

Abstract
We describe a general approach for identifying components of subcellular structures in a multicellular organism by exploiting the ability to generate thousands of independent transformants in Arabidopsis thaliana. A library of Arabidopsis cDNAs was constructed so that the cDNAs were inserted at the 3′ end of the green fluorescent protein (GFP) coding sequence. The library was introduced en masse into Arabidopsis by Agrobacterium-mediated transformation. Fluorescence imaging of 5,700 transgenic plants indicated that ≈2% of lines expressed a fusion protein with a different subcellular distribution than that of soluble GFP. About half of the markers identified were targeted to peroxisomes or other subcellular destinations by non-native coding sequence (i.e., out-of-frame cDNAs). This observation suggests that some targeting signals are of sufficiently low information content that they can be generated frequently by chance. The potential of the approach for identifying markers with unique dynamic processes is demonstrated by the identification of a GFP fusion protein that displays a cell-cycle regulated change in subcellular distribution. Our results indicate that screening GFP-fusion protein libraries is a useful approach for identifying and visualizing components of subcellular structures and their associated dynamics in higher plant cells.
Much of our current understanding of cellular structure has been derived from the use of methods that create static images or that obscure structure in individual cells, such as the analysis of fixed tissue specimens or fractionated cellular constituents. More representative kinds of information that allow cellular structures and dynamics to be observed in their native states can be obtained from observations of living cells. The use of green fluorescent protein (GFP) facilitates the construction of cytological markers for live cell biological studies as chimeric proteins composed of GFP and a protein of interest (1, 2). This approach can be highly successful but typically requires prior knowledge of protein localization or targeting signals (3).
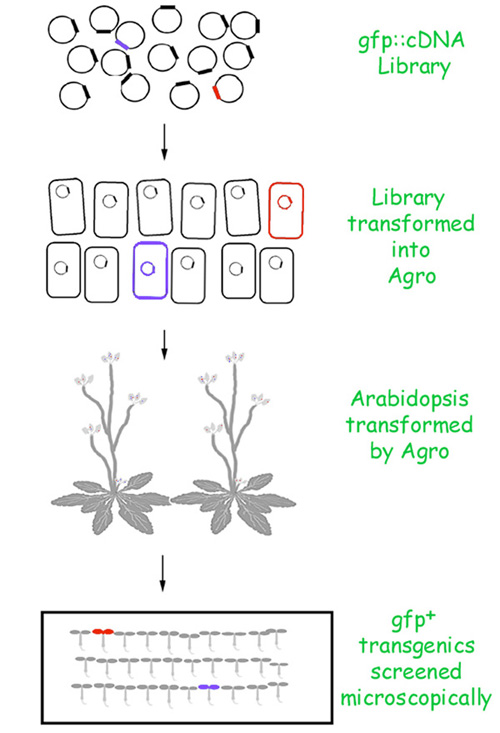
We have explored a high-throughput approach to examining subcellular organization by producing large numbers of transgenic plants that express random GFP∷cDNA fusions and observing them by using fluorescence microscopy. In principle, this strategy should facilitate the identification of large numbers of targeted GFP fusion proteins that can be used for the direct identification of novel features of subcellular organization. The ease with which Arabidopsis can be transformed by Agrobacterium facilitated the production of large numbers of independent transgenic lines. In addition, Arabidopsis has large cells that are readily amenable to microscopic observation of subcellular structure. To assess the utility of the approach, we have analyzed 5,700 transgenic plants to determine the frequency with which useful fusions can be isolated, the spectrum of structures marked, and the identity of the fusion protein sequences isolated.
The approach we have taken is similar, in principle, to a GFP marking strategy performed by using the yeast Saccharomyces pombe (4). In that study, random GFP∷genomic DNA fusions were prescreened for inducible cytotoxicity and then were examined microscopically for interesting localization features. In addition, during the course of this work, a similar study reported the use of GFP∷cDNA fusion libraries to mark subcellular structure in mammalian tissue culture cells (5).
Our approach is complementary to these studies in that the use of a multicellular organism permits the identification of fusion proteins that exhibit differential localization in different cells, intracellular structures, or other localization dependent on multicellularity. Also, by exploiting the extensive genome sequence information for Arabidopsis, we have observed that many out-of-frame fusions result in GFP localization to various subcellular structures because of the low information content of some targeting signals. Overall, our results indicate that random GFP∷cDNA fusion libraries can be used to efficiently mark a wide variety of subcellular structures and dynamic processes in plant cells. In addition to facilitating marker isolation, the approach is effective for exploring the existence of novel subcellular structures, domains, and dynamic processes in living cells.
