

Abstract
Limited neuromuscular input results in muscle weakness in neuromuscular disease either because of a reduction in the density of muscle innervation, the rate of neuromuscular junction activation or the efficiency of synaptic transmission1. We developed a small molecule fast skeletal troponin activator, CK-2017357, as a means to increase muscle strength by amplifying the response of muscle when neuromuscular input is diminished secondary to a neuromuscular disease. Binding selectively to the fast skeletal troponin complex, CK-2017357 slows the rate of calcium release from troponin C and sensitizes muscle to calcium. As a consequence, the force-calcium relationship of muscle fibers shifts leftwards as does the force-frequency relationship of a nerve-muscle pair. In vitro and in vivo, CK-2017357 increases the production of force at sub-maximal stimulation rates. Importantly, we show that sensitization of the fast skeletal troponin complex to calcium improves muscle force and grip strength immediately after single doses of CK-2017357 in a model of neuromuscular disease, myasthenia gravis. Troponin activation may provide a new therapeutic approach to improve physical activity in diseases where neuromuscular function is compromised.
Skeletal muscle contraction occurs following motor neuron firing and transmission of this impulse across the neuromuscular junction (NMJ). The ensuing action potential triggers the release of calcium from the sarcoplasmic reticulum (SR). As the frequency of nerve firing increases, the amount of calcium released from the SR increases, causing the force of muscle contraction to ramp proportionally from a twitch up to a maximal tetanic contraction2. The relationship of the frequency of motor neuron firing to muscle force is termed rate coding 3 and is one of the means by which muscle strength is controlled. Under normal conditions, skeletal muscle operates in a range of forces between 10% and 65% of maximum4.
In many diseases, muscle weakness is the result of limited neuromuscular input and can lead to significant disability and increased mortality. Peripheral motor neuropathies such as amyotrophic lateral sclerosis (ALS), spinal muscular atrophy (SMA), and Charcot-Marie-Tooth disease (CMT) cause motor neuron damage and death, straining the ability of surviving motor neurons to stimulate muscle effectively to generate force5-8. In myasthenia gravis (MG), weakness and fatigue result from failure of signal transmission at the NMJ which limits calcium release and force production9; treatment consists of acetylcholinesterase inhibitors and immunosuppression although weakness and fatigue are still common in these patients10. Therapeutic options for other neuropathies are limited or non-existent.
We hypothesized that amplifying the response of the sarcomere, the fundamental contractile unit in skeletal muscle, to inadequate motor neuron input would improve muscle force generation and physical function in patients with neuromuscular diseases. One means to do so is to increase the calcium sensitivity of the troponin-tropomyosin regulatory complex, the calcium sensor within the sarcomere that regulates the actin-myosin force-generating interaction. We identified a structural class of fast skeletal troponin activators following high-throughput screens of fast (Type II) and slow (Type I) skeletal myofibrils. This compound class was subsequently optimized for potency, physiochemical properties, and pharmacokinetics leading to the synthesis of CK-2017357 (molecular weight = 230.3, Fig. 1a).
FIGURE 1. CK-2017357 is a selective calcium sensitizer of the fast skeletal troponin complex.
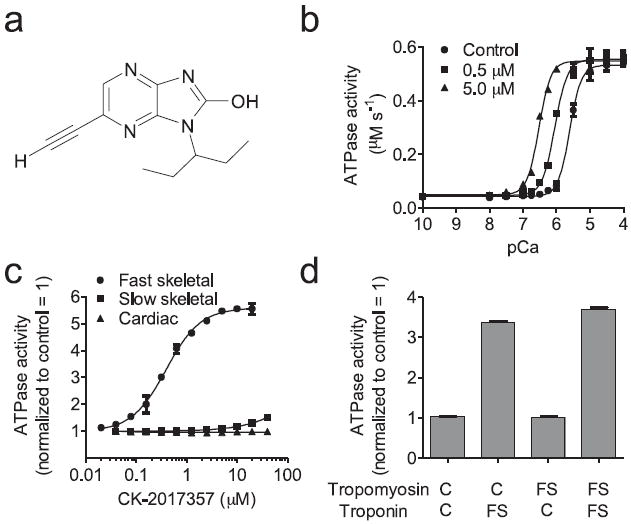
(a) The chemical structure of CK-2017357. (b) Calcium dependence of the fast skeletal myofibril ATPase at three concentrations of CK-2017357. (c) The dose response of CK-2017357 treated fast skeletal, slow skeletal and cardiac myofibrils at pCa 6.0. (d) Activation of heterologous, reconstituted thin filaments (source of regulatory complex isoform, C = cardiac, FS = fast skeletal) by CK-2017357 (20 μM) using cardiac myosin ATPase (mean ± SD) as a probe for thin filament activation at a pCa 6.75, approximately the pCa25 for all systems.
