

Abstract
This study describes the effects of long-chain fatty acids on inflammatory signaling in cultured astrocytes. Data show that the saturated fatty acid palmitic acid, as well as lauric acid and stearic acid, trigger the release of TNFα and IL-6 from astrocytes. Unsaturated fatty acids were unable to induce cytokine release from cultured astrocytes. Furthermore, the effects of palmitic acid on cytokine release require TLR4 rather than CD36 or TLR2, and do not depend on palmitic acid metabolism to palmitoyl-CoA. Inhibitor studies revealed that pharmacologic inhibition of p38 or p42/44 MAPK pathways prevents the pro-inflammatory effects of palmitic acid, while JNK and PI3K inhibition does not affect cytokine release. Depletion of microglia from primary astrocyte cultures using the lysosomotropic agent L-Leucine methyl ester (LME) revealed that the ability of palmitic acid to trigger cytokine release is not dependent on the presence of microglia. Finally, data show that the essential ω-3 fatty acid docosahexaenoic acid (DHA) acts in a dose-dependent manner to prevent the actions of palmitic acid on inflammatory signaling in astrocytes. Collectively, these data demonstrate the ability of saturated fatty acids to induce astrocyte inflammation in vitro. These data thus raise the possibility that high levels of circulating saturated fatty acids could cause reactive gliosis and brain inflammation in vivo, and could potentially participate in the reported adverse neurologic consequences of obesity and metabolic syndrome.
Keywords: brain inflammation, cytokines, dyslipidemia, obesity, reactive gliosis
INTRODUCTION
In the United States, the long-term consumption of diets high in fat and calories appears to be a primary cause of obesity, which is becoming increasingly prevalent in developed Western nations. This is an important public health concern, as obesity is closely associated with an enhanced risk for a myriad of diseases, including type 2 diabetes, cardiovascular disease, gastrointestinal and respiratory difficulties, stroke, and many types of cancer (reviewed in (Haslam & James 2005)). Furthermore, the physiologic complications of obesity are now known to include detrimental effects on brain physiology and function (reviewed in (Bruce-Keller et al. 2009), (Fillit et al. 2008), (Middleton & Yaffe 2009)). For example, studies have reported deficits in learning, memory, and executive function in obese as compared to nonobese patients (Elias et al. 2003), (Elias et al. 2005), (Waldstein & Katzel 2006), and regression studies have associated increased BMI with decreased brain volume (Ward et al. 2005). Other studies have confirmed alterations of brain morphology in overweight and obese young adults, and further show that clinical obesity is associated with reductions in focal gray matter volume and enlarged white matter, particularly in the frontal lobe (Pannacciulli et al. 2006). Experimental studies in animals have confirmed neurologic vulnerability to obesity and high fat diet and further show that diet-induced metabolic dysfunction leads to increased brain inflammation, reactive gliosis, and vulnerability to injury (Pistell et al. 2010), (Bruce-Keller et al. 2009), (Bruce-Keller et al. 2010), (White et al. 2009), (Studzinski et al. 2009), (Winocur & Greenwood 2005), (Stranahan et al. 2008), (Jurdak et al. 2008), (Molteni et al. 2002).
Although the physiologic mechanisms whereby obesity adversely affects the brain are poorly understood, both experimental and human studies have shown that increased inflammation is a key physiologic feature of obesity (reviewed in (Hotamisligil 2006)). The brain is highly sensitive to inflammatory mediators, and there is ample data to indicate that cytokines specifically can have clinically significant adverse effects on cognition and neuronal homeostasis (Wilson et al. 2002), (Rothwell 1999), (Akiyama et al. 2000). Relatively high levels of cytokine binding have been demonstrated in the cortex and hippocampus (Parnet et al. 2002), and cytokines such as TNFα, IL-1β and IL-6 can disrupt pathways involved in cognition and memory (Bellinger et al. 1995), (Jankowsky & Patterson 1999). Thus, the concerted elevations of these cytokines in the brains could likely be a major player in the development of adverse neurologic complications of obesity and high fat diet consumption. While there are likely multiple mechanisms through which enhanced adiposity could lead to widespread inflammation, it is established that plasma concentrations of fatty acids are significantly increased in close association with obesity and metabolic syndrome (Opie & Walfish 1963), (Reaven et al. 1988), and saturated long-chain fatty acids can activate inflammatory and innate immune responses in the body (Averill & Bornfeldt 2009), (Kennedy et al. 2009). The effects of serum free fatty acids on brain inflammation have not been directly evaluated, but there is compelling evidence that these lipids are indeed able to alter CNS function. For example, fatty acids have long been known to be blood-brain barrier permeable (Dhopeshwarkar & Mead 1973), (Smith & Nagura 2001), and data further suggest that high fat diets might increase the uptake of fatty acids into the brain from the plasma (Wang et al. 1994), (Karmi et al. 2010). Furthermore, the saturated free fatty acids palmitic acid and lauric acid have both been shown to trigger inflammation in cultured macrophages (Laine et al. 2007), and to modulate amyloid processing in neurons and astrocytes ((Patil & Chan 2005), (Patil et al. 2006). Finally, published data show that peripherally administered fatty acids accumulate primarily in astrocytes (Morand et al. 1979), (Bernoud et al. 1998), and the localization of astrocytes to the blood brain barrier and their established role in providing blood-derived nutrients to neurons makes astrocytes a likely and physiologically significant target for the actions of elevated fatty acids. This study was thus undertaken to determine how elevations in serum lipids might alter astrocyte physiology, and was designed to specifically delineate the degree to which, and the mechanisms by which, saturated and unsaturated fatty acids could modulate inflammatory signaling in cultured astrocytes.
MATERIALS AND METHODS
Materials
The saturated fatty acids lauric acid (C12:0 or dodecanoic acid), palmitic acid (C16:0 or hexadecanoic acid), and stearic acid (C18:0 or octadecanoic acid ); the unsaturated fatty acids myristoleic acid (14:1(n-5) or tetradecenoic acid), oleic acid (18:1(cis-9) or (9Z)-Octadec-9-enoic acid), and linoleic acid (18:2(n-6) or cis, cis-9,12-octadecadienoic acid); and the polyunsaturated ω-3 fatty acid docosahexaenoic acid (DHA, 22:6(n-3)) and ω-6 fatty acid arachidonic acid (20:4(n-6)) were all purchased from Sigma-Aldrich. Triacsin C was also obtained from Sigma-Aldrich. TAK-242, OxPAPC, and neutralizing antibodies to TLR2 (clone C9A12) were obtained from InvivoGen. UO126 was obtained from Calbiochem, and SB 239063, SP 600125, LY 294002, and UO124 were obtained from Tocris Bioscience. Neutralizing antibodies to CD36 (clone FA6.152) were obtained from Meridian Life Science, Inc., while neutralizing antibodies to TLR4 (clone MTS501) were obtained from Imgenex Corporation.
Cell culture and treatment
The Institutional Animal Care and Use Committee at the Pennington Center approved all experimental protocols, which were compliant with NIH guidelines on the use of experimental animals. Primary astrocytes were derived from male and female 1–3 day-old Sprague-Dawley (Harlan) rat pups as described previously (Turchan-Cholewo et al. 2008). Briefly, mixed glial cultures were generated from the brain (cerebral cortices without meninges) and maintained in Modified Eagle Medium (Gibco BRL) with 10% fetal bovine serum. After 7–10 days in vitro, microglia were removed from 70–80% confluent astrocyte cultures by orbital rotation (OR, 45 min at 200 rpm on an orbital shaker). In experiments designed to determine the role of contaminating microglia, orbital rotation was followed immediately by exposure to L-Leucine methyl ester (LME, 50 mM for 60 min), and the remaining cells were allowed to recover for 24–48 h before treatment.
Preparation of albumin-bound fatty acids
On the day of use, fatty acid stock solutions of 200 mM were prepared in 100% EtOH. Working water-soluble solutions of 5 mM fatty acids were then generated by incubating the fatty acids in PBS containing 10% endotoxin- andfatty acid-free BSA at 37°C for 60–90 min with occasional vortexing. This solution was then added to cells to obtain final fatty acid concentrations ranging from 50 – 400 μM. Equal volumes of the PBS/EtOH//10% fatty acid-free BSA vehicle were applied to control cells.