Materials and Methods
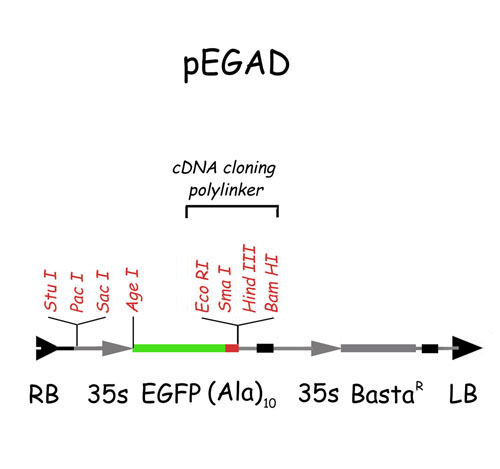
Vector Construction.
pEGAD was constructed in three major steps: (i) A binary Ti plasmid with the basta herbicide resistance locus was constructed by using a pBI121 derivative, pBIMC (Deanne Falcone, University of Kentucky), as a starting point. A SphI fragment containing the NPTII gene and the T-DNA right border was removed from pBIMC and was replaced with a PCR fragment containing the T-DNA right border and several unique cloning sites. A Basta herbicide resistance gene was isolated from pDHB321.1 (David Bouchez, Institut National de la Recherche Agronomique) as an EcoRI/ClaI fragment, was blunted with the Klenow fragment of DNA polymerase I, and was inserted into the nascent pEGAD precursor at a Klenowed EcoRI site adjacent to the T-DNA left border. The remaining EcoRI and HindIII sites in this derivative were destroyed, yielding pBasta. (ii) A DNA fragment containing mulitcloning sites (MCS) and an (Ala)10 flexible linker was synthesized with BamHI and BspEI overhangs and was ligated to pEGFP-C1 (CLONETECH) digested with BspEI and BamHI. The (Ala)10-MCS synthetic linker was made by annealing the following synthetic oligonucleotides in vitro [(Ala)10MCS(+) 5′-CCGGAGCTGCGGCCGCTGCCGCTGCGGCAGCGG- CCGAATTCCCCGGGCTCGAGAAGCTTG; (Ala)10MCS(−) 5′-GATCCAAGCTTCTCGAGCCCGGGGAATTCGGCC-GCTGCCGCAGCGGCAGCGGCCGCAGCT]. (iii) The final vector, pEGAD, was constructed by subcloning the EGFP-(Ala)10MCS cassette into pBasta, driving GFP expression from a CaMV 35S promoter present in the parent plasmid pBI121. The deduced sequence of pEGAD was deposited in GenBank (accession no. AF218816). A diagram of the vector is presented in the supplemental data on the PNAS web site, www.pnas.org. The (Ala)10 linker was modeled after that used by Doyle and Botstein (6). The EGFP gene used in these studies contains the chromophore described by Cormack et al. (7), and the codon optimization described by Haas et al. (8) and Chiu et al. (9). The expression plasmid and strains described here are available from the Arabidopsis Biological Resource Center.
cDNA Library Construction.
Two cDNA libraries were made by using poly(A) mRNA isolated from an equal weight mixture of mRNA isolated from 3-day-old etiolated and 3-day-old etiolated, 1-amino-cyclopropane-1-carboxylate (10 μM) treated seedlings and petri plate-grown callus tissue. First strand cDNA was primed by using an equimolar ratio of two phosphorylated primers: (T)15 and (N)6TT using 5 μg of mRNA with Superscript II reverse transcriptase (GIBCO/BRL). Reactions were preformed according to the manufacturer's instructions with the exception that methyl-dCTP (Boehringer Mannheim) was used in place of dCTP. After first strand synthesis, reverse transcriptase was heat-inactivated and second strand cDNA was synthesized by the addition of RNase H, Escherichia coli DNA polymerase I, E. coli DNA ligase, β-NAD, and ammonium sulfate to the first strand reaction. cDNA was ligated to a phosphorylated linker made by annealing the synthetic oligonucleotide 5′ PO4 GCTTGAATTCAAGC. When ligated to the cDNA, this linker generates a unique HindIII site at the 3′ end of the cDNA, enabling directional cloning. Second strand cDNA was digested with EcoRI and HindIII and was gel purified. cDNA fragments 0.3–1.5 kb were ligated to pEGAD digested with EcoRI and HindIII. Ligations were transformed into ultracompetent XL10-Gold E. coli (Stratagene). Each library had a complexity of ≈40,000 clones.
Transformation of Libraries into Arabidopsis.