CK-2017357 selectively sensitized fast skeletal muscle to calcium by binding to its troponin complex. We prepared nearly pure populations of fast, slow, and cardiac myofibrils from well characterized sources11-14: rabbit psoas (fast), bovine masseter (slow), and bovine cardiac muscle (muscle composition confirmed by myosin heavy chain analysis, Supplementary Fig. 1a). Addition of CK-2017357 to fast skeletal myofibrils resulted in a leftward shift in the calcium-dependent myosin adenosine triphosphatase (ATPase) relationship (Fig. 1b) with the pCa producing half-maximal force (pCa50) shifting from 5.61 ± 0.01 (control) to 6.52 ± 0.02 (CK-2017357, 5.0 μM). We characterized potency by measuring an increase in the ATPase rate at a fixed calcium concentration (pCa = 6.0) resulting in an EC50 of 390 ± 17 nM (Fig. 1c). There was little or no effect in myofibrils from slow skeletal and cardiac muscle (Fig. 1c), demonstrating its selectivity for fast skeletal muscle. Activation of myofibrils from bovine rectus abdominis muscle (EC50 = 770 ± 100 nM, Supplementary Fig. 1b), a mixed fiber type muscle that contains in part fast skeletal muscle, confirmed its selectivity profile in muscle derived from the same species.
We leveraged this selectivity to determine the target of CK-2017357 using heterologous reconstituted versions of troponin-tropomyosin–regulated actin-myosin15. CK-2017357 activated only those reconstitutions containing the fast skeletal troponin complex (Fig. 1d). Isothermal titration calorimetry further confirmed a direct interaction of CK-2017357 with fast skeletal troponin (Kd = 40 ± 6 nM). Addition of CK-2017357 to purified fast skeletal troponin complex resulted in an exothermic reaction that fit well to a single site binding model. Consistent with its myofibril selectivity, there was modest affinity for slow skeletal troponin (Kd = 3800 ± 700 nM) and none measureable for cardiac troponin, (Fig. 2a). This selectivity is not surprising given the substantially different amino acid composition (~50%) of the troponin complex in each muscle type.
FIGURE 2. CK-2017357 binds to the skeletal troponin complex and slows calcium release.
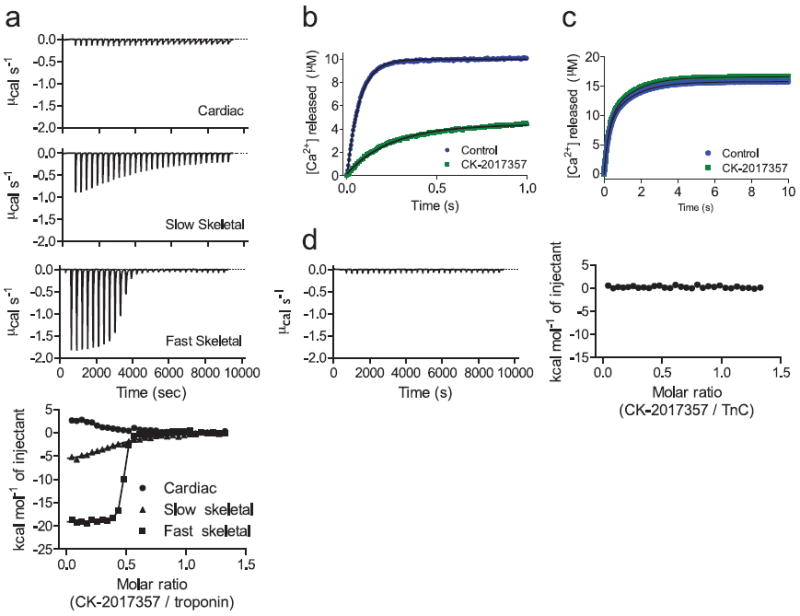
(a) Isothermal titration calorimetry of CK-2017357 added into purified cardiac, slow skeletal, and fast skeletal intact troponin complex (bottom panel). Shown above are representative experiments for the heats of addition to each isoform during the CK-2017357 titration. (b) Fluorescence intensity of the calcium chelator, Quin-2, following rapid mixing with intact fast skeletal troponin (10 μM final concentration) or (c) recombinant rabbit fast skeletal muscle troponin C (10 μM final concentration) in the presence or absence of CK-2017357 (20 μM final concentration). (d) Isothermal titration calorimetry of CK-2017357 added to recombinant rabbit fast skeletal troponin C (50 μM).