ELISA
Levels of TNFα, IL-6, and IL-1β in cell culture medium were measured by ELISA as described previously (Bruce-Keller et al. 2001).
Measures of protein expression by Western blot
Cells were homogenized in a Tris-buffered saline (pH 7.4) lysis buffer containing 0.1% Triton X-100, 5 mM EDTA, 1 mM sodium orthovanadate, and protease inhibitor cocktail (Sigma-Aldrich, Inc.). Homogenates were denatured in SDS, and equivalent amounts of protein were electrophoretically separated in polyacrylamide gels and blotted onto nitrocellulose. To ensure unfettered data interpretation, samples from all treatment groups were included in each individual gel. Blots were processed using the following primary antisera: anti-phospho p42/44 MAPK (1:2000, Cell Signaling Technology), anti-phospho p38 MAPK (1:1000, Cell Signaling Technology), anti-Iba-1 (1:500, Wako Chemicals USA Inc.), anti-TLR4 (1:500, Abcam Inc.); and anti-tubulin (1:1000, Wako Chemicals USA Inc.). After incubation with primary antibodies, blots were washed and exposed to horseradish peroxidase-conjugated secondary antibodies, and visualized using a chemiluminescence system (Amersham Biosciences).
Statistical analyses
All data are shown as mean ± standard error of measurement. Cytokine release values were analyzed with 1-way analyses of variance (ANOVA), followed by Bonferroni posttests to determine differences between treatment groups. Statistical significance for all analyses was accepted at p < 0.05, and *, **, and *** represent p < 0.05, p < 0.01, and p < 0.001, respectively.
RESULTS
1. Palmitic but not oleic acid increases cytokine release from astrocytes
Long-chain fatty acids have been shown to induce cytokine release from adipocytes (Ajuwon & Spurlock 2005), (Bradley et al. 2008), and from lymphocytes (Laine et al. 2007), (Stentz & Kitabchi 2006), (Håversen et al. 2009), but their proinflammatory potential has not been evaluated in brain cells. To determine if elevated serum lipids could trigger brain inflammation, the effects of the saturated fatty acid palmitic acid and the unsaturated fatty acid oleic acid on cytokine release from cultured primary astrocytes was evaluated. The fatty acids were conjugated to BSA as described in Methods, and applied to astrocytes for 24 h under serum-free conditions at concentrations of 50–400 μM. These concentrations have been shown to be effective at inducing cytokine release from cultured lymphocytes (Stentz & Kitabchi 2006), (Håversen et al. 2009), and are within a physiologic dose-range as serum non-esterified fatty acid levels in rat range from 100–200 μM in control, non-fasting conditions to 500–600 μM in fasted animals (Rustan et al. 1992), (Xu et al. 2010). While brain levels of soluble fatty acids have not been reported, fatty acids have long been known to be blood-brain barrier permeable (Dhopeshwarkar & Mead 1973), (Smith & Nagura 2001), and reports further suggest that high fat diets might actually increase the uptake of fatty acids into the brain from the plasma (Wang et al. 1994), (Karmi et al. 2010). Data show that palmitic acid induced significant, dose-dependent increases in TNFα and IL-6 release from cultured astrocytes (Fig. 1A and B). However, high doses of palmitic acid (400 μM) also significantly decreased MTT conversion to formazan (Fig. 1C), suggesting that this dose of palmitic acid induced some degree of mitochondrial dysfunction or cytotoxicity. Conversely, oleic acid did not cause cytokine release or alter MTT conversion at any concentration (Fig. 1A, B, and C). To determine if the effects of palmitic acid and oleic acid were representative of the effects of other long-chain fatty acids, additional cells were exposed to 100 μM concentrations of alternate saturated and unsaturated fatty acids. Specifically, the ability of the saturated fatty acids lauric acid and stearic acid, as well as the unsaturated fatty acids myristoleic acid, linoleic acid, and the ω-6 fatty acid arachidonic acid, to induce astrocytic release of the cytokine TNFα and IL-6, as well as IL-1β, was examined. Data show that stimulation of cells with the saturated fatty acids lauric acid or stearic acid resulted in levels of TNFα and IL-6 release generally similar to that induced by palmitic acid (Table 1), although stearic acid-induced release of IL-1β did not reach statistical significance (Table 1). Conversely, myristoleic acid, oleic acid, linoleic acid, and arachidonic acid were all unable to induce the release of significant levels of TNFα, IL-6, or IL-1β (Table 1), even when applied at concentrations of up to 400 μM (data not shown).
Figure 1. Effects of palmitic and oleic acid on TNFα and IL-6 release from cultured astrocytes.

Primary astrocyte cultures were established as described in Methods, and exposed to increasing concentrations of BSA-conjugated palmitic acid (PA) or oleic acid (OA). Release of (A) TNFα, (B) IL-6, and (C) MTT conversion to formazan was measured after 24 h. Data were compiled from 4 separate experiments, and are means and SEM of 20–40 dishes per group. ** and *** indicate significant (p<0.01, and p<0.001, respectively) increases in cytokine release from astrocytes treated with palmitic acid as compared to cells treated with BSA alone.
Table 1. Effects of long-chain unsaturated and saturated fatty acids on cytokine release from cultured astrocytes.
Primary astrocyte cultures were established as described in Methods, and exposed to either vehicle (BSA), or BSA-conjugated fatty acids (100 μM final concentration, see Methods). Release of TNFα, IL-6, and IL-1β was measured after 24 h. Data were compiled from 2 separate experiments, and are means and SEM of 10–30 dishes per group.
| TNFα (pg/ml) | IL-6 (pg/ml) | IL-1β (pg/ml) | ||
|---|---|---|---|---|
| BSA | 102.6 ± 8.7 | 999.8 ± 80.5 | 246.1 ± 29.1 | |
| Unsaturated FA (0.1 mM) | Myristolate | 32.0 ± 12.5 | 882.0 ± 242.6 | 26.2 ± 6.8 |
| Olate | 121.1 ± 21.5 | 673.4 ± 154.8 | 197.1 ± 41.5 | |
| Linolate | 146.1 ± 21.4 | 687.5 ± 101.7 | 175.0 ± 25.0 | |
| Arachidonate | 140.3 ± 18.3 | 1142.0 ± 276.8 | 347.0 ± 192.9 | |
| Saturated FA (0.1 mM) | Laurate | 521.3 ± 60.7*** | 6154.5 ± 724.9*** | 994.5 ± 153.1** |
| Palmatate | 849.0 ± 74.9*** | 3120.6 ± 173.0*** | 1229.0 ± 189.1*** | |
| Sterate | 430.7 ± 39.5*** | 3163.3 ± 394.4*** | 634.0 ± 118.6 | |
indicate significant (p<0.01, and p<0.001, respectively) increases in cytokine release from astrocytes treated with saturated fatty acids as compared to cells treated with BSA alone.
2. Inhibitors of TLR4, but not TLR2, CD36, or acyl-CoA synthetase, block the effects of palmitic acid on cytokine release
To begin to unravel the physiologic mechanisms whereby palmitic acid increases cytokine release in astrocytes, pharmacologic inhibitors of Toll-like receptor 4 (TLR4, TAK-242, 5 μM), and acyl-CoA synthetase (Triacsin C, 10 μM), and neutralizing antibodies to CD36 (FA6.152, 20 μg/ml), were applied to cells 45 min before palmitic acid treatment, and cytokine levels were measured after 24 h. Data show that only the TLR4 inhibitor (TAK-242), but not inhibitors of acyl-CoA synthetase or CD36, was able to prevent palmitic acid-induced cytokine release (Fig. 2A, 2B). Evaluation of MTT conversion did not indicate any cytotoxicity or mitochondrial dysfunction in astrocytes treated with TAK-242, or any of the other inhibitors (Fig. 2C). To confirm the role of TLR4, additional cells were exposed to 100 μM palmitic acid in the presence or absence of TLR4 inhibitor OxPAPC. OxPAPC is a mixture of oxidized phospholipids that inhibit TLR4 signaling by competing with CD14, LBP and MD2 (von Schlieffen et al. 2009). As OxPAPC also has activity towards TLR2 (Erridge et al. 2008), additional cells were treated with palmitic acid in the presence or absence of neutralizing antibodies directed against either TLR4 or TLR2. Data show that both OxPAPC and TLR4 neutralizing antibodies significantly attenuated palmitic acid-induced cytokine release without affecting MTT conversion (Table 2). Conversely, TLR2 neutralizing antibodies were unable to modulate the effects of palmitic acid on cytokine release (Table 2).
