

Short abstract
The subcellular localization of the entire proteome of the yeast Saccharomyces cerevisiae, and comparison with less comprehensive studies of mammalian cells, provides insights into the localization of the mammalian proteome.
Abstract
The subcellular localization of the entire proteome of an organism, the yeast Saccharomyces cerevisiae, has been revealed for the first time. Comparison with less comprehensive studies of mammalian cells provides insights into the localization of the mammalian proteome.
Maintaining the integrity of cellular processes and pathways requires the careful orchestration of individual proteins and entire protein complexes, so that they interact at defined sites at the correct time. One way in which this is achieved is through intracellular compartmentalization. Membrane-bounded organelles and distinct cytoskeletal elements are key features of eukaryotic cells and serve to sequester components into restricted spaces. Identifying all the proteins of any particular organelle or macromolecular structure is therefore a key step towards a comprehensive understanding of cellular biology. Systematic bioinformatic analysis of data available from genome-sequencing projects has been one strategy used in an attempt to achieve this goal [1]. Another approach has been to use proteomics, whereby individual organelles are isolated and their constituents identified on a large scale by mass-spectroscopy methods (reviewed in [2]). Finally, a parallel strategy to systematically localize proteins on a large scale has been the cellular expression of tagged versions of proteins followed by their visualization in cells, thereby providing a view in vivo of the proteins that reside in any particular compartment.
The first of such large-scale gene-tagging and localization projects were carried out in the yeasts Saccharomyces cerevisiae and Schizosaccharomyces pombe [3,4], because both organisms are genetically tractable, are single-celled and therefore have less functional specialization than multicellular organisms, and possess only a modest number of genes compared with higher eukaryotes. The use of green fluorescent protein (GFP [5]) as the protein tag significantly increased the efficiency by which localizations could be ascribed and proved to set a standard for many subsequent studies using various cell lines from other organisms and increasingly large genomic and cDNA libraries (reviewed in [6]). Similar tagging approaches have also been developed for plants, initially using random cDNA-GFP fusions in Arabidopsis [7] and more recently using a cDNA library in Nicotiana [8].
Although each of these conceptually similar projects had their own specific advantages, they all suffered from the common problem of a high degree of potential redundancy, as proteins were not identified before they were studied: identifying a localization of interest is only the first step, and the protein that is localized must still be identified. There is therefore a risk that the protein has been identified previously and is already well characterized. When proportionally fewer or no proteins have been localized to the organelle of interest, however, as was the case in the screen by Escobar and colleagues [8], this problem appears to be less critical.
The completion of sequencing a variety of genomes now provides a resource through which the systematic identification of proteins localizing to a specific organelle can be managed without such redundancy problems. As open reading frames (ORFs) are predicted by the available sequence data, they can now be amplified and fused to either the amino or the carboxyl terminus of the GFP gene, or both, and the localizations of the resulting fusion proteins can be observed in transfected cells [9-11]. In this way, not only is localization information for unknown proteins obtained, but the effects of the position of the GFP tag on the localization can also be considered [9], which increases the data quality significantly.
Although rapid recombination-based cloning systems to create tagged ORFs for expression are now available, extending them to determine the localization of all predicted human proteins remains an enormous task, largely because of the complexity of multicellular animals. Not only are there splice variants of many proteins, but there is also a huge assortment of cell types, each with its own specialized function and therefore its own protein components. Furthermore, determination of exactly how many ORFs exist in the human genome, a prerequisite for determining the subcellular localization of each of the proteins they encode, remains incomplete.
Attempts to find the localization of all proteins (the 'localizome') for an entire organism have therefore now returned to the yeast S. cerevisiae. Using a combination of directed high-throughput tagging of ORFs with the V5 epitope (derived from the P and V proteins of simian virus 5) and random transposon tagging with the hemagglutinin (HA) epitope, followed by immunofluorescence, the localizations of a total of 2,744 proteins, representing 44% of the genome, have been experimentally determined [10]. The authors of this study also integrated their results with previously reported localizations, thereby increasing the coverage to 55%. Finally, they used a Bayesian analysis to extrapolate from the results, thereby providing for the first time an overview of protein localization for an entire organism.