The troponin complex contains three subunits, troponin C, I, and T. Troponin C is a calcium sensor with four binding sites, two high affinity (Kd ~50 nM) and two low affinity (Kd ~5 μM) 16. Following calcium release from the SR, binding to the low affinity sites results in tropomyosin movement and allows the actin-myosin interaction to proceed17. As calcium is pumped back into the SR, calcium levels fall, the low affinity sites release calcium, and the muscle contraction ends. We measured calcium release from the low affinity sites of fast skeletal troponin using a fluorescent calcium indicator in a stopped flow apparatus. CK-2017357 (20 μM) slowed the rate of Ca2+ release from 14.7 s-1 [95% CI 14.5-14.8] to 4.0 s-1 [95% CI 3.9-4.1], consistent with an increase in the affinity of troponin for calcium (Fig. 2b). Similar experiments with isolated fast skeletal troponin C failed to demonstrate either a change in calcium release rate (Fig. 2c) or a binding interaction (Fig. 2d), suggesting the CK-2017357 binding site lies at an interface between two or more troponin subunits.
We sought to understand how an increase in troponin calcium affinity would translate to changes in muscle force. Chemically “skinned” human muscle fibers devoid of the plasma membrane and sarcoplasmic reticulum were prepared from muscle biopsies of the vastus lateralis (VL), a mixed fast (type IIa, IIx) and slow (type I) muscle. These fibers contract when exogenous calcium is added to the muscle fiber. As expected, treatment of fast fibers with CK-2017357 shifted the force-calcium relationship leftwards without increasing maximum force or the shape of the curve; in contrast, slow fibers were approximately ten fold less responsive to CK-2017357 (Fig. 3a). Skinned skeletal muscle fibers from animal sources demonstrated similar selectivity and notably skinned cardiac muscle fibers obtained from rat heart were unresponsive to CK-2017357 (60 μM) (Fig. 3b). These experiments confirmed the selectivity of CK-2107357 for fast skeletal muscle in an intact muscle system.
FIGURE 3. CK-2017357 shifts the force-calcium relationship in fast skeletal muscle leftwards and amplifies the response of muscle to nervous input.