Figure 2. Effects of inhibitors of TLR4, CD36, and acyl-CoA synthetase on palmitic acid-induced TNFα and IL-6 release.

Primary astrocytes were with vehicle (BSA) or 200 μM palmitic acid (PA) in the presence or absence of a pharmacologic inhibitor of TLR4 (TAK-242, 5 μM), neutralizing antibodies to CD36 (FA6.152, 20 μg/ml), or a pharmacologic inhibitor of acyl-CoA synthetase (Triacsin C, 10 μM). All inhibitors were applied to cells 45 min before administration of BSA or palmitic acid. Release of (A) TNFα, (B) IL-6, and (C) MTT conversion to formazan was measured after 24 h. Data were compiled from 2–4 separate experiments, and are means and SEM of 10–20 dishes per group. *** indicates significant (p<0.001) increases in cytokine release from astrocytes treated with palmitic acid as compared to control cells treated with just BSA, and ### indicates significant (p<0.001) decreases in cytokine release from astrocytes treated with palmitic acid in the presence of TAK-242 as compared to cells treated with palmitic acid alone.
Table 2. Effects of TLR4 and TLR4 inhibitors on palmitic acid-induced cytokine release from cultured astrocytes.
Primary astrocyte cultures were established as described in Methods, and exposed to BSA, or BSA-conjugated palmitic acid (100 μM final concentration, see Methods). Selected cells were also exposed to PA in the presence of increasing concentrations of a pharmacologic inhibitor of TLR2/4 (OxPAPC), a neutralizing antibody to TLR4 (FA6.152), or a neutralizing antibody to TLR2 (FA6.152). All inhibitors were applied to cells 45 min before administration of palmitic acid. Release of TNFα, IL-6, and IL-1β; and MTT conversion was measured after 24 h. Data were compiled from 2 separate experiments, and are means and SEM of 10–30 dishes per group.
| TNFα (pg/ml) | IL-6 (pg/ml) | IL-1β (pg/ml) | MTT (% BSA) | ||
|---|---|---|---|---|---|
| BSA | 125.7 ± 22.1 | 758.9 ± 75.6 | 128.5 ± 18.2 | 100.0 ± 5.3 | |
| 100 μM PA | 995.1 ± 46.5*** | 4632.2 ± 177.6*** | 1191.6 ± 61.5*** | 101.0 ± 6.9 | |
| PA with OxPAPC | 10 μM | 108.3 ± 13.6### | 2428.5 ± 159.3### | 129.3 ± 23.8### | 124.1 ± 14.3 |
| 30 μM | 98.1 ± 16.9### | 2382.2 ± 290.6### | 112.3 ± 40.1### | 125.0 ± 11.4 | |
| 50 μM | 110.0 ± 20.2### | 2165.5 ± 350.8### | 163.4 ± 47.8### | 120.8 ± 16.1 | |
| PA with TLR4 Ab | 0.5 μg | 677.2 ± 54.2## | 2774.2 ± 479.1### | 881.1 ± 116.6 | 116.6 ± 13.0 |
| 1 μg | 648.2 ± 67.5### | 2530.3 ± 502.0### | 752.5 ± 65.0## | 120.8 ± 14.3 | |
| 5 μg | 528.6 ± 38.9### | 1924.4 ± 341.0### | 619.3 ± 100.8### | 109.4 ± 15.7 | |
| PA with TLR2 Ab | 0.5 μg | 1004.9 ± 77.2 | 4083.2 ± 231.4 | 1180.0 ± 83.5 | 101.0 ± 5.2 |
| 1 μg | 953.5 ± 69.4 | 4379.2 ± 163.2 | 1249.7 ± 115.8 | 111.9 ± 4.4 | |
| 5 μg | 1155.9 ± 179.3 | 4705.8 ± 342.1 | 1727.7 ± 279.6 | 90.4 ± 6.6 | |
indicates significant (p<0.001) increases in cytokine release from astrocytes treated with palmitic acid as compared to control cells treated with just BSA, and
indicates significant (p < 0.01 and p<0.001, respectively) decreases in cytokine release from astrocytes treated with palmitic acid in the presence of inhibitors as compared to cells treated with palmitic acid alone.
3. Inhibitors of p42/44 MAPK and p38MAPK, but not JNK or PI3K, block the effects of palmitic acid on cytokine release
To next address the intracellular signal transduction pathways whereby palmitic acid increases cytokine release from astrocytes, pharmacologic inhibitors of p42/p44 MAPK (UO126, 5 μM), p38MAPK (SB239063, 5 μM), phosphatidylinositol 3-kinase (LY294002, 10 μM), or c-Jun N-terminal kinase (SP600124, 10 μM) were applied to cells 45 min before palmitic acid treatment, and cytokine levels were evaluated after 24 h. Data show that pretreatment with SB239063 and UO126, but not SB600125 or LY294002, significantly reduced palmitic acid-induced cytokine release (Fig. 3A, B). None of the inhibitors significantly affected MTT conversion (Fig. 3C), suggesting that the inhibitory effects of SB 239063 and UO126 were not related to cell death or injury. Application of UO124 (5 μM), an inactive structural enantomer of UO126, did not attenuate palmitic acid-induced cytokine release (data not shown). To further confirm the role of p38 and p42/p44 MAPK in the proinflammatory effects of fatty acids in cultured glia, cells were treated with palmitic acid, and then processed for Western blot as described in Methods. Data show that 10 and 30 minute exposures to palmitic acid increased phosphorylation of both p38 and p42/p44 MAPK in cultured astrocytes, but did not affect tubulin expression (Fig. 3C), or total pools of p38 and p42/p44 MAPK (data not shown). To determine if activation of NFkB might participate in the proinflammatory effects of fatty acids in cultured glia, cells were treated with 100 μM palmitic acid for 1 – 6 hrs and then processed for gel shift analyses. Analysis of nuclear proteins did not reveal any evidence of NFkB activation in cultured astrocytes following palmitic acid administration (data not shown).
Figure 3. Effects of signal transduction pathway inhibitors on palmitic acid-induced TNFα and IL-6 release.

Primary astrocytes were treated with vehicle (BSA) or 200 μM palmitic acid (PA) in the presence or absence of inhibitors of p42/44 MAPK (UO126,5 μM), p38MAPK (SB 239063, 5 μM), phosphatidylinositol 3-kinase (LY 294002, 10 μM), or c-Jun N-terminal kinase (SP 600124, 10 μM). All inhibitors were applied to cells 45 min before administration of BSA or palmitic acid. Release of (A) TNFα, (B) IL-6, and (C) MTT conversion to formazan in cells treated with inhibitors and BSA or palmitic acid was measured after 24 h. Data were compiled from 2–4 separate experiments, and are means and SEM of 10–20 dishes per group. *** indicates significant (p<0.001) increases in cytokine release from astrocytes treated with palmitic acid as compared to control cells treated with just BSA. #, ##, and ### indicate significant (p<0.05, p<0.01, and p<0.001, respectively) decreases in cytokine release from astrocytes treated with palmitic acid in the presence of UO126 or SB 239063 as compared to cells treated with palmitic acid alone. (D) Representative images of Western blots depicting increases in phosphorylation of p42/p44 MAPK (top panel) and p38 (middle panel) as compared to tubulin expression (bottom panel) in cultured astrocytes after 0, 10, and 30 minute-exposures to palmitic acid.