Very recent work has now extended the experimental determination of the yeast localizome. Using PCR amplification of every predicted ORF into a GFP-containing cassette followed by homologous recombination into the genome, the laboratories of Weissman and O'Shea have created a collection of yeast strains in which the products of 97% (6,029) of the ORFs are GFP-tagged at their carboxyl termini [11]. The real achievement of this work is not only that it provides a means to determine the localization of every protein, but also that proteins are expressed from their endogenous promoters and are therefore most likely to be present at physiological levels. The results [11] revealed the localizations of 4,156 proteins, 75% of all predicted ORFs, and these were assigned to 22 localization categories using a two-stage strategy: first by observing the GFP-tagged proteins alone, and then by using colocalization experiments with well-established proteins tagged with the red fluorescent protein (RFP). Some of these (2,526) had already been assigned localizations in the Saccharomyces genome database [12], and 80% of these were in agreement with the GFP tagging results [11]. More importantly, of the proteins that had not been previously localized (2,374), the data [11] provide localization information for 70%. For example, of the 164 proteins now identified as localizing in the nucleolus, 82 were newly identified by this work [11]. Finally, the authors [11] correlated their localization data with that contained in the GRID database, a repository of protein-protein interaction data [13,14]. This allowed them to look at the localizations of each interacting pair of proteins described in the GRID database and from this calculate the number of interactions occurring between each compartment compared with the number calculated for a random dataset. They found enrichments in interactions between different cellular locations - for example between actin structures and the bud neck of the dividing cell. This reflects the dynamic interchange of proteins between compartments, an essential feature of cell function.
Despite the impressive scale of the work by Huh et al. [11], a number of interesting questions arise. Firstly, almost 2,000 of the strains generated did not provide localization information. Do these represent proteins that are expressed at specific stages of the cell cycle, or only in response to external stimuli? Alternatively, are they regulatory proteins that are constitutively expressed at low (undetectable) levels, but that nevertheless perform vital functions? Secondly, GFP-tagging of ORFs at only one end undoubtedly means that the localizations of some protein classes will be incorrectly assigned and that certain categories will therefore be under-represented. For example, many proteins of the endoplasmic reticulum (ER) rely on a carboxy-terminal KDEL or KKXX motif (in the single-letter amino-acid code) for their sequestration in this organelle, and the masking of these signals by GFP may result in ER proteins being delivered to downstream compartments of the secretory pathway.
Overall, the experimental data of Kumar et al. [10] and Huh et al. [11] largely agree in terms of the distribution of proteins to various organelles (Figure 1), but do these datasets from yeast also provide insights into the localizome of mammalian cells? If, indeed, 7% and 2.5% of proteins are localized to the ER and Golgi complex, respectively, extrapolation of the yeast data [10,11] to a human genome containing approximately 30,000 ORFs would predict that there would be 2,100 and 750 proteins, respectively, in these two compartments in human cells. This would be consistent with a recent proteomic analysis of the Golgi complex from rat liver cells that identified 588 distinct protein spots, of which 394 represented probable residents [15]. Clearly, although proteomics is a valuable tool, it remains an open question whether current subcellular proteomic approaches allow the comprehensive identification of all proteins making up an organelle, and the number of proteins associated with the Golgi complex in the rat liver cell study [15] may thus be an underestimate.
Figure 1.
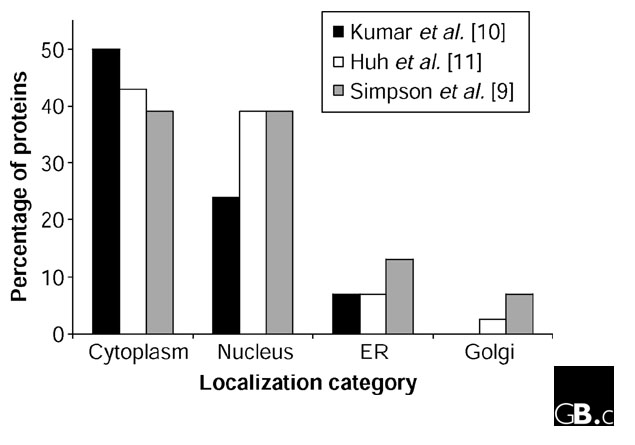
Comparison of the distribution of GFP-tagged proteins to different subcellular organelles as determined in three different studies [9-11]. The percentage of proteins localizing to the cytoplasm, nucleus, endoplasmic reticulum (ER) and Golgi are shown as example compartments. The data are taken for yeast cells [10,11] and for mammalian cells [9]. In [10] Golgi localization was not determined.
In this context, it is interesting to note that extrapolation of our localization data from almost 600 GFP-tagged novel human ORFs ([9]; see also [16] for an update) indicates that as many as 3,900 (13%) and 2,100 proteins (7%) encoded by the human genome may localize to the ER and Golgi, respectively (Figure 1). These numbers are much higher than those extrapolated from the yeast data [10,11]; one interpretation of the discrepancy could be that localization studies are easier in large mammalian cells, resulting in a greater accuracy in identifying structures, particularly those in the secretory pathway. Alternatively, it may be that because mammalian cells are more complex in terms of the molecules they secrete, the ER and Golgi require a greater protein diversity. In addition, the protein composition of the ER and Golgi may vary between different cell types in humans, depending on the cell specialization, thus necessitating a higher number of ER- and Golgi-localized proteins encoded by the human genome. Continued large-scale localization analysis of human proteins therefore seems essential if we are finally to establish a localizome for mammalian cells.
