat peptide to the FKBP domain. The MBP tag alone showed no interaction with HpSlyDΔC on the basis of an NMR experiment (data not shown).
PPIase active sites and activity of HpSlyD
The substrate binding sites are located at the highly conserved PPIase domain of FKBP and FKBP-like proteins. Since the active sites are highly conserved among different members of the family proteins, the corresponding binding pocket in HpSlyD was identified on the basis of structural alignments (Fig. 6), which consists of residues Tyr-17, Leu-28, Asp-29, Leu-37, Phe-39, Ile-46, Ile-47, Leu-50, Tyr-73, Leu-135, and Phe-137. Ile-90 and Ile-91 of HsFKBP12 are unique and there are no corresponding residues in other homologues. Comparing the active sites of FKBPs from three species (Fig. 6), we demonstrated that the active site of HpSlyDΔC is slightly different from that of both HsFKBP12 and EcSlyD. Specifically, Phe-39, Leu-37, and Leu-135 are situated at positions which could not be superimposed with their counterparts and the substrate binding pocket for HpSlyDΔC is much larger than that of HsFKBP12 and EcSlyD (Fig. 6).
Fig. 6.
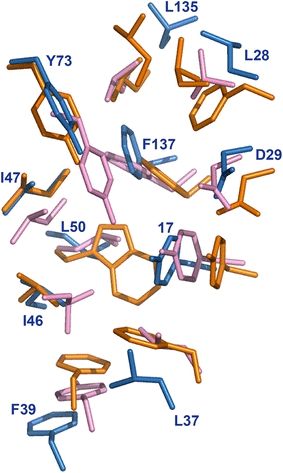
Superimposition of the side chains of the residues in catalytic sites of three FKBP homologues: HpSlyDΔC (sky blue), HsFKBP12 (orange), and EcSlyD (pink). The numbering of the residues corresponds to HpSlyDΔC
The PPIase activities (k cat/K m) obtained from the single-proline-limited RCM-T1 refolding are summarized in Fig. S5 and Table S2. The PPIase activity of HpSlyD (1.17 × 106 M−1 s−1) is comparable to that of EcSlyD (0.82 × 106 M−1 s−1 at 15 °C) [29] or TtSlyD (0.29 × 106 M−1 s−1 at 15 °C) [8], considering that our refolding assay was conducted at 20 °C. Deletion of the C-terminus only slightly reduced (approximately 20%) the PPIase activity. In contrast, the activities decreased by approximately 400- and 1,000-fold, respectively, for the loss of the IF domain (HpSlyDΔIF) and the loss of both the IF domain and the C-terminus (HpSlyDΔIFΔC). These results suggest that the metal-binding C-terminus has subtle effects on the catalytic activity of the protein and chaperone-active IF domain is likely to facilitate the functioning of the FKBP domain in proline-dependent refolding of protein substrates.
Previously, both FK506 and rapamycin have been shown to act as potent inhibitors of the PPIase activity of some of the FKBPs (e.g., HsFKBP12 [42] and MtFKBP17 [11]). The chemical shift perturbations induced by either direct binding or conformational changes were evaluated. Addition of FK506 caused shifts of several residues, e.g., Ile-15, Glu-18, Asp-29, Ile-41, Thr-43, Gln-45, Ile-47, Val-65, Val-66, Ile-67, Glu-70, Gly-74, Leu-130, Phe-137, and Arg-138. These residues are exclusively from the FKBP domain and are located either on or near the active sites (Fig. 5c), indicating a chemical shift variation on the fast equilibrium timescale. Similar results were obtained for rapamycin (Fig. 5d), except that perturbed residues experience chemical shift variation on the slow equilibrium timescale. These results demonstrated that both immunosuppressive agents are able to compete with the substrate for binding to the active sites, resulting in the inhibition of the PPIase activity of HpSlyD. In both cases, the residues for the IF domain remained unperturbed. As shown in Table S2, the drugs inhibit the PPIase activity only at a relatively high concentration (10 μM), probably attributed to the low affinity of the drugs for the active sites [9, 48].