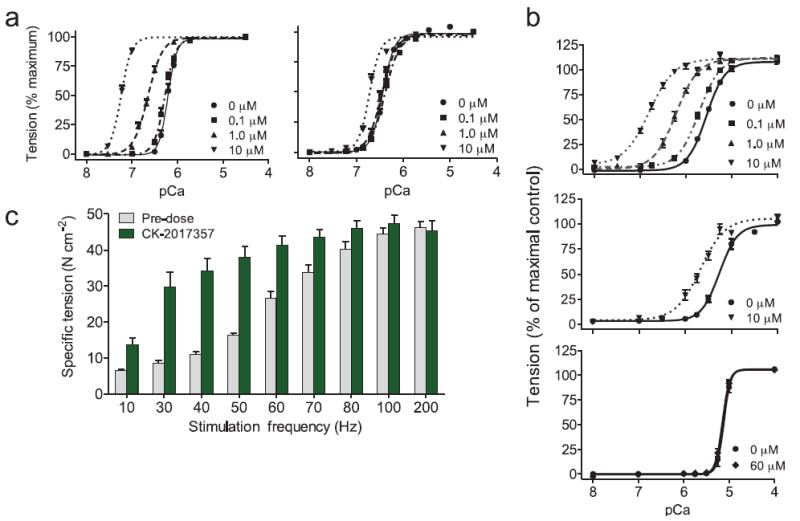
(a) The force-calcium relationship in human skinned fast (left) and slow (right) skeletal muscle fibers vs. calcium concentration at three concentrations of CK-2017357. Force measurements (each condition, n = 10) are displayed as a fraction of maximal force (Fo). (b) The force-calcium relationship of single skinned fibers from (top) rabbit fast skeletal psoas muscle fibers (n = 12), (middle) rat slow skeletal soleus muscle (n = 13), and (bottom) rat cardiac muscle fibers (n = 10) treated with CK-2017357. (c) The force-frequency relationship of rat EDL muscle in situ pre- and post-treatment with CK-2017357 (10 mg kg-1, administered into the gastrointestinal tract via a duodenal cannula to simulate oral administration, n = 6). For all graphs, mean ± SEM.
Given its effects on calcium sensitivity, CK-2017357 should result in an increase in the response of muscle to neuromuscular input. To test this hypothesis, we measured the response of living skeletal muscle to treatment with CK-2017357 using an in situ preparation of the extensor digitorum longus (EDL) muscle in the rat, where nerve and blood supply are intact18. In this fast fiber skeletal muscle, systemic infusion of CK-2017357 resulted in rapid, dose-dependent increases in muscle force (Supplementary Fig. 2). Examination of the force-frequency relationship demonstrated a substantial increase in muscle force at sub-maximal stimulation rates without increasing maximum tension at maximal tetanic stimulation rates (Fig. 3c). Tension in the absence of nerve stimulation did not change following treatment (data not shown). Thus, the change in the force-frequency relationship was consistent with the effect of CK-2017357 on the force-calcium relationship in skinned muscle fibers.
The leftward shift in the force-frequency relationship of fast skeletal muscle led us to test whether CK-2017357 could increase muscle force in a model of neuromuscular disease. Passive transfer experimental autoimmune myasthenia gravis (PT-EAMG) is a rat model of myasthenia gravis in which treatment with an inhibitory nicotinic acetylcholine receptor (nAChR) antibody leads to muscle weakness and fatigue19. Antibody treatment led to a substantial decline in grip strength (~50%), reaching a plateau 72 hours post-injection (Fig. 4a) that remained stable to 96 hours. Underlying this weakness was lower muscle force measured in situ across a wide range of nerve stimulation frequencies (Fig. 4b). In this model, administration of CK-2017357 rapidly increased muscle force in situ (Fig. 4c) and eliminated the decline in force produced by prolonged stimulation, so-called use dependent fatigue or sag (Fig. 4d,e).
FIGURE 4. CK-2017357 improves muscle and physical function in a model of neuromuscular disease.