4. Microglial depletion does not prevent the effects of palmitic acid on cytokine release
Studies next addressed the possible role of microglial contamination in the effects of palmitic acid on cytokine release from astrocytes. Initial experiments determined the extent of microglial contamination of astrocyte cultures, and the relative expression of TLR4 in cultures of increasing astrocyte purity. Cultured mixed glia were established as described in Methods and subjected to either no manipulation (NM), to orbital rotation (OR alone), or to orbital rotation followed immediately by exposure to the lysosomotropic agent LME (OR/LME) as described in Methods. LME-treated cells were allowed to recover for 48 hrs before harvesting for Western blot, and cell homogenates from all 3 treatment groups were processed and run together on the same gels. Blot images show that while OR significantly decreased the extent of microglial contamination in astrocyte cultures (as indicated by a marked decrease in Iba-1), the combination of OR /LME resulted in the seemingly complete elimination of microglia from astrocyte cultures (Fig. 4A). However, TLR4 expression was generally stable across treatments (Fig. 4A), confirming previous reports of astrocytic TLR4 expression (Gorina et al. 2011), (El-Hage et al. 2011). Experiments next determined the effects of palmitic acid on astrocyte cultures that had been completely depleted of microglia by the combination of orbital rotation and LME exposure. Data show that palmitic acid remained able to induce dose-dependent increases in TNFα and IL-6 release from microglial-depleted astrocytes (Fig. 4B and C). Data also show that pretreatment with SB239063 or UO126 significantly reduced palmitic acid-induced cytokine release from microglial-depleted astrocytes (Fig. 4B and C). These data indicate that the effects of palmitic acid on astrocyte inflammation are not dependent on the presence of microglia. However, macrophages are well-known to release cytokines in response to saturated fatty acid exposure (Tappia et al. 1995), (Laine et al. 2007), (Martins de Lima-Salgado et al. 2011), and thus it is nearly certain that the population of microglia remaining in mixed glial cultures subjected to orbital rotation alone contributes to cytokine release in response to palmitic acid under these conditions.
Figure 4. Effects of microglial depletion on palmitic acid-induced TNFα and IL-6 release.
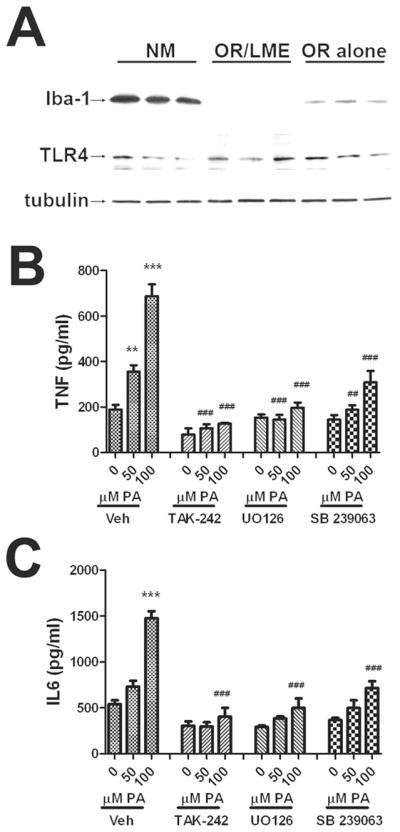
Cultured astrocytes were subjected to either no manipulation (NM), orbital rotation (OR alone), or orbital rotation followed immediately by exposure to LME (OR/LME) as described in Methods. (A) Representative images of Western blots depicting expression of the microglial marker Iba-1 (top panel), TLR4 (middle panel), and tubulin (bottom panel) in cultured cells subjected to the different manipulations. Release of (B) TNFα or (C) IL-6 from cells subjected to orbital rotation followed by LME that were treated with increasing does of palmitic acid in the presence or absence of inhibitors of TLR4 (TAK-242, 5 μM), p42/44 MAPK (UO126,5 μM), or p38MAPK (SB 239063, 5 μM). All inhibitors were applied to cells 45 min before administration of BSA or palmitic acid. Data were compiled from 2–4 separate experiments, and are means and SEM of 10–20 dishes per group. ** and *** indicate significant (p<0.01, and p<0.001, respectively) increases in cytokine release from microglia-depleted astrocytes treated with palmitic acid as compared to control cells treated with BSA. ## and ### indicate significant (p<0.01, and p<0.001, respectively) decreases in cytokine release from astrocytes treated with palmitic acid in the presence of TAK-242, UO126, or SB 239063 as compared to cells treated with the same dose of palmitic acid alone.
5. DHA prevents the effects of palmitic acid on cytokine release
The essential ω-3 fatty acid docosahexaenoic acid (DHA) is a critical contributor to cell structure and function in the nervous system (reviewed in (Bourre 2004)). Furthermore, several reports have shown that ω-3 fatty acids such as DHA have potent anti-inflammatory effects (reviewed in (Calder 2007)) and significant neurologic benefits as well (Dyall & Michael-Titus 2008), (McNamara 2010). To determine if DHA could modulate the proinflammatory effects of palmitic acid, cultured astrocytes were pretreated with increasing doses of DHA (10–100 μM) 30 min before application of BSA or 200 μM palmitic acid. Astrocytes in these experiments were not exposed to LME, as previous experiments (see Section 4, above) revealed that cytokine release was not dependent on microglial contaminates. Data show that increasing doses of DHA significantly decreased basal, unstimulated levels of IL-6, but not TNFα (Fig. 5A and B). Additionally, pretreatment with DHA significantly and dose-dependently attenuated palmitic acid-induced cytokine release (Fig. 5A and B), but did not affect MTT conversion (Fig. 5F). To examine more closely how DHA might prevent palmitic acid-induced cytokine release, cultured astrocytes were treated with vehicle (BSA) or 50 μM DHA for 45 min, and then stimulated with 200 μm PA or vehicle for exactly 10 min, after which cells were quickly rinsed and frozen, and processed for Western blot. Examination of blot images indicated that DHA attenuated the phosphorylation of both p38MAKP and p42/44 MAPK in cultured astrocytes, suggesting that the action of DHA is upstream of MAPK activation, and thus may involve modulation of TLR4 expression or activity. Indeed, recent publications show that in cultured leukocytes, DHA prevents LPS- or lauric acid-induced dimerization and activation of TLR4, thereby blunting TLR4-mediated signaling and consequent inflammatory responses (Wong et al. 2009).
Figure 5. Effects of DHA on palmitic acid-induced TNFα and IL-6 release.

Primary astrocytes were establis treated with DHA 45 min before administration of BSA or 200 μM palmitic acid (PA). Release of (A) TNF, (B) IL-6, and (C) MTT conversion to formazan was measured after 24 h. Data were compiled from 3 separate experiments, and are means and SEM of 10–25 dishes per group. *** indicates significant (p<0.001) increases in cytokine release from astrocytes treated with palmitic acid as compared to control cells. #, ##, and ### indicate significant (p<0.05, p<0.01, and p<0.001, respectively) decreases in cytokine release from astrocytes treated with palmitic acid or BSA in the presence of DHA as compared to cells treated with palmitic acid alone. (D) Representative images of Western blots depicting alterations in phosphorylation of p42/p44 MAPK (top panel) and p38 (middle panel) as compared to tubulin expression (bottom panel) in cultured astrocytes treated with palmitic acid in the presence or absence of DHA. Cells were treated with vehicle (BSA) or 50 μM DHA for 45 min, and then stimulated with 200 μM PA or vehicle for exactly 10 min. Data are representative of 2 separate experiments each with 2–5 samples per treatment.