Chaperone activity of the IF domain
The IF domain of HpSlyD is highly flexible and has large exposed hydrophobic surfaces, which results in its binding to unfolded hydrophobic protein or peptide patches. Such a chaperone activity of the protein was noticed previously in other FKBP family proteins [8–11]. To gain deep insight into the chaperone activity of HpSlyD, several unfolded proteins or peptides were used to interact with the protein (unlabeled or 15N-labeled HpSlyDΔC) monitored by either fluorescence spectroscopy or 2D [1H–15N] HSQC.
As expected, when well-folded proteins were added to 0.3 mM 15N-labeled HpSlyDΔC, no chemical shift perturbation was observed, indicating there was no interaction (data not shown). In contrast, when RCM-La, a permanently unfolded model substrate, was added to the protein solution, large chemical shift changes (larger than 0.04 ppm) were found for Leu-81, Phe-89, Ile-92, Glu-93, Val-100, Asp-106, Ile-110, and Ala-112 (Fig. 5e). In addition, Gly-102 completely disappeared. These residues are exclusively located at the IF domain (Fig. 5e). Similarly, the aggregation-prone insulin B chain binds to HpSlyDΔC and perturbed chemical shifts (larger than 0.04 ppm) of residues exclusively from the IF domain, e.g., Leu-81, Val-84, Phe-89, Val-100, Gly-102, Gln-103, Asp-106, Asn-107, Thr-109, Ile-110, Gln-111, His-128, and Leu-130 (Fig. 5f).
The chaperone activities were investigated for HpSlyD and its variants as shown in Fig. S6. At a molar ratio of proteins to insulin of about 0.5:1, HpSlyD reduced the intensity of the scattered light by almost a half owing to the aggregation of insulin, whereas HpSlyDΔC caused less reduction in the intensity of the scattered light. Further increase in the molar ratio of proteins to insulin to 1:1 resulted in complete inhibition of DTT-induced aggregation of insulin B for both HpSlyD and HpSlyDΔC, indicating that the C-terminus is not indispensable for chaperone function of the IF domain. However, HpSlyDΔIF and HpSlyDΔIFΔC, lacking the IF domain, exhibited almost no chaperone activity.
Discussion
Unique structural properties of SlyD
The solution structure of HpSlyDΔC reported here is similar to that of homologues such as EcSlyD [9, 10], TtSlyD [8], and MtFKBP17 [11] with two functional, spatially separated and orientation-independent domains, the FKBP and IF domains. The additional C-terminal α4 in the FKBP domain is likely present in full-length HpSlyD (Fig. 7), and is also found in EcSlyD [9, 10] and TtSlyD [8], and might be a common feature for SlyD proteins. α4 represents a novel extension to the PPIase fold and is probably coupled with the C-terminal metal-binding tail to regulate PPIase activity, which is manifested by conformational changes of the protein upon binding of Ni2+ (Fig. 5a), in agreement with the findings of a previous study on EcSlyD [10]. Similar to other SlyD-like FKBPs, the two domains of HpSlyD are self-oriented and lack interdomain contacts, and exhibit different rotational correlation times. Such an orientational arrangement of the two domains may facilitate mutual cooperation for protein functioning, which may be a common characteristic for SlyD proteins, distinct from other PPIases containing additional chaperone domains, e.g., trigger factor [49], periplasmic FkpA [50], and SurA [51]. The PPIase and chaperone activities of these proteins are normally independent owing to lack of internal mobility. The catalytic sites of SlyD proteins are highly conserved and consist of mainly hydrophobic residues to enable substrate binding (Figs. 6, 7). Differences in the properties of certain residues such as deeper penetration of Tyr-68 in EcSlyD [9] or the size of the hydrophobic pocket (smaller in TtSlyD [8] but larger in HpSlyD) may result in slight differences in the catalytic activity. In particular, the IF domain has an “incomplete” β-barrel structure with high conformational dynamics (Fig. S4), allowing it to adapt rapidly to different unfolded protein substrates. Binding of unfolded protein substrates would significantly increase the stability of this domain by the “completion” of the β-barrel [8]. Taken together, the unique structure properties of SlyD may provide a basis for multifunctionality of the protein.