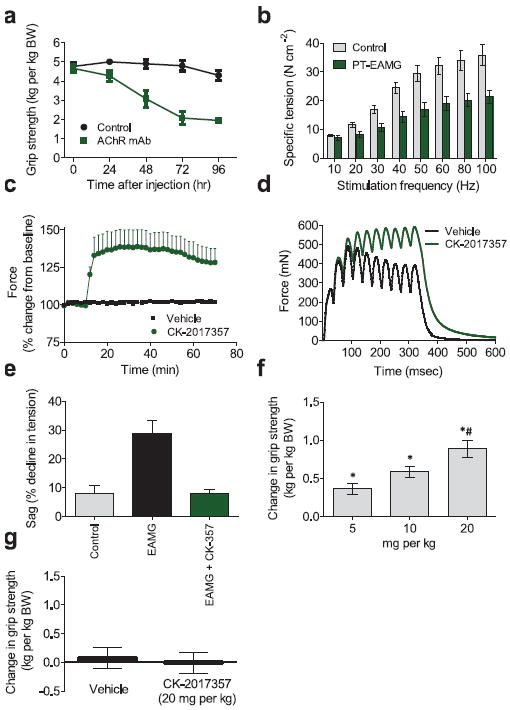
(a) Rat forelimb grip strength in PT-EAMG rats vs. time after administration of nAChR antibody α1/3/5 or phosphate buffered saline (control), n = 10 each group. (b) The force-frequency relationship of rat EDL muscle in situ 72-96 hours after intra-peritoneal injection (500 μg kg-1) of nAChRα1/3/5 antibody (PT-EAMG, n = 6) or phosphate buffered saline (control, n = 4). (c) In situ rat EDL peak muscle force vs. time after infusion of CK-2017357 (3 mg kg-1, n = 6) or vehicle (n = 4) in PT-EAMG rats. (d) In situ rat EDL muscle force vs. time for a single representative contraction (10 stimuli at 30 Hz) before and after administration of CK-2017357 (3 mg kg-1) in PT-EAMG rats. (e) Decline in muscle force (sag) under the same stimulation protocol in control rats (n = 4), PT-EAMG rats (n = 6), or PT-EAMG rats after infusion of CK-2017357 (n = 6). (f) Forelimb grip strength in PT-EAMG rats 60 minutes following a single oral dose of CK-2017357 (n = 20-22 in each dose group. * P < 0.001 vs. vehicle, # P < 0.01 vs. lower doses). (g) Rat forelimb grip strength change after oral administration of CK-2017357 (n = 14) or vehicle (n = 14) in healthy rats. For all graphs, mean ± SEM plotted.
We next tested whether these increases in the performance of individual, intact muscles could translate to enhanced physical performance in the same PT-EAMG model. Following randomization of the animals to two groups, CK-2017357 or vehicle was dosed orally in a blinded fashion at 72 hours followed by crossover to the opposite treatment at 96 hours with grip strength assessments at each time point. CK-2017357 treatment increased grip strength by up to 0.97 kg kg-1 body weight relative to placebo 60 minutes after dosing, representing more than a 50% increase in grip strength relative to baseline (Fig. 4f). Of note, control animals (no nAChR injection) did not show a change in grip strength with CK-2017357 (Fig. 4g) suggesting that increases in function in this setting required weakness produced by neuromuscular blockade.
CK-2017357 is a selective sensitizer of the fast skeletal troponin complex and a direct activator of skeletal muscle function. CK-2017357 may represent a therapeutic option for a range of serious neuromuscular disorders, by increasing muscle strength and reducing fatigability. Evidence also points to reduced efficiency of motorneuron excitation-contraction coupling in the sarcopenia of old age20-22, suggesting potentially broader applicability of this mechanism of action. Direct activators of the skeletal sarcomere, like the fast skeletal troponin activator, CK-2017357, may therefore hold promise in an array of patient conditions marked by muscle weakness.
METHODS
Materials
We used the method of Talmadge and Roy23 for glycerol SDS-PAGE analysis of myosin isoforms. We prepared rabbit fast skeletal myofibrils from psoas muscle13, slow skeletal myofibrils were prepared similarly from bovine masseter muscle12, and cardiac myofibrils from bovine heart11. Myofibrils were also prepared from bovine rectus abdominis muscle, a predominantly fast fiber muscle12. We purified myosin (as the S1 fragment produced by limited proteolytic treatment with chymotrypsin), actin, troponin and tropomyosin from rabbit psoas muscle for skeletal isoforms and bovine heart for cardiac isoforms24-27. We reconstituted versions of troponin-tropomyosin regulated actin-myosin according to published methods28,29. For in vitro skinned fiber studies, we prepared muscle as described in Claflin, et al30. Human skeletal muscle tissue was obtained by needle biopsy from the vastus lateralis muscles of volunteer subjects under a protocol reviewed and approved by the Institutional Review Boards of the University of Michigan Medical School. The Cytokinetics Institutional Animal Care and Use Committee reviewed and approved protocols related to rodent models. Fluorescent reagents were purchased from Molecular Probes. Cytokinetics synthesized CK-2017357 and stock solutions for in vitro testing prepared in DMSO. Final DMSO concentration in test solutions was 1-2%.