A modified version of in planta transformation was developed for Arabidopsis shotgun transformation. Agrobacterium tumefaciens (strain GV3101) was transformed with pEGAD cDNA libraries by electroporation, generating sufficient colonies to represent 3-fold library coverage. Bacteria were scraped off selective plates, were washed in infiltration medium, and were resuspended in infiltration medium to a final OD600 of 0.5. This plate amplification step was introduced to avoid misrepresentations of library diversity that could occur from growth in liquid culture. Arabidopsis plants were submerged in Agrobacterium solutions for 2–5 min. Use of plate-grown Agrobacterium for infections gave transformation rates similar to Agrobacterium grown in liquid culture; ≈0.5–4% of T1 plants were transgenic, and, of these, 0.1–1% expressed detectable levels of GFP. Three to five percent of the primary transgenics (T1) in these populations had a visible morphological phenotype, suggesting the populations could be used to identify phenotypes induced by dominant negative fusion proteins, cosuppression, or other mechanisms. Infiltration medium is 1× Murashige and Skoog salts, 5% sucrose, 0.02% Silwet detergent, and 10 μg/liter benzylaminopurine.
Isolation and Microscopic Screening of GFP∷cDNA Transgenic Seedlings.
Seed from plants inoculated with Agrobacterium were germinated on agar-solidified media containing Murashige and Skoog salts (MS agar) and were screened for plants expressing GFP by using a Leica (Deerfield, IL) dissecting microscope equipped with a mercury lamp and epifluorescence filter set. GFP+ T1 seedlings were transferred onto microscope coverslips and were imaged on a Nikon inverted fluorescence microscope equipped with a Nikon 60× 1.2 numerical aperture water immersion objective and a Bio-Rad MRC 1024 confocal head. Plants possessing non-wild-type GFP distributions were transferred from coverslips to MS agar Petri plates to rehabilitate from the imaging process. After 1–2 weeks, seedlings were transferred to soil for seed production. Seed isolated from these lines was germinated and retested for marker phenotypes by using confocal microscopy.
Sequence Analysis of GFP∷cDNA Fusions.
Primers derived from the pEGAD vector sequence [EGAD(+) GCGCGATCACATGGTCCT, EGAD(−) TCCTCGAGATCAGTTATCTAG] were used to amplify inserts from transgenic plants. Single insert lines produced a single major amplification product. Fusion sequences were determined by sequencing PCR products with a GFP primer [EGAD(seq+) CTCGGCATGGACGAGCTG] adjacent to the cDNA fusion junction and the EGAD(−) primer, which is adjacent to the 3′ cloning junction.
Whole-Mount Immunocytochemistry.
Whole-mount immunocytochemistry was performed as in the work by Boudronk et al. (10), with some modifications. Tissue samples were incubated overnight in a 1:50 dilution of primary antisera raised against cottonseed catalase [provided by Dick Trelease (University of Arizona)], Arabidopsis cofilin [Rose Biochemicals, (www.rosebiotech.com)], or no antibody. The primary antibody was detected by TRITC-conjugated secondary antibody (Vector Laboratories). Confocal images of GFP and Texas Red fluorescence were acquired sequentially, using identical instrument settings for experimental and control samples.
Results and Discussion
Libraries of plants expressing GFP∷cDNA fusions were constructed in a two-step process. Initially, cDNA was synthesized from Arabidopsis poly(A)+ mRNA and was ligated directionally into a plant transformation vector (pEGAD) downstream of the gene for an enhanced GFP variant lacking a stop codon; approximately one-third of cDNA fusions to GFP are expected to be translated in their native coding frame using this approach. This cloning strategy allows for the isolation of protein domains and carboxy terminal sequences sufficient to cause GFP relocalization. pEGAD cDNA libraries were transformed en masse into A. tumefaciens, and these Agrobacterium populations were used to infect the Columbia wild type of Arabidopsis. The progeny of infected plants were subsequently screened by fluorescence microscopy to identify transgenic plants expressing GFP.
A pilot screen of 5,700 transgenic seedlings was performed to establish the frequency with which GFP∷cDNA fusion proteins were directed to new subcellular locations. The GFP variant used in these studies normally localizes to the cytoplasm and nucleoplasm of Arabidopsis cells (Fig. 1 A and B). Conventional fluorescence microscopy was used to screen for seedlings differing from this wild-type pattern. When identified, these lines were subsequently imaged by using confocal microscopy for further characterization. Lines with altered GFP localization were rescued and retested for GFP localization patterns in the subsequent generation. More than 50 seedlings could be screened per hour, a high throughput facilitated by the ease with which Arabidopsis transgenics can be isolated and their small size, which allows 30–40 young seedlings to be mounted on a single microscope slide.
Figure 1.