Fig. 7.

Multiple sequence alignments of HpSlyD and its homologues using the MUSCLE algorithm [56] with default parameters. Secondary structure elements were annotated by Geneious 5.3.4 (http://www.geneious.com) according to the calculated NMR structure as shown in Fig. 2c and other resolved structures of SlyD homologues. The α2 helix (in red) is only present in FKBP from Methanothermococcus thermolithotrophicus (MtFKBP17). Identical and similar residues between the homologues are highlighted in black or gray. The conserved HGHXH sequence previously mentioned is encircled in a red box. Residues of peptidylprolyl isomerase (PPIase) catalytic sites of HpSlyD are highlighted with blue. The UniPort accession numbers are as follows: Q6MQW6 (Bdellovibrio bacteriovorus SlyD), A1VXJ8 (Campylobacter jejuni SlyD), P0A9K9 (EcSlyD), P44830 (Haemophilus influenza SlyD), O25748 (HpSlyD), Q0EWE3 (Mariprofundus ferrooxydans), Q2W0E9 (Magnetospirillum magneticum), A3CV43 (Methanoculleus marisnigri FKBP PPIase), O52980 (MtFKBP17), Q82U07 (Nitrosomonas europaea SlyD), C7N8E0 (Slackia heliotrinireducens PPIase), O83369 (Treponema pallidum SlyD), and Q5SLE7 (TtSlyD)
Metal binding properties
Different from EcSlyD [10, 17], no significant changes in the circular dichroism spectra of HpSlyD were observed upon addition of different metal ions, indicative of identical secondary structure of the protein. For EcSlyD, the binding of Ni2+ causes the conversion of about 10% of β-sheet structure into an irregular structure [10]. This may account for the different length and composition of the C-terminal tail, since the C-terminus of HpSlyD possesses no negatively charged residues (Glu and Asp), unlike in EcSlyD. Furthermore, on the basis of sequence alignment, HpSlyD lacks the conserved HGHXH sequence motif at the beginning of the C-terminus, which was shown to tightly bind Ni2+ [8] and is shared by other non-epsilonproteobacterial SlyD homologues (Fig. 7). Our 2D [1H–15N] HSQC data demonstrated that binding of Ni2+ (only the first stoichiometry) to the protein perturbed several residues in the active sites of the PPIase domain, suggesting that residues in this region either directly interact with metal ions or undergo certain conformational changes. In contrast, no such perturbation was observed upon Zn2+ binding, probably owing to the binding of metal at the C-terminus, which is too flexible to be observed in the 2D [1H–15N] HSQC spectrum. No histidines are involved in nickel binding except at the C-terminus, again different from EcSlyD (His-36 and His-38 bind nickel ions) [10]. Unlike EcSlyD (most residues at β5b), two important residues for catalysis (Leu-37 and Phe-39 of HpSlyD) were not affected upon nickel binding owing to the absence of histidine residues. We hypothesized that the effect of nickel binding on PPIase activity of HpSlyD is different from that for EcSlyD, possibly owing to the β-sheet β5b being undisrupted upon binding. The inhibition of the PPIase activity upon nickel binding [17] is mainly due to the unnecessity of the activity during the maturation processes of [NiFe] hydrogenase [52].
Interestingly, zinc ions induce the dimerization of the protein but not nickel, as shown in Fig. 1. The loss of zinc from HpSlyD may favor the normal function of SlyD.