ATPase Assays
We measured steady state ATPase activity at 25°C using a pyruvate kinase and lactate dehydrogenase-coupled enzyme system31 and a SpectraMax plate reader (Molecular Devices) to monitor the change in absorbance as a function of time. The buffer used was 12 mM Pipes, 2 mM MgCl2, 1 mM DTT at pH 6.8, with 60 mM KCl added for fast and slow skeletal myofibrils.
Isothermal titration calorimetry
We did isothermal titration calorimetry experiments in a Micro-Cal VP-ITC microcalorimeter (General Electric, Inc.) using 50 μM rabbit fast skeletal troponin complex or purified recombinant rabbit fast TnC in Pipes buffer (12 mM K-Pipes pH 6.8, 100 mM KCl, 250 μM CaCl2, 5 mM β-mercaptoethanol and 3% DMSO). The reference chamber contained Pipes buffer and 3% DMSO. The ligand solution injected into the sample chamber contained CK-2017357 (300 μM). Ligand injections were made every 300 s (10 μL). To correct for the heats of dilutions of CK-2017357, we subtracted the stable heat signal from the injections near the end of the experiment from the remaining values and used the instrument software package for data collection and analysis using a single binding site model.
Measurement of calcium release from troponin
We measured calcium release from recombinant troponin C or the troponin complex using the fluorescent calcium chelator Quin-232 in a SF-61DX stopped-flow fluorimeter (TgK Scientific) with excitation provided by a monochromator (337 nm, 10 mm slit width) and emission measured through a glass filter (495 nm long pass). Calcium standard curves prepared in the presence of the equivalent concentrations of DMSO and CK-2017357 allowed for the translation of fluorescence intensities into calcium concentrations. Final reaction conditions were: 10 μM troponin C or troponin complex (preincubated with equimolar supplemental CaCl2), 1% DMSO (± 20 μM CK-2017357), 75 μM Quin-2, 12 mM K-Pipes pH 6.8, 2 mM MgCl2, 1 mM DTT, 25°C.
Muscle force measurements
We performed skinned fiber studies as described in Claflin, et al30. After attaching human fibers with 10-0 monofilament nylon, we measured force with a Model 403A force transducer and Model 322C servomotor (Aurora Scientific, Inc.) at a sarcomere length of ~2.7 μm. In human fiber studies, we distinguished fiber type based on shortening speed before measurement of isometric force30. Studies of rat and rabbit skinned fibers used a 400A force transducer (Aurora Scientific, Inc) and 5 % methylcellulose in acetone to attach fibers.
We measured in situ muscle force in the EDL muscle of rats as described by Brooks18 using an in situ muscle apparatus (Aurora Inc., Model 806C). Administration of CK-2017357 occurred via one of two different routes depending on experiment: intra-aortic delivery by a cannula advanced into the aorta via the contralateral femoral artery or duodenal delivery via an indwelling cannula in the duodenum (to simulate an oral dose which was not easily otherwise administered to animals under anesthesia).
Rodent Model of Passive Experimental Autoimmune Myasthenia Gravis (PT-EAMG)
We developed a rodent model (PT-EAMG) of myasthenia gravis based on the protocols described by V. A. Lennon et al19. Weakness developed following a single intra-peritoneal injection of 500 μg kg-1 of nAChRα1/3/5 antibody (SC-58604, Santa Cruz Biotechnology) as confirmed by forelimb grip measurements (Columbus Instruments). To make the study groups more homogeneous, we randomized animals with a 40-70% drop in grip strength from baseline at 72 hours into blinded vehicle (0.5% HPMC & 0.2% Tween-80) and compound treatment groups (CK-2017357 at 5, 10 and 20 mg/kg). Following baseline grip assessment, rats were dosed orally and grip strength measured at 60 minutes. The efficacy of the alternative treatment was examined 96 hours after antibody injection, in a crossover design.
Statistical Analysis
Based on the crossover design of the grip strength measurements in the PT-EAMG model, we performed the statistical analysis of change from baseline for grip strength between active treatment and vehicle using an analysis of covariance (ANCOVA) procedure with animals included in the model as a random effect, baseline grip strength as a covariate, and treatment, sequence, and period as fixed factors. Unless otherwise noted, values in text are reported as mean ± standard error.