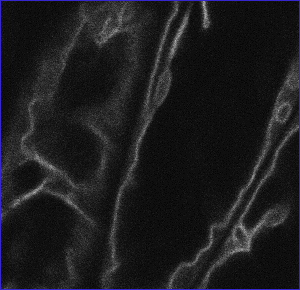
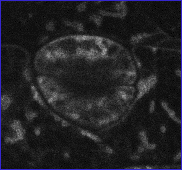
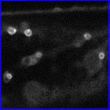
Canonical images of marker classes. (A and B) Wild type: Hypocotyl epidermal cells of transgenic seedlings expressing pEGAD GFP; nuclei and cytoplasmic strands are evident. Shown for comparison are a single confocal optical section (A) and a brightest point projection of several optical sections (B) of the same plant cells. (C) Cell surface: Cotyledon epidermal cells of EGAD line Q8, which expresses a GFP fusion to the plasma membrane channel protein PIP2A. In plasmolysis experiments, GFP fluorescence associates with the membrane of plasmolysed cells, indicating the marker is not cell wall localized. (D) Cell contact junctions: Leaf petiole epidermal cells in line LEEZ, which expresses an out-of-frame fusion protein. Markers of this class highlight both plasma membranes and membrane contact zones. Contact zones are specific to the “cell contact junction group” and are not observed in members of the cell surface group. (E) Endoplasmic reticulum: Cotyledon epidermal cells in line Q4, which express an in-frame fusion to a predicted protein containing a carboxy terminal membrane anchor rich in lysine residues. This marker colocalizes with fluorescent BODIPY-Ceramide, an endoplasmic reticulum (ER) membrane marker dye (data not shown), and is similar in appearance to KDEL-tagged GFP (20). (F) Vacuolar membrane: Hypocotyl epidermal cells in line Q5, which expresses a fusion to delta-TIP, a vacuolar membrane channel protein. Vacuolar membrane encasing trans-vacuolar cytosolic strands and invaginations creates a complicated pattern of fluorescence. The vacuolar membrane can be seen to flow over organelles at the cell periphery (see supplemental data) (G) Tiny bubbles: Hypocotyl epidermal cell of line Q10, which expresses a fusion protein to a novel glycine rich protein. In addition to the bubble-like structures, a reticulate ER-like pattern is also faintly marked in this line. The identity of the bubble-like structures is unknown but may be ER associated (see supplemental data). (H) Nuclear exclusion. Line Q1 expresses a fusion protein to a predicted small acidic ribosomal protein. Markers in this group do not show the nuclear localization characteristic of wild-type GFP (see B). (I) Nuclear: Root meristem cells in line N7, which expresses a GFP fusion to a transcription factor-like protein. (J) Nucleolar: Hypocotyl epidermal cells in line expressing a fusion protein targeted to the nucleus (K). Chromosomes: A dividing root cell in line M253, showing accumulation of the GFP∷CRY2 fusion protein on anaphase chromosomes. Before mitosis, the marker appears to localize to the nuclear lumen, as seen in several adjacent cells. These interphase nuclei are overexposed to better show the chromosomal pattern in the dividing cell. (L) Q-balls. Shown are structures of unknown identity illuminated by an out-of-frame fusion protein, F2. (M) Streaming dots: Hypocotyl cell of EGAD line V6, which expresses a fusion to an EST of unknown function. The images shown in B–E and G–I are brightest-point projections of confocal Z-series. The remaining images are single optical sections acquired by using confocal microscopy. (Bars = 20 μm.)
Of the 5,700 lines screened, 120 displayed heritable, non-wild-type GFP distribution patterns, representing a success rate of 1 per 45 plants screened. These markers were grouped into 17 phenotypic classes defining similar patterns of subcellular GFP localization, as visualized by confocal microscopy (Table 1). Fig. 1 shows canonical images for many of the phenotypic classes identified. Because it is conceivable that different subcellular compartments may be visually indistinguishable at the level of confocal imaging, descriptive aspects of microscopic phenotypes were frequently chosen to name phenotypic classes rather than inferred subcellular localizations. Localization patterns consistent with several major subcellular compartments or domains were identified: plasma membrane (Fig. 1C), sites of contact between neighboring cells (Fig. 1D), endoplasmic reticulum (Fig. 1E), vacuolar membrane (Fig. 1F), the nucleus (Fig. 1I), the nucleolus (Fig. 1J), condensed chromosomes (Fig. 1K), the peroxisome (Fig. 2A), and preferential retention in the cytosol (exclusion from the nucleoplasm) (Figs. 1 H and 4A). In addition, several structures were marked that will require further characterization to establish their identity or authenticity as native subcellular structures (Fig. 1 G, L, and M).
Table 1.