Implication of HpSlyD in maturation of urease and hydrogenase
Both [NiFe] hydrogenase and urease are crucial for H. pylori and their maturations are complicated, involving multiple chaperones and maturation factors [1]. SlyD has been shown to participate in the maturation of both urease and hydrogenase in H. pylori [6, 15]. A recent interactomics study in H. pylori showed that SlyD interacts with HypB (a nickel-binding accessory protein) [6]. Such an interaction was also observed in E. coli [13], and the deletion of the IF domain of SlyD or the mutation of the proline-rich stretch between the N-terminal CXXCGC nickel-binding motif and the GTPase domain of HypB disrupted the interaction [53]. HypB from H. pylori (HpHypB; UniProt accession number O25560) lacks both the N-terminal nickel binding site and the proline-rich region. Nevertheless, our preliminary work indicated that HpSlyD binds to HpHypB (data not shown). The molecular and structural basis for such an interaction might be distinct from that for E. coli and is currently under investigation in our laboratory.
We demonstrated that HpSlyD binds the Tat signal peptide of the H. pylori [NiFe] hydrogenase small subunit for the first time (Fig. 5b). In E. coli, two chaperone proteins, HyaE and HybE, were shown to be able to interact with Tat-signal-peptide-bearing hydrogenase ([NiFe] hydrogenases 1 and 2) precursors to proofread the assembly of hydrogenase complexes and prevent the export of immature Tat-dependent hydrogenases [54]. However, there is no homologue of the two proteins in H. pylori; SlyD may therefore serve a similar role to HyaE and HybE and participate in the maturation process of hydrogenase in H. pylori by binding to Tat signal peptide of [NiFe] hydrogenase and escorting it to the Tat translocon as proposed previously [6]. It remains unclear how SlyD senses the immature [NiFe] hydrogenase and ensures insertion of the cofactor and transports the maturated hydrogenase complex across the cytoplasmic membrane. Further study is necessary to clarify whether the unique C-terminus of SlyD plays such a role. The physiological relevance of the nickel binding and the associated conformational changes of SlyD in H. pylori remain to be defined.
Sequence comparison of HpHypB with its counterparts in other species showed that HpHypB is deficient in nickel binding residues such as a polyhistidine stretch, which might be compensated by the C-terminal extended HpSlyD. The binding of nickel ions to SlyD impaired the PPIase activity of SlyD [17], which is not required for hydrogenase assembly in E. coli as demonstrated previously [53], through conformational changes. We propose that SlyD, once bound to nickel ions, will donate the metal ions by working together with HypA/HypB in urease maturation [15]. Prior to insertion of nickel ions into hydrogenase, HpSlyD will prohibit the transport of the immature, cofactor-deficient [NiFe] hydrogenase across the cytoplasmic membrane by binding to the hydrogenase small subunit Tat signal peptide. Recently, SlyD in E. coli was shown to have the capability to interact with the Tat-signal-peptide-free large-subunit [NiFe] hydrogenase 3 (HycE) through its IF domain [55], which complicated the role of SlyD in the maturation process of hydrogenase. The present study might at least shed light on the role of SlyD in the hydrogenase assembly and translocation.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This work was supported by the Research Grants Council of Hong Kong (HKU107C, HKU704207P, HKU703808P, HKU704909P, N_HKU75209), the Croucher Foundation, and the University of Hong Kong. Electrospray ionization mass spectrometry was performed at the High Performance Tandem Mass Spectrometry Facility (HKU), partially supported by a Special Equipment Grant from the University Grants Committee of the Hong Kong Special Administrative Region, China (project code SEG_HKU02). The coordinates and chemical shifts of HpSlyD have been deposited in the Protein Data Bank (2KR7) and BioMagResBank (16629).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- DTT
dl-Dithiothreitol
- EcSlyD
SlyD from Escherichia coli
- FKBP
FK506-binding protein
- HpHydA SP
Twin-arginine translocation signal peptide with the sequence taken from the [NiFe] hydrogenase small subunit (pre-HydA) of Helicobacter pylori