DISCUSSION
Glial cells, consisting of microglia, astrocytes, and oligodendrocytes, constitute more than 70% of the total cell population in the central nervous system. Once thought of as only supportive systems for neurons, glial cells are now regarded as key modulatory, trophic, and immune elements in brain. Astrocytes in particular adopt organized and essentially non-overlapping sentinel positions throughout the entire brain with no brain regions devoid of astrocytes or closely related cells (reviewed in (Sofroniew & Vinters 2010)). Furthermore, astrocytes have intimate contact with both vascular and synaptic elements, and thus likely serve as key links between peripheral disease processes and detrimental brain responses. Thus, astrocytes may be pivotal in the effects of obesity and metabolic disease on the brain, although this has never been directly explored. In this study, cultured primary astrocytes were treated with saturated or unsaturated fatty acids, and the resulting inflammatory responses were evaluated. Data show that saturated, but not unsaturated, fatty acids trigger cytokine release from astrocytes in a dose-dependent manner. These effects appear to require TLR4 and activation of p38 and p42/44 MAPK pathways, but not JNK or PI3K activation. Finally, data also show that the essential ω-3 fatty acid DHA acts in a dose-dependent manner to prevent the actions of saturated fatty acids on astrocytic inflammatory signaling. Overall, these data demonstrate the ability of saturated fatty acids to induce astrocyte inflammation, and thus suggest a potential role for this phenomenon in the reported adverse neurologic consequences of obesity and metabolic syndrome.
Fatty acids serve both as energy substrates and integral membrane lipids that maintain the structure and function of neuronal membranes and membrane associated proteins/protein complexes. Fatty acids cross the blood–brain barrier mainly by simple diffusion in the unbound form, so that penetration of circulating free fatty acids to the CNS is generally proportional to their plasma concentration (Dhopeshwarkar & Mead 1973), (Miller et al. 1987), (Rapoport 1996), (Smith & Nagura 2001), even though a minor proportion of fatty acid uptake into the brain may also occur through direct uptake of lipoprotein particles mediated by lipoprotein receptors (Qi et al. 2002), (Rapoport 2001). Thus, while the maintenance of physiologic levels of polyunsaturated fatty acids is critical for normal brain function, there is ample evidence that alterations or elevations in saturated fatty acids can disrupt and undermine brain function. For example, epidemiological studies suggest that high-fat diet is a significant risk factor for the development of Alzheimer’s disease (AD) and the degree of saturation of fatty acids is critical in determining AD risk (Solfrizzi et al. 2005), (Scarmeas et al. 2006), (Grant 1999). Plasma concentrations of fatty acids are significantly increased in association with obesity and metabolic syndrome (Opie & Walfish 1963), (Reaven et al. 1988), and diets high in saturated fats may actually increase brain uptake of fatty acids from the plasma (Wang et al. 1994), (Karmi et al. 2010). Furthermore, the saturated fatty acids palmitic and lauric acid have both been shown to trigger inflammation in cultured macrophages (Tappia et al. 1995), (Laine et al. 2007), (Martins de Lima-Salgado et al. 2011), and to modulate amyloid processing in neurons and astrocytes ((Patil & Chan 2005), (Patil et al. 2006).
Data in this manuscript complement a growing body of literature describing the sensitivity of the brain to obesity and obesity-induced metabolic dysfunction (reviewed in (Middleton & Yaffe 2009), (Bruce-Keller et al. 2009), (Fillit et al. 2008)), and further support a potentially significant role for astrocytes in these effects. Indeed, the adverse effects of metabolic syndrome on brain physiology in rodents have been very well established by our labs (Pistell et al. 2010, Bruce-Keller et al. 2009), (Bruce-Keller et al. 2010), (White et al. 2009), (Zhang et al. 2009), (Studzinski et al. 2009) and by others (Farr et al. 2008), (Winocur & Greenwood 2005), (Stranahan et al. 2008), (Jurdak et al. 2008), (Molteni et al. 2002), and increased gliosis is a very common finding in rodent studies of diet-induced metabolic dysfunction. Reactive gliosis is strongly associated with brain inflammation, and increased inflammation is a key physiologic feature of obesity (reviewed in (Hotamisligil 2006). The brain is exquisitely sensitive to inflammatory pathways and mediators, and metabolic dysfunction in both humans and laboratory animals is associated with reactive gliosis and increased brain inflammation (Bruce-Keller et al. 2010), (White et al. 2009), (Mattson et al. 2003), (Zhang et al. 2005), (Souza et al. 2007), (Pistell et al. 2010). Cytokines, particularly TNFα, IL-1β, and IL-6, are major effectors of the neuroinflammatory cascade (Allan & Rothwell 2003), (Rothwell & Hopkins 1995), (Rothwell 1999), and can disrupt neurophysiologic mechanisms involved in cognition and memory (Bellinger et al. 1995), (Jankowsky & Patterson 1999) (Gemma & Bickford 2007). Indeed, the highest levels of cytokine binding have been demonstrated in certain areas associated with learning and memory, including regions of the cortex and hippocampus (Parnet et al. 2002). Furthermore, reports suggest that the detrimental effects of radiation therapy on cognitive function may be based in part on TNFα, IL-6, and IL-1β (Monje et al. 2003). It should also be noted that saturated long-chain fatty acids are known to activate inflammatory and innate immune responses in peripheral cells (Kennedy et al. 2009), (Averill & Bornfeldt 2009), (Tappia et al. 1995), (Laine et al. 2007), (Martins de Lima-Salgado et al. 2011), and that brain inflammation in animal models of metabolic syndrome is associated with increased serum lipids (Mattson et al. 2003), (Perry et al. 2003).
The essential ω-3 fatty acid DHA is a critical contributor to cell structure and function in the nervous system (reviewed in (Palacios-Pelaez et al. 2010), (Bourre 2004)), and data in this report show that DHA is able to prevent the proinflammatory actions of saturated fatty acids on astrocytes. These data are in keeping with numerous reports that have shown that ω-3 fatty acids such as DHA have potent anti-inflammatory and protective effects (reviewed in (Calder 2007)), and can indeed prevent or reverse some aspects of metabolic syndrome (Rossmeisl et al. 2009), (Fedor & Kelley 2009), (Hassanali et al. 2010 ). DHA is the most abundant omega-3 PUFA in the mammalian brain and retina, and is thought to preserve plasma membrane flexibility and resistance to membrane lipid oxidation (Eddy & Harman 1977). Indeed, a double-blind, randomized clinical trial using healthy adult volunteers given a 35 day regimen of 4 g fish oil/day (800 mg DHA and 1,600 mg EPA) or 4 g olive oil as placebo showed that ω-3 fatty acid supplementation significantly improved performance on several neurologic parameters, including anxiety, fatigue, depression, and confusion (Fontani et al. 2005). Furthermore, ω-3 fatty acid supplementation may reduce the number and severity of MS relapses in patients with MS (Nordvik et al. 2000), (Weinstock-Guttman et al. 2005). With further regards to the data presented in this manuscript, published experimental data indicate that DHA may limit inflammatory signaling by altering TLR4 presentation (Wong et al. 2009), (Lee et al. 2003), (De Smedt-Peyrusse et al. 2008). Toll-like receptors (TLRs) play a key role in inflammation and specifically in the recognition of products from pathogenic organisms, and our data corroborate other reports indicating that saturated fatty acids can serve as ligands for TLR4 (Lee et al. 2001), (Shi et al. 2006), (Kleinridders et al. 2009). Indeed, mice with either genetically engineered or naturally occurring mutations of TLR4 are protected from obesity-associated insulin resistance (Poggi et al. 2007), (Shi et al. 2006), (Tsukumo et al. 2007), and TLR4 disruption selectively prevents insulin resistance caused by diets high in saturated, but not unsaturated, fatty acids (Davis. et al. 2008). Collectively, these data support a potential role for astrocyte cytokine release, mediated by TL4 activation by increased circulating saturated fatty acids, in driving CNS inflammation in the context of obesity/metabolic syndrome.
Acknowledgments
The authors are grateful to Taryn Parino for expert technical assistance. This work was supported by grants from the NIH (NS46267 and AG05119).
Footnotes
The authors have no competing financial or other conflicts of interest.