Marker classes identified by random GFP∷cDNA screening
| Localization phenotype | Number isolated | Correct frame |
|---|---|---|
| Torus | 43 | 1 /14 |
| Nuclear exclusion | 20 | 8 /10 |
| Q-balls | 8 | 0 /2 |
| Nuclear localization | 7 | 2 /3 |
| Streaming dots | 7 | 1 /3 |
| Cell surface | 5 | 5 /5 |
| Nucleolus | 6 | 2 /4 |
| Bright nuclei | 5 | 0 /2 |
| Blobs | 3 | 1 /2 |
| Vacuolar membrane | 4 | 3 /3 |
| ER membrane | 2 | 1 /1 |
| Cell contact junctions | 3 | 0 /1 |
| Tiny bubbles | 3 | 2 /2 |
| Chromosomes | 1 | 1 /1 |
| Regulated nuclear exclusion | 1 | 1 /1 |
| Wound induced granulation | 1 | 1 /1 |
| Darts | 2 | 0 /1 |
| Total | 120 | 29 /56 |
Each of 120 transgenic lines was classified into one of 17 types (left column) based on the appearance of GFP localization by confocal microscopy. The number of lines isolated in each phenotypic group is shown in the middle column. The frequency with which fusion proteins of a given phenotypic group were in the same translational reading frame as their native proteins is shown in the right column. Thus, for instance, the correct reading frame could be inferred for 14 of the fusion proteins in the “Torus” class, but only 1 of these was in frame with GFP. These estimates were inferred by sequence analysis of the subset of single insert transgenic plants containing markers with homology to previously characterized genes (see text for description).
Figure 2.

Dynamics of torus marker. Sequential images were acquired at 0.75-second intervals from a hypocotyl cell of EGAD line C2, which expresses an out-of-frame fusion protein. The time series shows a Torus structure adopting a tubular morphology (marked by an arrow). (Bar = 1 μM.)
Figure 4.

GFP∷GF14 shows dynamic nuclear localization. (A) Confocal image of hypocotyl cells expressing GFP∷GF14. GFP∷GF14 localizes to the nuclei of cells undergoing cytokinesis (yellow arrows) but not to the nuclei of most interphase cells (red arrow). (B) Confocal time series of hypocotyl cell undergoing cytokinesis. GFP∷GF14 accumulates in the nuclear lumen early in cytokinesis (1, yellow arrow) and dissipates as cytokinesis proceeds (2 and 3, yellow arrows). Part of the phragmosome, the site of synthesis of the new cell wall, is visible as a mass of cytosolic fluorescence (red arrows). Note that the intensity of signal decreases in the nucleus, but fluorescence remains relatively constant in the cytoplasm at the periphery of the cell. The phragmosome, brightly labeled at the start of the sequence, dissipates by the third frame. The frames were acquired at t = 0, 17, and 22 min, respectively. (Bar = 20 microns.)
In principle, the screening approach described here may be useful for identifying components of diverse subcellular compartments. Because the full genome sequence of Arabidopsis will soon be available (11), it will be possible to access the complete coding sequence of each fusion protein by simply sequencing the cDNA in the corresponding fusion constructs. However, the utility for this purpose depends on the frequency with which random GFP∷cDNA fusions retain localization patterns faithful to their native states. To gain insight into this aspect of the method, we sequenced the cDNAs responsible for the localization patterns in 109 of the 120 marker lines. PCR amplification using primers that flank the pEGAD cDNA cloning site was used to isolate cDNAs from marker lines. Twenty-three lines contained more than a single PCR amplification product and were excluded from this analysis. Fifty-six of the eighty-six cDNAs from single insert transgenic lines were found to have significant homology to characterized Arabidopsis gene products by using blastn and blastx searches of GenBank. This set of 56 cDNAs with homology to characterized genes was used as a dataset to make inferences about the general molecular features of the GFP-fusion protein markers identified.
One generalization that can be made from this analysis is that GFP can be directed to many subcellular locations by fusion to non-native protein sequences that are created by out-of frame translation. This is illustrated by markers in the “Torus” phenotypic class, where 13 of the 14 markers with homology to characterized genes were generated by out-of-frame cDNA fusions to GFP (Table 1). In the Torus lines, GFP is targeted to a torus-shaped structure ≈1 μm in diameter (Fig. 2). This organelle contains a central region lacking GFP fluorescence and is remarkably dynamic, frequently adopting transient tubular morphologies (Fig. 2; supplemental data). One Torus marker, J5, was created by an in-frame translational fusion to the carboxy terminal 30 amino acids of an Arabidopsis homolog of a Cucumis sativa protein purified from peroxisomes (12) and a related sequence from Brassica napus (GenBank accession no. AJ000886) (Table 4) (11). The carboxy terminal three amino acids of these three proteins and most of the Torus markers resemble the canonical tri-amino acid peroxisomal targeting sequence SKL* (Table 2) (13). In addition, one of the carboxy terminal sequences identified, SRL*, has been shown to be required for peroxisomal localization of a short-chain dehydrogenase/reductase in mammalian cells (14). Collectively, these observations suggest that the organelle tagged by the Torus class markers is the plant peroxisome. This conclusion is supported by immunolocalization experiments that show that catalase, a peroxisome resident, and GFP colocalize in one of the Torus marker lines (Fig. 3). Because some of the carboxy terminal tripeptide sequences in Table 2 are highly divergent from proposed peroxisomal targeting sequences (13, 14), it may be that sequences internal to the carboxy terminus may act as peroxisomal targeting sequences (13).