- HpHypB
HypB from Helicobacter pylori
- HpSlyD
SlyD in Helicobacter pylori
- HpSlyDΔC
C-terminus-truncated SlyD from Helicobacter pylori
- HpSlyDΔIF
Insert-in-flap-domain-deleted SlyD from Helicobacter pylori
- HpSlyDΔIFΔC
C-terminus-truncated insert-in-flap-domain-deleted SlyD from Helicobacter pylori
- HsFKBP12
Human FK506-binding protein
- HSQC
Heteronuclear single quantum coherence
- IF
Insert-in-flap
- IMAC
Immobilized metal ion affinity chromatography
- MBP
Maltose-binding protein
- MtFKBP17
FK506-binding protein from Methanothermococcus thermolithotrophicus
- NOE
Nuclear Overhauser effect
- PPIase
Peptidylprolyl isomerase
- RCM-La
Reduced and carboxymethylated α-lactalbumin
- RCM-T1
Reduced and carboxymethylated (Ser54Gly, Pro55Asn) RNase T1
- RMSD
Root mean square deviation
- SP-less MalE
Signal-peptide-less maltose-binding protein
- Tat
Twin-arginine translocation
- TCEP
Tris(2-carboxyethyl)phosphine hydrochloride
- Tris
Tris(hydroxymethyl)aminomethane
- TtSlyD
SlyD from Thermus thermophilus
Footnotes
T. Cheng and H. Li contributed equally to the study.
References
- 1.Kusters JG, van Vliet AHM, Kuipers EJ. Clin Microbiol Rev. 2006;19:449–490. doi: 10.1128/CMR.00054-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Evans DJ, Jr, Evans DG, Kirkpatrick SS, Graham DY. Microb Pathog. 1991;10:15–26. doi: 10.1016/0882-4010(91)90062-F. [DOI] [PubMed] [Google Scholar]
- 3.Maier RJ, Fu C, Gilbert J, Moshiri F, Olson J, Plaut AG. FEMS Microbiol Lett. 1996;141:71–76. doi: 10.1111/j.1574-6968.1996.tb08365.x. [DOI] [PubMed] [Google Scholar]
- 4.Benoit S, Maier RJ. J Bacteriol. 2003;185:4787–4795. doi: 10.1128/JB.185.16.4787-4795.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mehta N, Olson JW, Maier RJ. J Bacteriol. 2003;185:726–734. doi: 10.1128/JB.185.3.726-734.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stingl K, Schauer K, Ecobichon C, Labigne A, Lenormand P, Rousselle JC, Namane A, de Reuse H. Mol Cell Proteomics. 2008;7:2429–2441. doi: 10.1074/mcp.M800160-MCP200. [DOI] [PubMed] [Google Scholar]
- 7.Olson JW, Mehta NS, Maier RJ. Mol Microbiol. 2001;39:176–182. doi: 10.1046/j.1365-2958.2001.02244.x. [DOI] [PubMed] [Google Scholar]
- 8.Löw C, Neumann P, Tidow H, Weininger U, Haupt C, Friedrich-Epler B, Scholz C, Stubbs MT, Balbach J. J Mol Biol. 2010;398:375–390. doi: 10.1016/j.jmb.2010.03.014. [DOI] [PubMed] [Google Scholar]
- 9.Weininger U, Haupt C, Schweimer K, Graubner W, Kovermann M, Brüser T, Scholz C, Schaarschmidt P, Zoldak G, Schmid FX, Balbach J. J Mol Biol. 2009;387:295–305. doi: 10.1016/j.jmb.2009.01.034. [DOI] [PubMed] [Google Scholar]
- 10.Martino L, He Y, Hands-Taylor KL, Valentine ER, Kelly G, Giancola C, Conte MR. FEBS J. 2009;276:4529–4544. doi: 10.1111/j.1742-4658.2009.07159.x. [DOI] [PubMed] [Google Scholar]
- 11.Suzuki R, Nagata K, Yumoto F, Kawakami M, Nemoto N, Furutani M, Adachi K, Maruyama T, Tanokura M. J Mol Biol. 2003;328:1149–1160. doi: 10.1016/S0022-2836(03)00379-6. [DOI] [PubMed] [Google Scholar]