Supplementary Material
Acknowledgments
The project described was supported by Cytokinetics, Inc. and by Award Number RC3NS070670 (F.I.M.) from the National Institute of Neurological Disorders And Stroke. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Neurological Disorders and Stroke or the National Institutes of Health.
Footnotes
AUTHORS CONTRIBUTIONS All authors contributed extensively to the work presented in this paper.
COMPETING FINANCIAL INTERESTS AJR, JJH, ACH, ARM, RK, LD, GG, KHL, DM, WFB, MMC, DC, SEC, MG, RH, ZJ, PL, HR, KGS, JS, VV, DJM, BPM, FIM are past or present employees of Cytokinetics Inc. and own stock or stock options in Cytokinetics. DLA and DRC received research support from Cytokinetics, Inc.
References
- 1.Classification of the neuromuscular disorders. Lancet. 1968;1:1020–1021. [PubMed] [Google Scholar]
- 2.Pollard T, Earnshaw W. Cell Biology. Saunders/Elsevier; Philadelphia: 2008. [Google Scholar]
- 3.Freund HJ. Motor unit and muscle activity in voluntary motor control. Physiol Rev. 1983;63:387–436. doi: 10.1152/physrev.1983.63.2.387. [DOI] [PubMed] [Google Scholar]
- 4.Jasmin BJ, Gardiner PF. Patterns of EMG activity of rat plantaris muscle during swimming and other locomotor activities. J Appl Physiol. 1987;63:713–718. doi: 10.1152/jappl.1987.63.2.713. [DOI] [PubMed] [Google Scholar]
- 5.Sharma KR, Miller RG. Electrical and mechanical properties of skeletal muscle underlying increased fatigue in patients with amyotrophic lateral sclerosis. Muscle Nerve. 1996;19:1391–1400. doi: 10.1002/(SICI)1097-4598(199611)19:11<1391::AID-MUS3>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 6.Kent-Braun JA, Miller RG. Central fatigue during isometric exercise in amyotrophic lateral sclerosis. Muscle Nerve. 2000;23:909–914. doi: 10.1002/(sici)1097-4598(200006)23:6<909::aid-mus10>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 7.Patzko A, Shy ME. Update on Charcot-Marie-Tooth disease. Curr Neurol Neurosci Rep. 2010;11:78–88. doi: 10.1007/s11910-010-0158-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Monani UR. Spinal muscular atrophy: a deficiency in a ubiquitous protein; a motor neuron-specific disease. Neuron. 2005;48:885–896. doi: 10.1016/j.neuron.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 9.Drachman DB. Myasthenia gravis. N Engl J Med. 1994;330:1797–1810. doi: 10.1056/NEJM199406233302507. [DOI] [PubMed] [Google Scholar]
- 10.Conti-Fine BM, Milani M, Kaminski HJ. Myasthenia gravis: past, present, and future. J Clin Invest. 2006;116:2843–2854. doi: 10.1172/JCI29894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Solaro RJ, Pang DC, Briggs FN. The purification of cardiac myofibrils with Triton X-100. Biochim Biophys Acta. 1971;245:259–262. doi: 10.1016/0005-2728(71)90033-8. [DOI] [PubMed] [Google Scholar]
- 12.Young OA, Davey CL. Electrophoretic analysis of proteins from single bovine muscle fibres. Biochem J. 1981;195:317–327. doi: 10.1042/bj1950317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herrmann C, Sleep J, Chaussepied P, Travers F, Barman T. A structural and kinetic study on myofibrils prevented from shortening by chemical cross-linking. Biochemistry. 1993;32:7255–7263. doi: 10.1021/bi00079a023. [DOI] [PubMed] [Google Scholar]
- 14.Bloemink MJ, Adamek N, Reggiani C, Geeves MA. Kinetic analysis of the slow skeletal myosin MHC-1 isoform from bovine masseter muscle. J Mol Biol. 2007;373:1184–1197. doi: 10.1016/j.jmb.2007.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malik FI, et al. Cardiac myosin activation: a potential therapeutic approach for systolic heart failure. Science. 2011;331:1439–1443. doi: 10.1126/science.1200113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosenfeld SS, Taylor EW. Kinetic studies of calcium and magnesium binding to troponin C. J Biol Chem. 1985;260:242–251. [PubMed] [Google Scholar]