Table 2.
Carboxyterminal sequence of Torus markers
| Torus line | COOH sequence | |||
|---|---|---|---|---|
| A5 | S | R | L | ∗ |
| C4 | S | R | L | ∗ |
| C5 | S | R | L | ∗ |
| C2 | S | Q | L | ∗ |
| D5 | S | Q | L | ∗ |
| D4 | S | S | L | ∗ |
| B1 | S | S | L | ∗ |
| A2 | S | Y | L | ∗ |
| 305 | S | N | L | ∗ |
| D2 | S | C | L | ∗ |
| B5 | S | P | L | ∗ |
| 301b | S | P | L | ∗ |
| D1 | S | S | I | ∗ |
| D2 | S | K | I | ∗ |
| B4 | S | R | F | ∗ |
| E5 | S | L | W | ∗ |
| 37-25 | S | H | R | ∗ |
| V5 | F | K | L | ∗ |
| 37-21 | Q | K | L | ∗ |
| A4 | W | R | L | ∗ |
| B3 | L | R | L | ∗ |
| E1 | P | R | L | ∗ |
| V16 | P | S | L | ∗ |
| C3 | T | I | Y | ∗ |
| D3 | T | N | L | ∗ |
| V8 | T | R | I | ∗ |
| E4 | C | M | M | ∗ |
| A1 | G | K | M | ∗ |
| V26 | E | G | P | ∗ |
Deduced carboxy terminal 3 amino acids of the Torus marker fusion proteins isolated from marker lines transgenic for single GFP∷cDNA fusion proteins.
Figure 3.

Torus structures react with an antibody directed against a peroxisomal protein. Shown are confocal images of whole-mount Arabidopsis seedling tissue probed with anticatalase serum (catalase, upper row) and serum from a control rabbit (serum control, lower row). EGFP fluorescence was detected at 510–532 nm with excitation at 488 nm (GFP, shown in green pseudocolor). Texas Red-conjugated secondary antibody was detected at 595–615 nm with excitation at 568 nm (Texas Red, shown in red pseudocolor). Correspondence of the fluorescence patterns is shown in color-merged images (Merged).
The high frequency with which non-native coding sequences direct GFP to this organelle suggest that some of its targeting sequences are degenerate in that they can be generated at a high frequency by chance. Interestingly, several other subcellular destinations were also labeled by non-native protein fusions (Table 1), suggesting the possibility that minimal targeting sequences for some other cellular destinations may also be of low information content. A larger collection of out-of-frame fusions will be required to explore further the question of chance targeting to nonperoxisomal destinations in Arabidopsis. It was previously reported that, in Saccharomyces cerevisiae, replacement of the signal sequence of invertase with random peptides led to correct targeting in ≈20% of the cases (15). Although our analysis suggests that a high percentage of GFP∷cDNA fusion proteins display artifactual localization, they can frequently be recognized by comparing the reading frame of the fusion protein to the reading frame deduced from the results of the Arabidopsis genome sequencing project.
Excluding the Torus class, 29 of 42 markers were caused by in-frame fusion proteins. Where sequence homology was available, the general trend among this class was that in-frame markers displayed localization patterns consistent with published data or suggested by sequence homology (Table 3). For example, some members of the cell surface group are homologous to water channels (PIPs) purified from plasma membranes (16). Members of the vacuolar membrane phenotypic class are homologous to proteins experimentally localized to vacuolar membranes (17–19), and members of the nuclear group are similar to described nuclear proteins (20) (Table 3). These observations imply that random screening can be used to isolate both markers of structures and components native to those structures. Ultimately, an accurate estimate of the percentage of faithfully localized proteins will require detailed analyses of many additional lines.
Table 3.