References
- Ajuwon KM, Spurlock ME. Palmitate activates the NF-kappaB transcription factor and induces IL-6 and TNFalpha expression in 3T3-L1 adipocytes. J Nutr. 2005;135:1841–1846. doi: 10.1093/jn/135.8.1841. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan SM, Rothwell NJ. Inflammation in central nervous system injury. Philos Trans R Soc Lond B Biol Sci. 2003;358:1669–1677. doi: 10.1098/rstb.2003.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Averill MM, Bornfeldt KE. Lipids versus glucose in inflammation and the pathogenesis of macrovascular disease in diabetes. Curr Diab Rep. 2009;9:18–25. doi: 10.1007/s11892-009-0005-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellinger FP, Madamba SG, Campbell IL, Siggins GR. Reduced long-term potentiation in the dentate gyrus of transgenic mice with cerebral overexpression of interleukin-6. Neurosci Lett. 1995;198:95–98. doi: 10.1016/0304-3940(95)11976-4. [DOI] [PubMed] [Google Scholar]
- Bernoud N, Fenart L, Bénistant C, Pageaux JF, Dehouck MP, Molière P, Lagarde M, Cecchelli R, Lecerf J. Astrocytes are mainly responsible for the polyunsaturated fatty acid enrichment in blood-brain barrier endothelial cells in vitro. J Lipid Res. 1998;39:1816–1824. [PubMed] [Google Scholar]
- Bourre JM. Roles of unsaturated fatty acids (especially omega-3 fatty acids) in the brain at various ages and during ageing. J Nutr Health Aging. 2004;8:163–174. [PubMed] [Google Scholar]
- Bradley RL, Fisher FF, Maratos-Flier E. Dietary fatty acids differentially regulate production of TNF-alpha and IL-10 by murine 3T3-L1 adipocytes. Obesity (Silver Spring) 2008;16:938–944. doi: 10.1038/oby.2008.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce-Keller AJ, Barger SW, Moss NI, Pham JT, Keller JN, Nath A. Pro-inflammatory and pro-oxidant properties of the HIV protein Tat in a microglial cell line: attenuation by 17beta-estradiol. J Neurochem. 2001;78:1315–1324. doi: 10.1046/j.1471-4159.2001.00511.x. [DOI] [PubMed] [Google Scholar]
- Bruce-Keller AJ, Keller JN, Morrison CD. Obesity and vulnerability of the CNS. Biochim Biophys Acta. 2009;1792:395–400. doi: 10.1016/j.bbadis.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce-Keller AJ, White CL, Gupta S, Knight AG, Pistell PJ, Ingram DK, Morrison CD, Keller JN. NOX activity in brain aging: Exacerbation by high fat diet. Free Radic Biol Med. 2010;49:22–30. doi: 10.1016/j.freeradbiomed.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calder PC. Immunomodulation by omega-3 fatty acids. Prostaglandins Leukot Essent Fatty Acids. 2007;77:327–335. doi: 10.1016/j.plefa.2007.10.015. [DOI] [PubMed] [Google Scholar]
- Davis JE, Gabler NK, Walker-Daniels J, Spurlock ME. Tlr-4 deficiency selectively protects against obesity induced by diets high in saturated fat. Obesity (Silver Spring) 2008;16:1248–1255. doi: 10.1038/oby.2008.210. [DOI] [PubMed] [Google Scholar]
- De Smedt-Peyrusse V, Sarqueil F, Moranis A, Harizi H, Mongrand S, Layé S. Docosahexaenoic acid prevents lipopolysaccharide-induced cytokine production in microglial cells by inhibiting lipopolysaccharide receptor presentation but not its membrane subdomain localization. J Neurochem. 2008;105:296–307. doi: 10.1111/j.1471-4159.2007.05129.x. [DOI] [PubMed] [Google Scholar]
- Dhopeshwarkar GA, Mead JF. Uptake and transport of fatty acids into the brain and role of the blood-brain barrier system. Adv Lipid Res. 1973;11:109–142. doi: 10.1016/b978-0-12-024911-4.50010-6. [DOI] [PubMed] [Google Scholar]
- Dyall SC, Michael-Titus AT. Neurological benefits of omega-3 fatty acids. Neuromolecular Med. 2008;10:219–235. doi: 10.1007/s12017-008-8036-z. [DOI] [PubMed] [Google Scholar]
- Eddy DE, Harman D. Free radical theory of aging: effect of age, sex and dietary precursors on rat-brain docosahexanoic acid. J Am Geriatr Soc. 1977;25:220–229. doi: 10.1111/j.1532-5415.1977.tb00303.x. [DOI] [PubMed] [Google Scholar]
- El-Hage N, Podhaizer EM, Sturgill J, Hauser KF. Toll-like Receptor Expression and Activation in Astroglia: Differential Regulation by HIV-1 Tat, gp120, and Morphine. Immunol Invest. 2011;40:498–522. doi: 10.3109/08820139.2011.561904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias MF, Elias PK, Sullivan LM, Wolf PA, D’Agostino RB. Lower cognitive function in the presence of obesity and hypertension: the Framingham Heart Study. Int J Obes Relat Metab Disord. 2003;27:260–268. doi: 10.1038/sj.ijo.802225. [DOI] [PubMed] [Google Scholar]
- Elias MF, Elias PK, Sullivan LM, Wolf PA, D’Agostino RB. Obesity, diabetes and cognitive deficit: the Framingham Heart Study. Neurobiol Aging. 2005;26:11–16. doi: 10.1016/j.neurobiolaging.2005.08.019. [DOI] [PubMed] [Google Scholar]
- Erridge C, Kennedy S, Spickett CM, Webb DJ. Oxidized Phospholipid Inhibition of Toll-like Receptor (TLR) Signaling Is Restricted to TLR2 and TLR4: roles for CD14, LPS-binding protein, and MD2 as targets for specificity of inhibition. J Biol Chem. 2008;283:24748–24759. doi: 10.1074/jbc.M800352200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farr SA, Yamada KA, Butterfield DA, Abdul HM, Xu L, Miller NE, Banks WA, Morley JE. Obesity and hypertriglyceridemia produce cognitive impairment. Endocrinology. 2008;149:2628–2636. doi: 10.1210/en.2007-1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedor D, Kelley DS. Prevention of insulin resistance by n-3 polyunsaturated fatty acids. Curr Opin Clin Nutr Metab Care. 2009;12:138–146. doi: 10.1097/MCO.0b013e3283218299. [DOI] [PubMed] [Google Scholar]
- Fillit H, Nash DT, Rundek T, Zuckerman A. Cardiovascular risk factors and dementia. Am J Geriatr Pharmacother. 2008;6:100–118. doi: 10.1016/j.amjopharm.2008.06.004. [DOI] [PubMed] [Google Scholar]
- Fontani G, Corradeschi F, Felici A, Alfatti F, Migliorini S, Lodi L. Cognitive and physiological effects of omega-3 polyunsaturated fatty acid supplementation in healthy subjects. Eur J Clin Invest. 2005;35:691–699. doi: 10.1111/j.1365-2362.2005.01570.x. [DOI] [PubMed] [Google Scholar]
- Gemma C, Bickford PC. Interleukin-1beta and caspase-1: players in the regulation of age-related cognitive dysfunction. Rev Neurosci. 2007;18:137–148. doi: 10.1515/revneuro.2007.18.2.137. [DOI] [PubMed] [Google Scholar]
- Gorina R, Font-Nieves M, Márquez-Kisinousky L, Santalucia T, Planas AM. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFκB signaling, MAPK, and Jak1/Stat1 pathways. Glia. 2011;59:242–255. doi: 10.1002/glia.21094. [DOI] [PubMed] [Google Scholar]
- Grant WB. Dietary links to Alzheimer’s disease: 1999 update. J Alzheimer’s Dis. 1999;1:197–201. doi: 10.3233/jad-1999-14-501. [DOI] [PubMed] [Google Scholar]
- Haslam DW, James WP. Obesity. Lancet Neurol. 2005;366:1197–1209. doi: 10.1016/S0140-6736(05)67483-1. [DOI] [PubMed] [Google Scholar]
- Hassanali Z, Ametaj BN, Field CJ, Proctor SD, Vine DF. Dietary supplementation of n-3 PUFA reduces weight gain and improves postprandial lipaemia and the associated inflammatory response in the obese JCR:LA-cp rat. Diabetes Obes Metab. 2010;12:139–147. doi: 10.1111/j.1463-1326.2009.01130.x. [DOI] [PubMed] [Google Scholar]