- 12.Kay JE. Biochem J. 1996;314:361–385. [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang JW, Butland G, Greenblatt JF, Emili A, Zamble DB. J Biol Chem. 2005;280:4360–4366. doi: 10.1074/jbc.M411799200. [DOI] [PubMed] [Google Scholar]
- 14.Roof WD, Fang HQ, Young KD, Sun JL, Young R. Mol Microbiol. 1997;25:1031–1046. doi: 10.1046/j.1365-2958.1997.5201884.x. [DOI] [PubMed] [Google Scholar]
- 15.Benanti EL, Chivers PT. J Bacteriol. 2009;191:2405–2408. doi: 10.1128/JB.01657-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wülfing C, Lombardero J, Plückthun A. J Biol Chem. 1994;269:2895–2901. [PubMed] [Google Scholar]
- 17.Hottenrott S, Schumann T, Plückthun A, Fischer G, Rahfeld JU. J Biol Chem. 1997;272:15697–15701. doi: 10.1074/jbc.272.25.15697. [DOI] [PubMed] [Google Scholar]
- 18.Mitterauer T, Nanoff C, Ahorn H, Freissmuth M, Hohenegger M. Biochem J. 1999;342:33–39. doi: 10.1042/0264-6021:3420033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Graubner W, Schierhorn A, Brüser T. J Biol Chem. 2007;282:7116–7124. doi: 10.1074/jbc.M608235200. [DOI] [PubMed] [Google Scholar]
- 20.Ilver D, Rappuoli R, Telford JL (2001) In: Mobley HLT, Mendz GL, Hazell SL (eds) Helicobacter pylori: physiology and genetics. ASM Press, Washington, pp 219–226 [PubMed]
- 21.Dalbey RE, Robinson C. Trends Biochem Sci. 1999;24:17–22. doi: 10.1016/S0968-0004(98)01333-4. [DOI] [PubMed] [Google Scholar]
- 22.Wickner W, Schekman R. Science. 2005;310:1452–1456. doi: 10.1126/science.1113752. [DOI] [PubMed] [Google Scholar]
- 23.Ge RG, Zhang Y, Sun XS, Watt RM, He QY, Huang JD, Wilcox DE, Sun HZ. J Am Chem Soc. 2006;128:11330–11331. doi: 10.1021/ja062589t. [DOI] [PubMed] [Google Scholar]
- 24.Ge RG, Watt RM, Sun XS, Tanner JA, He QY, Huang JD, Sun HZ. Biochem J. 2006;393:285–293. doi: 10.1042/BJ20051160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ge RG, Sun HZ. Acc Chem Res. 2007;40:267–274. doi: 10.1021/ar600001b. [DOI] [PubMed] [Google Scholar]
- 26.Zeng YB, Zhang DM, Li HY, Sun HZ. J Biol Inorg Chem. 2008;13:1121–1131. doi: 10.1007/s00775-008-0397-0. [DOI] [PubMed] [Google Scholar]
- 27.Cun SJ, Li HY, Ge RG, Lin MCM, Sun HZ. J Biol Chem. 2008;283:15142–15151. doi: 10.1074/jbc.M800591200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xia W, Li H, Sze KH, Sun HZ. J Am Chem Soc. 2009;131:10031–10040. doi: 10.1021/ja900543y. [DOI] [PubMed] [Google Scholar]
- 29.Knappe TA, Eckert B, Schaarschmidt P, Scholz C, Schmid FX. J Mol Biol. 2007;368:1458–1468. doi: 10.1016/j.jmb.2007.02.097. [DOI] [PubMed] [Google Scholar]
- 30.Maillard J, Spronk CAEM, Buchanan G, Lyall V, Richardson DJ, Palmer T, Vuister GW, Sargent F. Proc Natl Acad Sci USA. 2007;104:15641–15646. doi: 10.1073/pnas.0703967104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gill SC, von Hippel PH. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 32.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 33.Goddard TD, Kneller DG. SPARKY 3. University of California, San Francisco