- 17.Poole KJ, et al. A comparison of muscle thin filament models obtained from electron microscopy reconstructions and low-angle X-ray fibre diagrams from non-overlap muscle. J Struct Biol. 2006;155:273–284. doi: 10.1016/j.jsb.2006.02.020. [DOI] [PubMed] [Google Scholar]
- 18.Brooks SV, Faulkner JA. Contraction-induced injury: recovery of skeletal muscles in young and old mice. Am J Physiol. 1990;258:C436–442. doi: 10.1152/ajpcell.1990.258.3.C436. [DOI] [PubMed] [Google Scholar]
- 19.Lindstrom JM, Engel AG, Seybold ME, Lennon VA, Lambert EH. Pathological mechanisms in experimental autoimmune myasthenia gravis. II. Passive transfer of experimental autoimmune myasthenia gravis in rats with anti-acetylcholine receptor antibodies. J Exp Med. 1976;144:739–753. doi: 10.1084/jem.144.3.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jimenez-Moreno R, Wang ZM, Gerring RC, Delbono O. Sarcoplasmic reticulum Ca2+ release declines in muscle fibers from aging mice. Biophys J. 2008;94:3178–3188. doi: 10.1529/biophysj.107.118786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Erim Z, Beg MF, Burke DT, de Luca CJ. Effects of aging on motor-unit control properties. J Neurophysiol. 1999;82:2081–2091. doi: 10.1152/jn.1999.82.5.2081. [DOI] [PubMed] [Google Scholar]
- 22.Ling SM, Conwit RA, Ferrucci L, Metter EJ. Age-associated changes in motor unit physiology: observations from the Baltimore Longitudinal Study of Aging. Arch Phys Med Rehabil. 2009;90:1237–1240. doi: 10.1016/j.apmr.2008.09.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Talmadge RJ, Roy RR. Electrophoretic separation of rat skeletal muscle myosin heavy-chain isoforms. J Appl Physiol. 1993;75:2337–2340. doi: 10.1152/jappl.1993.75.5.2337. [DOI] [PubMed] [Google Scholar]
- 24.Margossian SS, Lowey S. Preparation of myosin and its subfragments from rabbit skeletal muscle. Methods Enzymol. 1982;85(Pt B):55–71. doi: 10.1016/0076-6879(82)85009-x. [DOI] [PubMed] [Google Scholar]
- 25.Pardee JD, Spudich JA. Purification of muscle actin. Methods Enzymol. 1982;85(Pt B):164–181. doi: 10.1016/0076-6879(82)85020-9. [DOI] [PubMed] [Google Scholar]
- 26.Potter JD. Preparation of troponin and its subunits. Methods Enzymol. 1982;85(Pt B):241–263. doi: 10.1016/0076-6879(82)85024-6. [DOI] [PubMed] [Google Scholar]
- 27.Smillie LB. Preparation and identification of alpha- and beta-tropomyosins. Methods Enzymol. 1982;85(Pt B):234–241. doi: 10.1016/0076-6879(82)85023-4. [DOI] [PubMed] [Google Scholar]
- 28.Ebashi S, Ebashi F, Kodama A. Troponin as the Ca++-receptive protein in the contractile system. J Biochem. 1967;62:137–138. doi: 10.1093/oxfordjournals.jbchem.a128628. [DOI] [PubMed] [Google Scholar]
- 29.Spudich JA, Watt S. The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J Biol Chem. 1971;246:4866–4871. [PubMed] [Google Scholar]
- 30.Claflin DR, et al. Effects of high- and low-velocity resistance training on the contractile properties of skeletal muscle fibers from young and older humans. Journal of Applied Physiology. 2011;111:1021–1030. doi: 10.1152/japplphysiol.01119.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De La Cruz EM, Ostap EM. Kinetic and equilibrium analysis of the myosin ATPase. Methods Enzymol. 2009;455:157–192. doi: 10.1016/S0076-6879(08)04206-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rosenfeld SS, Taylor EW. Kinetic studies of calcium binding to regulatory complexes from skeletal muscle. J Biol Chem. 1985;260:252–261. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.