The localization of an entire proteome [11] marks a milestone in our quest to understand cell function. This is just the first step towards the goal, however, because only the integration of large localization datasets with other functional data will ultimately provide a biological atlas of function [17]. For example, the combination of the new localization information with genome-wide protein-protein interaction data [18] should both serve to corroborate predicted functions and improve the confidence in each dataset. Other new resources, such as the collection of yeast strains expressing tandem affinity purification (TAP)-tagged proteins from their endogenous chromosomal locations, are also ready for exploitation [19]. Finally, all these experimental data need to be combined so that they can be visualized in parallel, both for individual proteins of interest and for whole subcellular structures. This will then allow further refinement of bioinformatic tools that can be used to 'harvest' similar information from more complex organisms. Future genome-wide projects will undoubtedly use more functional assays with resources such as those described here, and it should now be possible to tailor the assays to proteins residing in a specific location. In this way we will be able to observe the effect of manipulating one protein on the entire protein population of the organelle or structure of interest. Ultimately, localizome information will be instrumental in establishing the interrelationships between the proteins that determine biological function.
Acknowledgments
Acknowledgements
The Pepperkok laboratory is supported by the BMBF with grants 01GR0101 in the National Genome Research Network (NGFN) and 01KW0013 (EMBL) within the German Genome Project (DHGP).
References
- Drawid A, Gerstein M. A Bayesian system integrating expression data with sequence patterns for localizing proteins: comprehensive application to the yeast genome. J Mol Biol. 2000;301:1059–1075. doi: 10.1006/jmbi.2000.3968. [DOI] [PubMed] [Google Scholar]
- Dreger M. Proteome analysis at the level of subcellular structures. Eur J Biochem. 2003;270:589–599. doi: 10.1046/j.1432-1033.2003.03426.x. [DOI] [PubMed] [Google Scholar]
- Burns N, Grimwade B, Ross-Macdonald PB, Choi EY, Finberg K, Roeder GS, Snyder M. Large-scale analysis of gene expression, protein localization, and gene disruption in Saccharomyces cerevisiae. Genes Dev. 1994;8:1087–1105. doi: 10.1101/gad.8.9.1087. [DOI] [PubMed] [Google Scholar]
- Sawin KE, Nurse P. Identification of fission yeast nuclear markers using random polypeptide fusions with green fluorescent protein. Proc Natl Acad Sci USA. 1996;93:15146–15151. doi: 10.1073/pnas.93.26.15146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene expression. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- Pepperkok R, Simpson JC, Wiemann S. Being in the right location at the right time. Genome Biol. 2001;2:reviews1024.1–1024.4. doi: 10.1186/gb-2001-2-9-reviews1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler SR, Ehrhardt DW, Griffitts JS, Somerville CR. Random GFP-cDNA fusions enable visualization of subcellular structures in cells of Arabidopsis at a high frequency. Proc Natl Acad Sci USA. 2000;97:3718–3723. doi: 10.1073/pnas.97.7.3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar NM, Haupt S, Thow G, Boevink P, Chapman S, Oparka K. High-throughput viral expression of cDNA-green fluorescent protein fusions reveals novel subcellular addresses and identifies unique proteins that interact with plasmodesmata. Plant Cell. 2003;15:1507–1523. doi: 10.1105/tpc.013284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson JC, Wellenreuther R, Poustka A, Pepperkok R, Wiemann S. Systematic subcellular localisation of novel proteins identified by large-scale cDNA sequencing. EMBO Rep. 2000;1:287–292. doi: 10.1093/embo-reports/kvd058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Agarwal S, Heyman JA, Matson S, Heidtman M, Piccirillo S, Umansky L, Drawid A, Jansen R, Liu Y, et al. Subcellular localization of the yeast proteome. Genes Dev. 2002;16:707–719. doi: 10.1101/gad.970902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O'Shea EK. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–691. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- Saccharomyces Genome Database http://www.yeastgenome.org/
- Breitkreutz BJ, Stark C, Tyers M. The GRID: the General Repository for Interaction Datasets. Genome Biol. 2003;4:R23. doi: 10.1186/gb-2003-4-3-r23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The GRID: the General Repository for Interaction Datasets http://biodata.mshri.on.ca/grid
- Taylor RS, Wu CC, Hays LG, Eng JK, Yates JR, 3rd, Howell KE. Proteomics of rat liver Golgi complex: minor proteins are identified through sequential fractionation. Electrophoresis. 2000;21:3441–3459. doi: 10.1002/1522-2683(20001001)21:16<3441::AID-ELPS3441>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- The cDNA-GFP Localisation Project http://www.embl-heidelberg.de/gfp-cdna/
- Vidal M. A biological atlas of functional maps. Cell. 2001;104:333–339. doi: 10.1016/s0092-8674(01)00221-5. [DOI] [PubMed] [Google Scholar]
- Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O'Shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]