- 34.Cornilescu G, Delaglio F, Bax A. J Biomol NMR. 1999;13:289–302. doi: 10.1023/A:1008392405740. [DOI] [PubMed] [Google Scholar]
- 35.Grzesiek S, Bax A, Clore GM, Gronenborn AM, Hu JS, Kaufman J, Palmer I, Stahl SJ, Wingfield PT. Nat Struct Biol. 1996;3:340–345. doi: 10.1038/nsb0496-340. [DOI] [PubMed] [Google Scholar]
- 36.Herrmann T, Güntert P, Wüthrich K. J Mol Biol. 2002;319:209–227. doi: 10.1016/S0022-2836(02)00241-3. [DOI] [PubMed] [Google Scholar]
- 37.Pearlman DA, Case DA, Caldwell JW, Ross WS, Cheatham TE, Debolt S, Ferguson D, Seibel G, Kollman P. Comput Phys Commun. 1995;91:1–41. doi: 10.1016/0010-4655(95)00041-D. [DOI] [Google Scholar]
- 38.Laskowski RA, Rullmann JAC, MacArthur MW, Kaptein R, Thornton JM. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 39.Koradi R, Billeter M, Wüthrich K. J Mol Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 40.DeLano WL. PyMOL, release 0.99rc6. Palo Alto: DeLano Scientific; 2006. [Google Scholar]
- 41.Scholz C, Eckert B, Hagn F, Schaarschmidt P, Balbach J, Schmid FX. Biochemistry. 2006;45:20–33. doi: 10.1021/bi051922n. [DOI] [PubMed] [Google Scholar]
- 42.Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J. Science. 1991;252:839–842. doi: 10.1126/science.1709302. [DOI] [PubMed] [Google Scholar]
- 43.Holm L, Park J. Bioinformatics. 2000;16:566–567. doi: 10.1093/bioinformatics/16.6.566. [DOI] [PubMed] [Google Scholar]
- 44.Stibrany RT, Fox S, Bharadwaj PK, Schugar HJ, Potenza JA. Inorg Chem. 2005;44:8234–8242. doi: 10.1021/ic050114h. [DOI] [PubMed] [Google Scholar]
- 45.Lever ABP. Inorganic electronic spectroscopy. 2. Amsterdam: Elsevier; 1984. [Google Scholar]
- 46.Chivers PT, Sauer RT. J Biol Chem. 2000;275:19735–19741. doi: 10.1074/jbc.M002232200. [DOI] [PubMed] [Google Scholar]
- 47.Cristóbal S, de Gier JW, Nielsen H, von Heijne G. EMBO J. 1999;18:2982–2990. doi: 10.1093/emboj/18.11.2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zoldák G, Schmid FX. J Mol Biol. 2011;406:176–194. doi: 10.1016/j.jmb.2010.12.010. [DOI] [PubMed] [Google Scholar]
- 49.Ludlam AV, Moore BA, Xu ZH. Proc Natl Acad Sci USA. 2004;101:13436–13441. doi: 10.1073/pnas.0405868101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saul FA, Arié JP, Vulliez-le Normand B, Kahn R, Betton JM, Bentley GA. J Mol Biol. 2004;335:595–608. doi: 10.1016/j.jmb.2003.10.056. [DOI] [PubMed] [Google Scholar]
- 51.Bitto E, McKay DB. Structure. 2002;10:1489–1498. doi: 10.1016/S0969-2126(02)00877-8. [DOI] [PubMed] [Google Scholar]
- 52.Zhang JW, Leach MR, Zamble DB. J Bacteriol. 2007;189:7942–7944. doi: 10.1128/JB.00922-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leach MR, Zhang JW, Zamble DB. J Biol Chem. 2007;282:16177–16186. doi: 10.1074/jbc.M610834200. [DOI] [PubMed] [Google Scholar]
- 54.Dubini A, Sargent F. FEBS Lett. 2003;549:141–146. doi: 10.1016/S0014-5793(03)00802-0. [DOI] [PubMed] [Google Scholar]
- 55.Chung KCC, Zamble DB. FEBS Lett. 2011;585:291–294. doi: 10.1016/j.febslet.2010.12.024. [DOI] [PubMed] [Google Scholar]
- 56.Edgar RC. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.