- Håversen L, Danielsson KN, Fogelstrand L, Wiklund O. Induction of proinflammatory cytokines by long-chain saturated fatty acids in human macrophages. Atherosclerosis. 2009;202:382–393. doi: 10.1016/j.atherosclerosis.2008.05.033. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Patterson PH. Cytokine and growth factor involvement in long-term potentiation. Mol Cell Neurosci. 1999:273–286. [PubMed] [Google Scholar]
- Jurdak N, Lichtenstein AH, Kanarek RB. Diet-induced obesity and spatial cognition in young male rats. Nutr Neurosci. 2008;11:48–54. doi: 10.1179/147683008X301333. [DOI] [PubMed] [Google Scholar]
- Karmi A, Iozzo P, Viljanen A, et al. Increased brain fatty acid uptake in metabolic syndrome. Diabetes. 2010;59:2171–2177. doi: 10.2337/db09-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy A, Martinez K, Chuang CC, LaPoint K, McIntosh M. Saturated fatty acid-mediated inflammation and insulin resistance in adipose tissue: mechanisms of action and implications. J Nutr. 2009;139:1–4. doi: 10.3945/jn.108.098269. [DOI] [PubMed] [Google Scholar]
- Kleinridders A, Schenten D, Könner AC, Belgardt BF, Mauer J, Okamura T, Wunderlich FT, Medzhitov R, Brüning JC. MyD88 signaling in the CNS is required for development of fatty acid-induced leptin resistance and diet-induced obesity. Cell Metab. 2009;10:249–259. doi: 10.1016/j.cmet.2009.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine PS, Schwartz EA, Wang Y, Zhang WY, Karnik SK, Musi N, Reaven PD. Palmitic acid induces IP-10 expression in human macrophages via NF-kappaB activation. Biochem Biophys Res Commun. 2007;358:150–155. doi: 10.1016/j.bbrc.2007.04.092. [DOI] [PubMed] [Google Scholar]
- Lee JY, Plakidas A, Lee WH, Heikkinen A, Chanmugam P, Bray G, Hwang DH. Differential modulation of Toll-like receptors by fatty acids: preferential inhibition by n-3 polyunsaturated fatty acids. J Lipid Res. 2003;44:479–486. doi: 10.1194/jlr.M200361-JLR200. [DOI] [PubMed] [Google Scholar]
- Lee JY, Sohn KH, Rhee SH, Hwang D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J Biol Chem. 2001;276:16683–16689. doi: 10.1074/jbc.M011695200. [DOI] [PubMed] [Google Scholar]
- Martins de Lima-Salgado T, Coccuzzo Sampaio S, Cury-Boaventura MF, Curi R. Modulatory effect of fatty acids on fungicidal activity, respiratory burst and TNF-α and IL-6 production in J774 murine macrophages. Br J Nutr. 2011;105:1173–1179. doi: 10.1017/S0007114510004873. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Duan W, Guo Z. Meal size and frequency affect neuronal plasticity and vulnerability to disease: cellular and molecular mechanisms. J Neurochem. 2003;84:417–431. doi: 10.1046/j.1471-4159.2003.01586.x. [DOI] [PubMed] [Google Scholar]
- McNamara RK. DHA deficiency and prefrontal cortex neuropathology in recurrent affective disorders. J Nutr. 2010;140:864–868. doi: 10.3945/jn.109.113233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton LE, Yaffe K. Promising strategies for the prevention of dementia. Arch Neurol. 2009;66:1210–1215. doi: 10.1001/archneurol.2009.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JC, Gnaedinger JM, Rapoport SI. Utilization of plasma fatty acid in rat brain: distribution of [14C]palmitate between oxidative and synthetic pathways. J Neurochem. 1987;49:1507–1514. doi: 10.1111/j.1471-4159.1987.tb01021.x. [DOI] [PubMed] [Google Scholar]
- Molteni R, Barnard R, Ying Z, Roberts C, Gómez-Pinilla F. A high-fat, refined sugar diet reduces hippocampal brain-derived neurotrophic factor, neuronal plasticity, and learning. Neuroscience. 2002;112:803–814. doi: 10.1016/s0306-4522(02)00123-9. [DOI] [PubMed] [Google Scholar]
- Monje ML, Toda H, Palmer TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science. 2003;302:1760–1765. doi: 10.1126/science.1088417. [DOI] [PubMed] [Google Scholar]
- Morand O, Baumann N, Bourre JM. In vivo incorporation of exogenous [1-14C]stearic acid into neurons and astrocytes. Neurosci Lett. 1979;13:177–181. doi: 10.1016/0304-3940(79)90038-7. [DOI] [PubMed] [Google Scholar]
- Nordvik I, Myhr KM, Nyland H, Bjerve KS. Effect of dietary advice and n-3 supplementation in newly diagnosed MS patients. Acta Neurol Scand. 2000;102:143–149. doi: 10.1034/j.1600-0404.2000.102003143.x. [DOI] [PubMed] [Google Scholar]
- Opie LH, Walfish PG. Plasma free fatty acid concentrations in obesity. N Engl J Med. 1963;268:757–760. doi: 10.1056/NEJM196304042681404. [DOI] [PubMed] [Google Scholar]
- Palacios-Pelaez R, Lukiw WJ, Bazan NG. Omega-3 essential fatty acids modulate initiation and progression of neurodegenerative disease. Mol Neurobiol. 2010;41:367–374. doi: 10.1007/s12035-010-8139-z. [DOI] [PubMed] [Google Scholar]
- Pannacciulli N, Del Parigi A, Chen K, Le DS, Reiman EM, Tataranni PA. Brain abnormalities in human obesity: a voxel-based morphometric study. Neuroimage. 2006;31:1419–1425. doi: 10.1016/j.neuroimage.2006.01.047. [DOI] [PubMed] [Google Scholar]
- Parnet P, Kelley KW, Bluthé RM, Dantzer R. Expression and regulation of interleukin-1 receptors in the brain. Role in cytokines-induced sickness behavior. J Neuroimmunol. 2002;125:5–14. doi: 10.1016/s0165-5728(02)00022-x. [DOI] [PubMed] [Google Scholar]
- Patil S, Chan C. Palmitic and stearic fatty acids induce Alzheimer-like hyperphosphorylation of tau in primary rat cortical neurons. Neurosci Lett. 2005;384:288–293. doi: 10.1016/j.neulet.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Patil S, Sheng L, Masserang A, Chan C. Palmitic acid-treated astrocytes induce BACE1 upregulation and accumulation of C-terminal fragment of APP in primary cortical neurons. Neurosci Lett. 2006;406:55–59. doi: 10.1016/j.neulet.2006.07.015. [DOI] [PubMed] [Google Scholar]
- Perry G, Nunomura A, Raina AK, et al. A metabolic basis for Alzheimer disease. Neurochem Res. 2003;28:1549–1552. doi: 10.1023/a:1025678510480. [DOI] [PubMed] [Google Scholar]
- Pistell PJ, Morrison CD, Gupta S, Knight AG, Keller JN, Ingram DK, Bruce-Keller AJ. Cognitive impairment following high fat diet consumption is associated with brain inflammation. J Neuroimmunol. 2010;219:25–32. doi: 10.1016/j.jneuroim.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poggi M, Bastelica D, Gual P, et al. C3H/HeJ mice carrying a toll-like receptor 4 mutation are protected against the development of insulin resistance in white adipose tissue in response to a high-fat diet. Diabetologia. 2007;50:1267–1276. doi: 10.1007/s00125-007-0654-8. [DOI] [PubMed] [Google Scholar]
- Qi K, Hall M, Deckelbaum RJ. Long-chain polyunsaturated fatty acid accretion in brain. Curr Opin Clin Nutr Metab Care. 2002;5:133–138. doi: 10.1097/00075197-200203000-00003. [DOI] [PubMed] [Google Scholar]
- Rapoport SI. In vivo labeling of brain phospholipids by long-chain fatty acids: relation to turnover and function. Lipids. 1996;31(Suppl):S97–S101. doi: 10.1007/BF02637059. [DOI] [PubMed] [Google Scholar]
- Rapoport SI. In vivo fatty acid incorporation into brain phosholipids in relation to plasma availability, signal transduction and membrane remodeling. J Mol Neurosci. 2001;16:243–261. doi: 10.1385/JMN:16:2-3:243. [DOI] [PubMed] [Google Scholar]