In-frame fusion proteins identified as markers using random GFP∷cDNA fusion screening
| Phenotypic class | Line | Identity* | Gene name | Gene function | GenBank accession no. |
|---|---|---|---|---|---|
| Torus | A5 | 1 | Tetrafunctional protein | Peroxisomal β-oxidation | AJ000886 |
| Nuclear exclusion | 29-5 | 2 | Bfn1 homolog | Putative DNA/RNA nuclease | U90264 |
| Nuclear exclusion | 36-30 | 2 | Cor47 | Cold inducible dehydration-related protein | X59814 |
| Nuclear exclusion | 270898A | 2 | Cor47 | Cold inducible dehydration-related protein | X59814 |
| Nuclear exclusion | 020698E | 2 | Cor47 | Cold inducible dehydration-related protein | X59814 |
| Nuclear exclusion | 060798C | 2 | GRF2, 14-3-3 like protein | Component of DNA binding complex | U09376 |
| Nuclear exclusion | 180898A | 3 | OEP8-like | Unknown | CAB10358 |
| Nuclear exclusion | 030898E | 3 | Novel | Unknown | C005287_21 |
| Nuclear exclusion | Q1 | 3 | Acidic ribosomal protein | Ribosomal protein | CAB39610 |
| Nuclear localization | N7 | 3 | Ankyrin-like protein | Transcription | CAA16704 |
| Nuclear localization | N9 | 3 | Ribosomal Protein S31 | Ribosomal protein | AAC27163 |
| Chromosomes | m253 | 2 | Cry2 | Light regulated nuclear factor | U43397 |
| Streaming dots | 39-18 | 1 | Acyl CoA binding protein | Unknown | X77134 |
| Cell surface | 160698D | 2 | PIP1b | Plasma membrane water channel | Z17399 |
| Cell surface | Q8 | 2 | PIP2a | Plasma membrane water channel | X75883 |
| Cell surface | PM | 2 | SIMIP | Salt induced water channel | AF003728 |
| Cell surface | 29-1 | 2 | LTI6b | Low temperature induced protein | AF104221 |
| Cell surface | 37-26 | 2 | RCI2A | Low temperature induced protein | AF122005 |
| Nucleolus | mc-1 | 2 | Ribosomal protein S11 | Ribosomal protein | J05216 |
| Nucleolus | 100898D | 1 | Ribosomal protein S41 | Ribosomal protein | X75423 |
| Blobs | 200598B | 3 | Enoyl CoA hydratase | Peroxisomal β-oxidation | CAB10400 |
| Vacuolar membrane | 230898A | 3 | DIP aquaporin homolog | Vacuolar membrane transporter | Z97343 |
| Vacuolar membrane | Q5 | 2 | Delta TIP | Vacuolar membrane water channel | U39485 |
| Vacuolar membrane | 160698C | 3 | Gamma TIP-like | Vacuolar membrane water channel | CAB10515 |
| ER membrane | Q4 | 3 | Novel | Unknown | AAB71445 |
| Tiny bubbles | Q10 | 2 | Glycine-rich protein 32 | Unknown | AF104330 |
| Tiny bubbles | 38-3 | 3 | Novel | Unknown | AC006420.2 |
| Regulated nuclear exclusion | LE8 | 2 | GF14A, 14-3-3 protein | Component of DNA binding complex | M96855 |
| Wound induced granules | 020798A | 2 | Nit1 | Nitrilase | U47114 |
Gene identity was determined by sequencing of the fusion proteins and comparison to gene sequences deposited in GenBank.
Sufficiently large collections of markers generated by this and future screens could be used to help extract protein targeting information by searching for peptide motifs shared by similar markers. The limited collection of markers isolated in this initial study allowed identification of sequence similarity shared by the majority of torus markers, the largest class we recovered. We have also noticed sequence similarities in other marker classes. For example, some of the nucleolar tags are short peptides rich in arginine and lysine (results not presented). The diversity of markers that can be identified with the method explored in this paper should allow similar analyses to be performed for a wide variety of subcellular locations.
Perhaps the greatest utility of the approach described here is its ability to facilitate the identification of novel features of subcellular structure and dynamic processes. Although our collection of markers represents a small percentage of the localization classes one might expect to find by microscopic screening, they have revealed many intriguing aspects of plant cell biology and have been useful for exploring aspects of plant subcellular dynamics. A number of examples illustrating this point are provided as supplemental data. One interesting observation we have made is that plant cells expressing a fusion between GFP and the carboxy terminus of GF14, a 14-3-3 protein, show differential accumulation in the cell nucleus. This pattern of localization is regulated in part by cell cycle state. The fusion protein is excluded from most nuclei; however, it accumulates in nuclei just after completion of nuclear division and again departs from the nuclei shortly before cytokinesis is complete (Fig. 4; D.W.E. and S.R.C., unpublished work). The function of this redistribution in the context of cytokinesis remains to be determined; however, its identification suggests the feasibility of future screens aimed directly at identifying proteins with regulated changes in subcellular distribution. Directed screens for these kinds of markers may be particularly valuable because changes in subcellular distribution are a frequent form of regulation in many signaling events.
Collectively, our results demonstrate that random GFP∷cDNA fusions efficiently generate novel in vivo subcellular tags for Arabidopsis. This approach should be applicable to other organisms in which large scale transformation is possible. The spectrum of the method could be enhanced by using normalized cDNA or constructing libraries of amino terminal fusions to GFP, modifications that could allow rare markers or markers requiring amino terminal targeting information to be identified. The expression of random localization tags in living cells may enable screens for proteins on the basis of predefined dynamic properties such as the redistribution of markers in response to signals like wounding, infection, or phytohormones. Kinetic images, technical details, and additional information is available in the supplemental data and at our web site (deepgreen.stanford.edu).