- Reaven GM, Hollenbeck C, Jeng CY, Wu MS, Chen YD. Measurement of plasma glucose, free fatty acid, lactate, and insulin for 24 h in patients with NIDDM. Diabetes. 1988;37:1020–1024. doi: 10.2337/diab.37.8.1020. [DOI] [PubMed] [Google Scholar]
- Rossmeisl M, Jelenik T, Jilkova Z, et al. Prevention and reversal of obesity and glucose intolerance in mice by DHA derivatives. Obesity (Silver Spring) 2009;17:1023–1031. doi: 10.1038/oby.2008.602. [DOI] [PubMed] [Google Scholar]
- Rothwell NJ. Annual review prize lecture cytokines - killers in the brain? J Physiol Pharmacol(London) 1999;514:3–17. doi: 10.1111/j.1469-7793.1999.003af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell NJ, Hopkins SJ. Cytokines and the nervous system II: actions and mechanisms of action. TINS. 1995;18(3):130–136. doi: 10.1016/0166-2236(95)93890-a. [DOI] [PubMed] [Google Scholar]
- Rustan AC, Christiansen EN, Drevon CA. Serum lipids, hepatic glycerolipid metabolism and peroxisomal fatty acid oxidation in rats fed omega-3 and omega-6 fatty acids. Biochem J. 1992;283:333–339. doi: 10.1042/bj2830333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarmeas N, Stern Y, Tang MX, Mayeux R, Luchsinger JA. Mediterranean diet and risk for Alzheimer’s disease. Ann Neurol. 2006;59:912–921. doi: 10.1002/ana.20854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116:3015–3025. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith QR, Nagura H. Fatty acid uptake and incorporation in brain: studies with the perfusion model. J Mol Neurosci. 2001;16:167–172. doi: 10.1385/JMN:16:2-3:167. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solfrizzi V, D’Introno A, Colacicco AM, Capurso C, Parigi AD, Capurso S, Gadaleta A, Capurso A, Panza F. Dietary fatty acids intake: possible role in cognitive decline and dementia. Exp Gerontol. 2005;40:257–270. doi: 10.1016/j.exger.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Souza CG, Moreira JD, Siqueira IR, Pereira AG, Rieger DK, Souza DO, Souza TM, Portela LV, Perry ML. Highly palatable diet consumption increases protein oxidation in rat frontal cortex and anxiety-like behavior. Life Sci. 2007;81:198–203. doi: 10.1016/j.lfs.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Stentz FB, Kitabchi AE. Palmitic acid-induced activation of human T-lymphocytes and aortic endothelial cells with production of insulin receptors, reactive oxygen species, cytokines, and lipid peroxidation. Biochem Biophys Res Commun. 2006;346:721–726. doi: 10.1016/j.bbrc.2006.05.159. [DOI] [PubMed] [Google Scholar]
- Stranahan AM, Norman ED, Lee K, Cutler RG, Telljohann RS, Egan JM, Mattson MP. Diet-induced insulin resistance impairs hippocampal synaptic plasticity and cognition in middle-aged rats. Hippocampus. 2008;18:1085–1088. doi: 10.1002/hipo.20470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studzinski CM, Li F, Bruce-Keller AJ, Fernandez-Kim SO, Zhang L, Weidner AM, Markesbery WR, Murphy MP, Keller JN. Effects of short-term Western diet on cerebral oxidative stress and diabetes related factors in APP x PS1 knock-in mice. J Neurochem. 2009;108:860–866. doi: 10.1111/j.1471-4159.2008.05798.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tappia PS, Man WJ, Grimble RF. Influence of unsaturated fatty acids on the production of tumour necrosis factor and interleukin-6 by rat peritoneal macrophages. Mol Cell Biochem. 1995;14:89–98. doi: 10.1007/BF01816941. [DOI] [PubMed] [Google Scholar]
- Tsukumo DM, Carvalho-Filho MA, Carvalheira JB, et al. Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes. 2007;56:1986–1998. doi: 10.2337/db06-1595. [DOI] [PubMed] [Google Scholar]
- Turchan-Cholewo J, Dimayuga FO, Ding Q, Keller JN, Hauser KF, Knapp PE, Bruce-Keller AJ. Cell-specific actions of HIV-Tat and morphine on opioid receptor expression in glia. J Neurosci Res. 2008;86:2100–2110. doi: 10.1002/jnr.21653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Schlieffen E, Oskolkova OV, Schabbauer G, et al. Multi-Hit Inhibition of Circulating and Cell-Associated Components of the Toll-Like Receptor 4 Pathway by Oxidized Phospholipids. Arterioscler Thromb Vasc Biol. 2009;29:356–362. doi: 10.1161/ATVBAHA.108.173799. [DOI] [PubMed] [Google Scholar]
- Waldstein SR, Katzel LI. Interactive relations of central versus total obesity and blood pressure to cognitive function. Int J Obes (Lond) 2006;30:201–207. doi: 10.1038/sj.ijo.0803114. [DOI] [PubMed] [Google Scholar]
- Wang SW, Wang M, Grossman BM, Martin RJ. Effects of dietary fat on food intake and brain uptake and oxidation of fatty acids. Physiol Behav. 1994;56:517–522. doi: 10.1016/0031-9384(94)90295-x. [DOI] [PubMed] [Google Scholar]
- Ward MA, Carlsson CM, Trivedi MA, Sager MA, Johnson SC. The effect of body mass index on global brain volume in middle-aged adults: a cross sectional study. BMC Neurol. 2005:23. doi: 10.1186/1471-2377-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstock-Guttman B, Baier M, Park Y, et al. Low fat dietary intervention with omega-3 fatty acid supplementation in multiple sclerosis patients. Prostaglandins, Leukot Essent Fat Acids. 2005;73:397–404. doi: 10.1016/j.plefa.2005.05.024. [DOI] [PubMed] [Google Scholar]
- White CL, Pistell PJ, Purpera MN, et al. Effects of high fat diet on Morris maze performance, oxidative stress, and inflammation in rats: contributions of maternal diet. Neurobiol Dis. 2009;35:3–13. doi: 10.1016/j.nbd.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CJ, Finch CE, Cohen HJ. Cytokines and cognition--the case for a head-to-toe inflammatory paradigm. J Am Geriatr Soc. 2002;50:2041–2056. doi: 10.1046/j.1532-5415.2002.50619.x. [DOI] [PubMed] [Google Scholar]
- Winocur G, Greenwood CE. Studies of the effects of high fat diets on cognitive function in a rat model. Neurobiol Aging. 2005;26:46–49. doi: 10.1016/j.neurobiolaging.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Wong SW, Kwon MJ, Choi AM, Kim HP, Nakahira K, Hwang DH. Fatty acids modulate Toll-like receptor 4 activation through regulation of receptor dimerization and recruitment into lipid rafts in a reactive oxygen species-dependent manner. J Biol Chem. 2009;284:27384–27392. doi: 10.1074/jbc.M109.044065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu ZJ, Fan JG, Ding XD, Qiao L, Wang GL. Characterization of High-Fat, Diet-Induced, Non-alcoholic Steatohepatitis with Fibrosis in Rats. Dig Dis Sci. 2010;55:931–940. doi: 10.1007/s10620-009-0815-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Bruce-Keller AJ, Dasuri K, Nguyen A, Liu Y, Keller JN. Diet-induced metabolic disturbances as modulators of brain homeostasis. Biochim Biophys Acta. 2009;1792:417–422. doi: 10.1016/j.bbadis.2008.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Dong F, Ren J, Driscoll MJ, Culver B. High dietary fat induces NADPH oxidase-associated oxidative stress and inflammation in rat cerebral cortex. Exp Neurol. 2005;191:318–325. doi: 10.1016/j.expneurol.2004.10.011. [DOI] [PubMed] [Google Scholar]