Supplementary Material
Acknowledgments
We thank Stephan Schmidheiny for the generous gift of the Leica dissecting microscope that made this work possible. We thank Marie-Theres Hauser for advice on immunocytochemistry and Farhah Assaad, Wolfgang Lukow-itz, Joe Ogas, Stewart Gillmor, and Seung Yung Rhee for valuable discussion during the course of this work. We also thank Dick Trelease for the catalase antibody, Deanne Falcone for pBIMC, and David Bouchez for pDHB321.1. S.R.C. was partially supported by a U.S. Department of Energy/National Science Foundation/U.S. Department of Agriculture TRI-Agency Training Grant. This work was supported in part by a grant (DE-FG02-97ER20133) from the U.S. Department of Energy Biological Energy Research Program.
Abbreviations
- GFP
green fluorescent protein
- MCS
multicloning site
- TBS
Tris-buffered saline
- ER
endoplasmic reticulum
Footnotes
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. AF218816).
References
- 1.Chalfie M, Tu Y, Euskirchen G, Ward W W, Prasher D C. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- 2.Tsien R Y. Annu Rev Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- 3.Cubitt A B, Heim R, Adams S R, Boyd A E, Gross L A, Tsien R Y. Trends Biochem Sci. 1995;20:448–455. doi: 10.1016/s0968-0004(00)89099-4. [DOI] [PubMed] [Google Scholar]
- 4.Sawin K, E, Nurse P. Proc Natl Acad Sci USA. 1996;93:15146–15151. doi: 10.1073/pnas.93.26.15146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rolls M M, Stein P A, Taylor S S, Ha E, McKeon F, Rapoport T A. J Cell Biol. 1999;146:29–43. doi: 10.1083/jcb.146.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doyle T, Botstein D. Proc Natl Acad Sci USA. 1996;93:3886–3891. doi: 10.1073/pnas.93.9.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cormack B P, Valdivia R H, Falkow S. Gene. 1996;173:33–38. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- 8.Haas J, Park E C, Seed B. Curr Biol. 1996;6:315–324. doi: 10.1016/s0960-9822(02)00482-7. [DOI] [PubMed] [Google Scholar]
- 9.Chiu W L, Niwa Y, Zeng W, Hirano T, Kobayashi H, Sheen J. Curr Biol. 1996;6:325–330. doi: 10.1016/s0960-9822(02)00483-9. [DOI] [PubMed] [Google Scholar]
- 10.Boudronk K, Dolan L, Shaw P J. J Cell Sci. 1998;111:3687–3694. doi: 10.1242/jcs.111.24.3687. [DOI] [PubMed] [Google Scholar]
- 11.Somerville C, Somerville S. Science. 1999;285:380–383. doi: 10.1126/science.285.5426.380. [DOI] [PubMed] [Google Scholar]
- 12.Preisigmuller R, Guhnemannschafer K, Kindl H. J Biol Chem. 1994;269:20475–20481. [PubMed] [Google Scholar]
- 13.Subramani S. Annu Rev Cell Biol. 1993;9:445–478. doi: 10.1146/annurev.cb.09.110193.002305. [DOI] [PubMed] [Google Scholar]
- 14.Fransen M, VanVeldhoven P P, Subramani S. Biochem J. 1999;340:561–568. [PMC free article] [PubMed] [Google Scholar]
- 15.Kaiser C A, Preuss D, Grisafi P, Botstein D. Science. 1987;235:312–317. doi: 10.1126/science.3541205. [DOI] [PubMed] [Google Scholar]
- 16.Kammerloher W, Fischer U, Piechottka G P, Schaffner A R. Plant J. 1994;6:187–199. doi: 10.1046/j.1365-313x.1994.6020187.x. [DOI] [PubMed] [Google Scholar]
- 17.Johnson K D, Hofte H, Chrispeels M J. Plant Cell. 1990;2:525–532. doi: 10.1105/tpc.2.6.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hofte H, Hubbard L, Reizer J, Ludevid D, Herman E M, Chrispeels M J. Plant Physiol. 1992;99:561–570. doi: 10.1104/pp.99.2.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chrispeels M J, Agre P. Trends Biochem Sci. 1994;19:421–425. doi: 10.1016/0968-0004(94)90091-4. [DOI] [PubMed] [Google Scholar]
- 20.Kobayashi K, Kanno S, Smit R, Vanderhorst G T J, Takao M, Yasui A. Nucleic Acids Res. 1998;26:5086–5092. doi: 10.1093/nar/26.22.5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haseloff J, Siemering K R, Prasher D C, Hodge S. Proc Natl Acad Sci USA. 1997;94:2122–2127. doi: 10.1073/pnas.94.6.2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}